
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular modeling and DFT studies on the antioxidant activity of Centaurea scoparia flavonoids and molecular dynamics simulation of their interaction with β-lactoglobulin†
Emadeldin M. Kamel a,
Albandari Bin-Ammarb,
Ashraf A. El-Bassuonya,
Mohammed M. Alanazic,
Ali Altharawid,
Ahmad F. Ahmedaef,
Ashwag S. Alanazig,
Al Mokhtar Lamsabhihi and
Ayman M. Mahmoud*jk
a,
Albandari Bin-Ammarb,
Ashraf A. El-Bassuonya,
Mohammed M. Alanazic,
Ali Altharawid,
Ahmad F. Ahmedaef,
Ashwag S. Alanazig,
Al Mokhtar Lamsabhihi and
Ayman M. Mahmoud*jk
aChemistry Department, Faculty of Science, Beni-Suef University, Beni-Suef 62514, Egypt
bDepartment of Clinical Nutrition, College of Applied Medical Sciences, University of Hail, Saudi Arabia
cDepartment of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia
dDepartment of Pharmaceutical Chemistry, College of Pharmacy, Prince Sattam Bin Abdulaziz University, Al-Kharj 11942, Saudi Arabia
eDepartment of Basic Medical Sciences, College of Medicine, Ajman University, Ajman 346, United Arab Emirates
fCenter of Medical and Bio-allied Health Sciences Research, Ajman University, Ajman 346, United Arab Emirates
gDepartment of Pharmaceutical Sciences, College of Pharmacy, Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia
hDepartamento de Química, Módulo 13, Universidad Autónoma de Madrid, Campus de Excelencia UAM-CSIC Cantoblanco, Madrid 28049, Spain
iInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, Madrid 28049, Spain
jPhysiology Division, Zoology Department, Faculty of Science, Beni-Suef University, Salah Salim St., Beni-Suef 62514, Egypt. E-mail: ayman.mahmoud@science.bsu.edu.eg
kDepartment of Life Sciences, Faculty of Science and Engineering, Manchester Metropolitan University, Manchester M1 5GD, UK
First published on 19th April 2023
Abstract
Plants of the genus Centaurea have been widely used as natural therapeutics in different countries. This study investigated the antioxidant–structure activity relationship of eight flavonoids isolated from Centaurea scoparia using DFT studies and in vitro radical scavenging and xanthine oxidase (XO) inhibition assays, and to correlate the theoretical values with the experimental findings. Docking analysis was carried out to explore the binding modes of the isolated phytochemicals with XO and bovine β-lactoglobulin (BLG). Interactions of the isolated compounds with BLG were studied using molecular dynamics (MD) simulations which revealed the involvement of hydrogen bonding. The root-mean-square deviation (RMSD) of BLG and BLG-flavonoid complexes reached equilibrium and fluctuated during the 10 ns MD simulations. The radius of gyration (Rg) and solvent accessible surface area (SASA) revealed that various systems were stabilized at approximately 2500 ps. In addition, the RMS fluctuations profile indicated that the ligand's active site exerted rigidity behavior during the simulation. The hydrogen atom transfer (HAT) and the energies of hydrogen abstractions were estimated by calculating the bond dissociation enthalpy (BDE) of O–H in gas phase and water. The isolated compounds showed radical scavenging and XO inhibitory activities along with binding affinity with XO as revealed in silico. The BDE was linked to the radical scavenging processes occurring in polar solvents. These processes are single electron transfer followed by proton transfer (SET-PT) and sequential proton loss electron transfer (SPLET). Our calculations indicated the agreement between the calculated results and the experimentally measured antioxidant activity of the flavonoids isolated from C. scoparia.
1. Introduction
The genus Centaurea (Family Asteraceae) comprises hundreds of species mainly distributed in Western Asia and the Middle East.1 Plants of this genus showed broad spectrum biological activities such as anti-microbial,2,3 anti-proliferative,4,5 antidiabetic,6–8 diuretic6 and antirheumatic.9 Centaurea scoparia (C. scoparia) is an annual or biannual plant distributed in the Eastern desert in Egypt.10 Previous phytochemical screening of C. scoparia afforded several phenolics and flavonoids, including 3′,4′-dihydroxy-(3′′,4′′-dihydro-3′′-hydroxy-4′′-acetoxy)-2′′,2′′-dimethylpyrano-(5′′,6′′:7,8)-flavone-3-O-β-D-gulcopyranoside, 3,3′,4′-trihydroxy- (3′′,4′′-dihydro-3′′,4′′-dihydroxy)-2′′,2′′-dimethylpyrano-(5′′,6′′:7,8)-flavone, cynaroside, apigetrin, centaureidin, oroxylin A, 5,7-dihydroxy-3′,4′,5′-trimethoxyflavone, atalantoflavone, 5-hydroxy-3′,4′,8-trimethoxy-2′′,2′′-dimethylpyrano (5′′,6′′:6,7)-flavone, 3′,4′,5,8-tetramethoxy-2′′,2′′-dimethylpyrano (5′′,6′′:6,7)-flavone,5 vanillin, luteolin, apigenin, cirsimaritin, hispidulin, (−)-matairesinol, salvigenin, (−)-arctigenin, and omega-hydroxypropioguaiacone.11 C. scoparia has also been shown to contain chlorinated and non-chlorinated guaianolides, including diain, janerin, cynaropicrin, deacylcynaropicrin,12,13 and other sesquiterpenes lactones.14,15 Despite the health promoting effects of flavonoids, their bioavailability is generally low and varies significantly between different classes and among members of a particular class.16 The mechanism explaining the transportation of flavonoids and their metabolites is still a matter of debate. Therefore, investigating their action mechanisms with a transport protein such as bovine β-lactoglobulin (BLG) would provide more trustworthy predictions to find tools for controlling their transport to the target active site. BLG is a major whey protein belonging to the lipocalin family. It consists of nine antiparallel β-sheets and one α-helix and has a molecular weight of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 Da. This protein was proven to show a high tendency towards hydrophobic ligands.17,18 As a result of being biocompatible and biodegradable, BLG is considered to be the main protein responsible for carrying the lipid-soluble drugs. Consequently, BLG would be an appropriate carrier for flavonoids and might enhance the solubility and bioavailability of these compounds.19 Given that BLG can influence various polyphenols and that its sensitization and stability are strongly influenced by binding to flavonoids, the study of their interaction is important.
400 Da. This protein was proven to show a high tendency towards hydrophobic ligands.17,18 As a result of being biocompatible and biodegradable, BLG is considered to be the main protein responsible for carrying the lipid-soluble drugs. Consequently, BLG would be an appropriate carrier for flavonoids and might enhance the solubility and bioavailability of these compounds.19 Given that BLG can influence various polyphenols and that its sensitization and stability are strongly influenced by binding to flavonoids, the study of their interaction is important.
Studies on the antioxidant activity of flavonoids revealed interesting biological activities and highlighted the structure–antioxidant activity relationship of these compounds. Molecular docking and molecular dynamics (MD) simulations are in silico techniques that foretell the particle interactions in a specific system in which the initial parameters and conditions are specified. MD tools are generally employed by researchers to figure out drug–enzyme interactions and reactivities. Given the tremendous development in DFT, the preferred method for examining electronic properties of flavonoids, theoretical studies have raised the curiosity for greater understanding of the mechanism of action of flavonoids as antioxidants. The antioxidant action of flavonoids strongly depends on its chemical structure as previously reported.20 Generally, the activity of flavonoids as antioxidants was agreed to proceed via one-step H-atom transfer (HAT), single-electron transfer followed by proton transfer (SET-PT) or sequential proton loss electron transfer (SPLET) mechanisms.21–23 In the HAT pathway, the radical accepts one hydrogen atom from a hydroxyl (OH) of the flavonoid to be stabilized and afford a comparatively stable phenoxy radical which may donate another hydrogen atom to form a stable quinoid structure.24 The homolytic bond dissociation enthalpy (BDE) of the OH group is the main parameter representing the HAT mechanism. BDE value is a good indicator for the flavonoid radical scavenging activity. A lower BDE value denotes a higher ability to loss hydrogen and consequently more facile radical scavenging action. According to the SET-PT mechanism, the flavonoid donates an electron to the radical forming Flav-OH˙+ and consequently the interaction of both Flav-O˙ and Flav-OH˙+ causes the stabilization of radicals and inhibition of the radical chain reaction.24 This electron transfer process is characterized by calculating the ionization potential (IP). Deprotonation of the previously formed flavonoid radical cation occurs after the single electron transfer process. This step can be described as the hydroxyl proton dissociation enthalpy (PDE). On the other hand, the SPLET mechanism proceeds via deprotonation of the flavonoids to afford a phenoxide anion in a first step depicting the anion proton affinity (PA). The next step is the electron transfer from the phenoxide anion that reveals the electron transfer enthalpy (ETE). Although the overall result of SET-PT and SPLET mechanisms is equivalent to HAT, these routes are thermodynamically favorable in polar media because of the charge separation. In addition, SET-PT and SPLET are preferred for radicals with higher electron affinities.25,26 Based on the output of these mechanisms, the structure-antioxidant activity relationship estimation of the isolated phenolics could be assessed by calculating BDE, IP, PDE and ETE.22,27
This study aimed to isolate and characterize flavonoids of C. scoparia aerial parts and to investigate their antioxidant activity. DFT calculations to compute the BDE values and compare the outputs with the experimental findings of the antioxidant activity of the isolated flavonoids were carried out. In addition, the radical scavenging mechanisms (HAT, SPLET and SET-PT) of the isolated flavonoids were characterized by calculating BDE, IP, PDE, PA and ETE. Furthermore, we assessed the interaction of BLG with the isolated flavonoids by molecular docking and MD simulations.
2. Experimental
2.1. Phytochemical investigation
A silica gel column (60 × 3 cm; 230–400 mesh) was employed for the chromatographic fractionation of fraction 3 using an n-hexane/acetone solvent system of increasing polarity (v/v = 50:1 to 0:1) to afford 34 sub-fractions (A1–A34). The sub-fractions A6–A14 were shown to possess similar TLC behavior and were collected together and further purified using silica gel (50 × 1.5 cm; 230–400 mesh) using the solvent mixture n-hexane:ethyl acetate (3:1) to afford 9 sub-fractions (R1–R9). The subfractions R4 and R5 were added on a top of a Sephadex LH-20 (30 × 1 cm) column to be purified using methanol as an eluent to give the pure compounds 5 (23 mg) and 6 (18 mg). Compound 1 (14 mg) was isolated from sub-fractions R8 and R9 by repeated silica gel column (60 × 3 cm; 230–400 mesh) using a mixture of hexane–dichloromethane-acetone of increasing polarity, and further applied to Sephadex LH-20 (30 × 1.5 cm) column using methanol. Fraction 5 was purified over a polyamide column (60 × 2 cm) and eluted with an ethanol/water system to yield ten sub-fractions (E1–E10). E2, E3 and E4 were shown to have similar TLC profiles, and consequently combined and placed over Sephadex LH-20 (35 × 2 cm) column using methanol to yield compounds 2 (21 mg) and 3 (17 mg). E6, E7 and E8 were combined and further chromatographed on reverse-phased column chromatography to afford 19 sub-fractions (S1–S19) using H2O/MeCN 9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 3:7, 2:8, 1:9 and 0:10. Sub-fractions S7–S15 were combined and subjected to radial chromatography using dichloromethane/ethyl acetate 9:1 (2 mm thickness) to produce compound 7 (23 mg). Fraction 6 was subjected to fractionation using silica gel (100 × 4 cm; 230–400 mesh) and then eluted using the solvent system chloroform/methanol mixture of increasing polarity (v/v = 50:1 to 0:1) to 24 sub-fractions (M1–M24). The sub-fractions M9–M19 were pooled and further chromatographed over silica gel column (60 × 2 cm; 230–400 mesh) using the mixture n-hexane/ethyl acetate 8.5:1.5 to afford 15 sub-fractions (C1–C15). C7 and C8 were collected and applied to Sephadex LH-20 column (20 × 1 cm) be means of the eluent methanol to afford the purified flavonoids 4 (16 mg) and 8 (15 mg).
2.2. DPPH radical scavenging activity
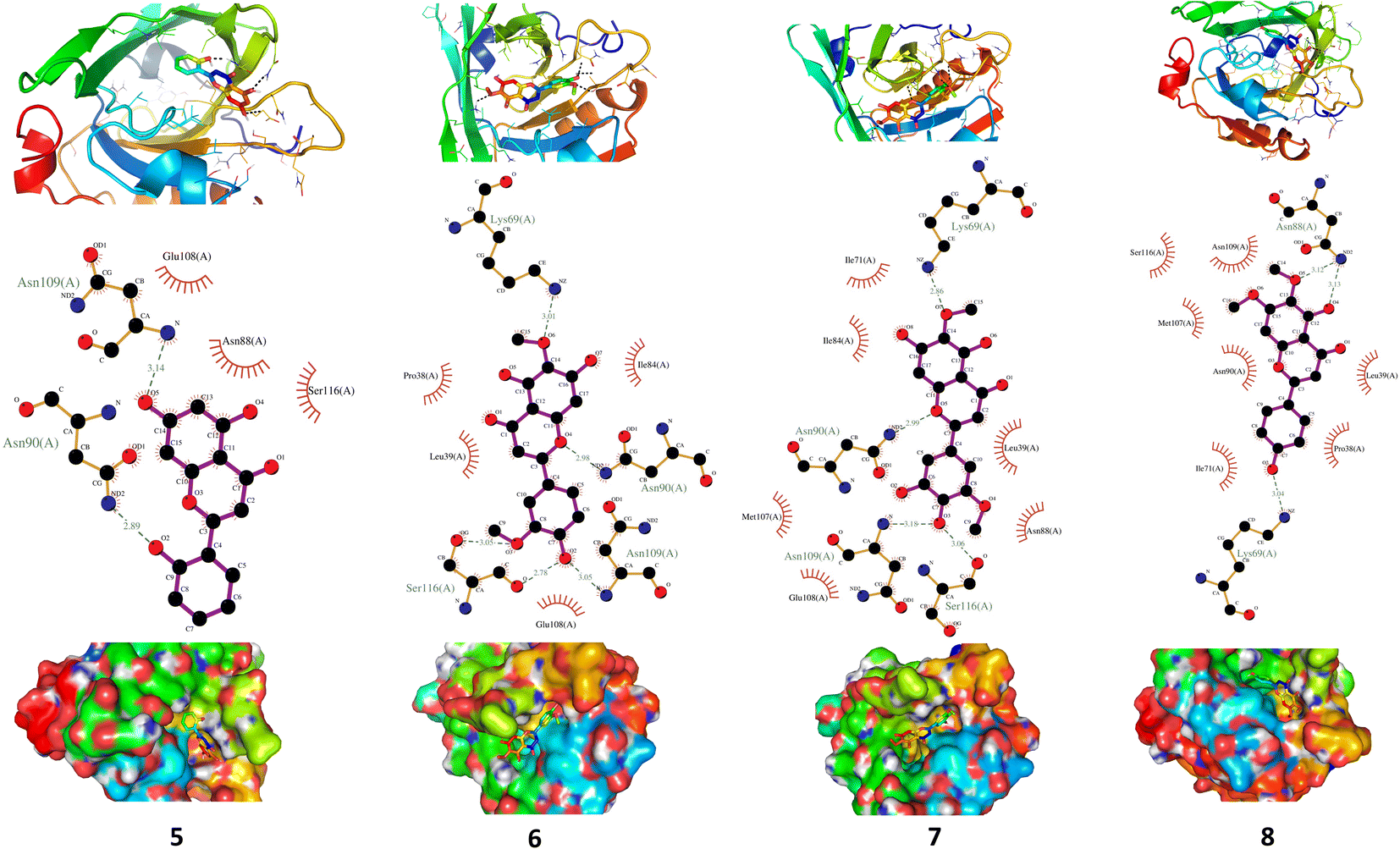
The antioxidant activity of the isolated flavonoids was evaluated by the DPPH free radical-scavenging assay as previously described28 with slight modifications. Different concentrations (1, 3, 10, 30 and 100 μg ml−1) of the isolated flavonoids were added to 0.08 mM DPPH. solution in ethanol and the mixture was actively stirred and incubated at room temperature for 30 min. Rutin was used as a standard antioxidant and the absorbance was recorded spectrophotometrically at 570 nm. The DPPH radical scavenging activity was calculated using the following equation: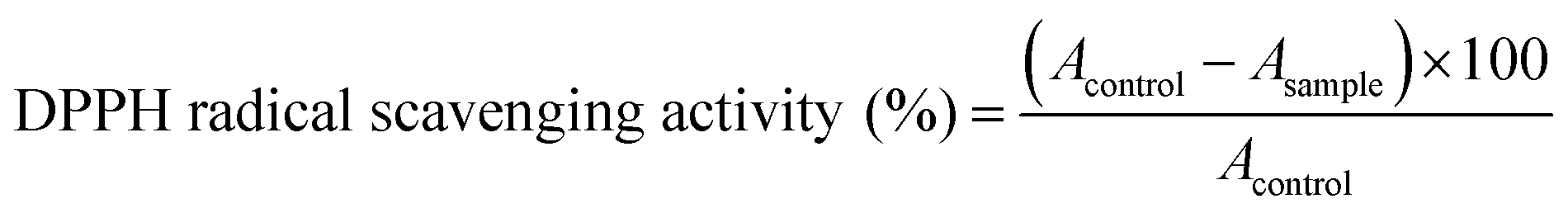
2.3. Assay of xanthine oxidase (XO) inhibitory activity
XO inhibitory activity of C. scoparia flavonoids was determined according to the method described by Özyürek et al.29 Different concentrations (10–200 μg ml−1) the compounds and AP were mixed with 0.5 mM xanthine, 50 mM sodium phosphate buffer, and XO, and the mixture was kept at 37 °C for 30 min. The reaction was stopped using 3.2% perchloric acid. 0.2 ml of the reaction mixture was mixed with 0.2 ml copper(II) chloride dihydrate (10 mM), 0.2 ml neocuproine (7.5 mM) and 0.4 ml ammonium acetate (1 M), and the absorbance was measured after 30 min at 450 nm.2.4. Computational details
3. Results and discussion
3.1. Phytochemical investigation
The dichloromethane extract of C. scoparia aerial parts was subjected to intensive chromatographic fractionation to afford eight known flavonoids reported for the first time in this species. The chemical structure of isolated phytochemicals (Fig. 1) was elucidated according to data obtained from spectroscopic tools (MS, UV, 1D and 2D NMR) (ESI Fig. S1–S16†). By comparison with data in the literature, the isolated compounds were identified as 3′-geranyl-5,7,4′-trihydroxyisoflavone (1),54,55 6-prenyl-5,7,4′-trihydroxyisoflavone (2),56,57 3′-geranyl-5,7,2′,4′-tetrahydroxyisoflavone (3),58 6-prenyl-3,5,7,4′-tetrahydroxyflavone (4),59 5,7,2′-trihydroxyflavone (5),60,61 5,7,4′-trihydroxy-6,3′-dimethoxyflavone (6),62 5,7,4′,5′-tetrahydroxy-6,3′-dimethoxyflavone (7)63 and 5,4′-dihydroxy-6,7-dimethoxyflavone (8).64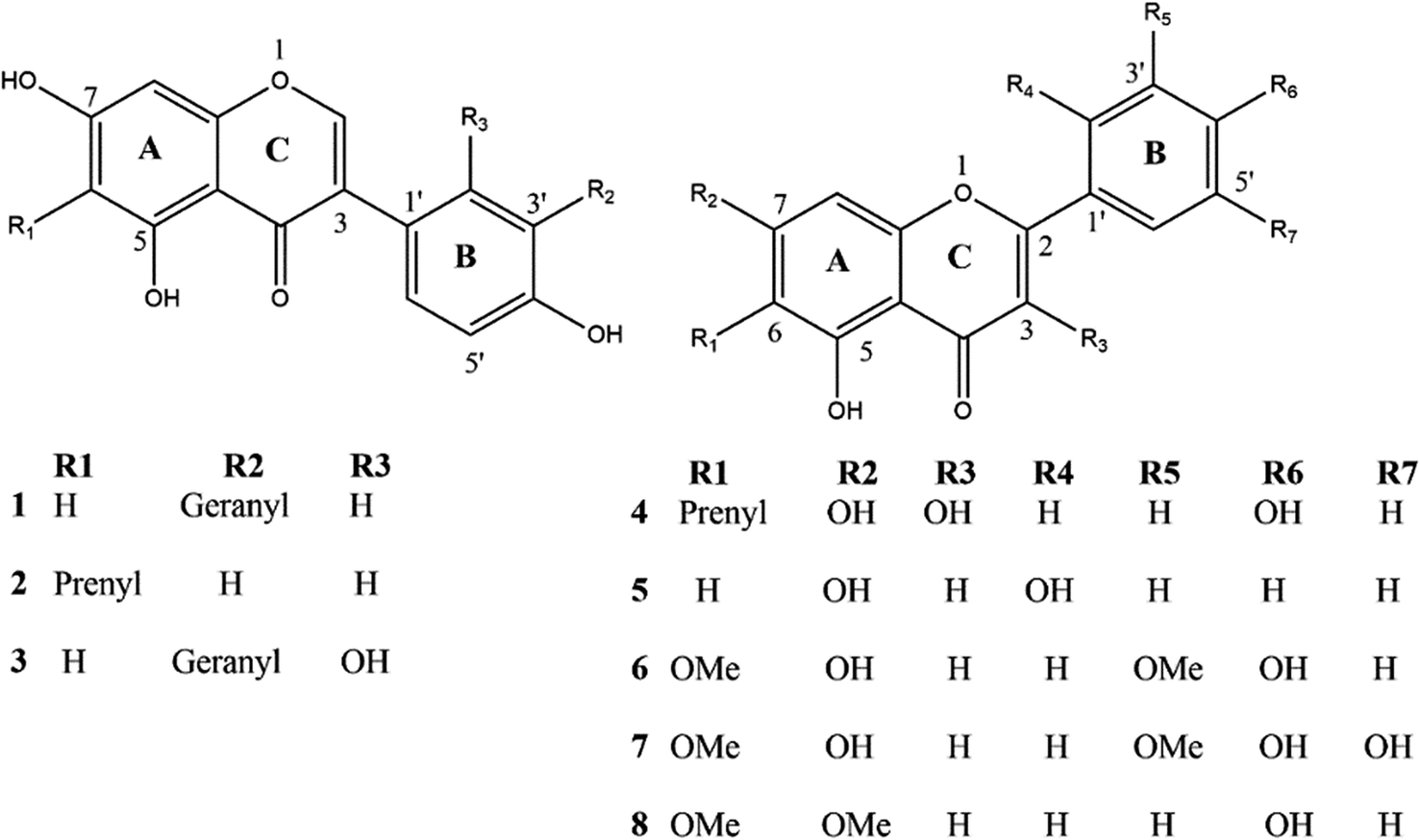 | ||
| Fig. 1 Chemical structure of the isolated compounds. | ||
3.2. The binding modes of C. scoparia flavonoids with BLG
We carried out molecular docking investigations to assess the potential binding affinity of C. scoparia flavonoids (1–8) with BLG. The binding affinities, polar bonding, and probable hydrophobic interactions of the isolated phytochemicals with BLG are represented in Table 1, and the binding modes are represented in Fig. 2 and 3.| Flavonoid | Binding affinity (kcal mol−1) | Polar bonds | Hydrophobic interactions |
|---|---|---|---|
| 1 | −7.8 | Lys69, Asn109 and Ser116 | Pro38, Ile71, Ile84, Ala86 and Asn90 |
| 2 | −6.9 | Ser116 | Leu39, Lys69 Ile84, Asp85 Ala86, Asn90 and Leu117 |
| 3 | −7.8 | Asn109 | Pro38, Leu39, Val41, Val43, Ile56, Leu59, Lys69, Ile71, Asn88, Val92, Phe105, Met107 and Ser116 |
| 4 | −6.9 | Lys69 | Leu31, Leu39, Ile71, Ile84, Asn90 and Asn109 |
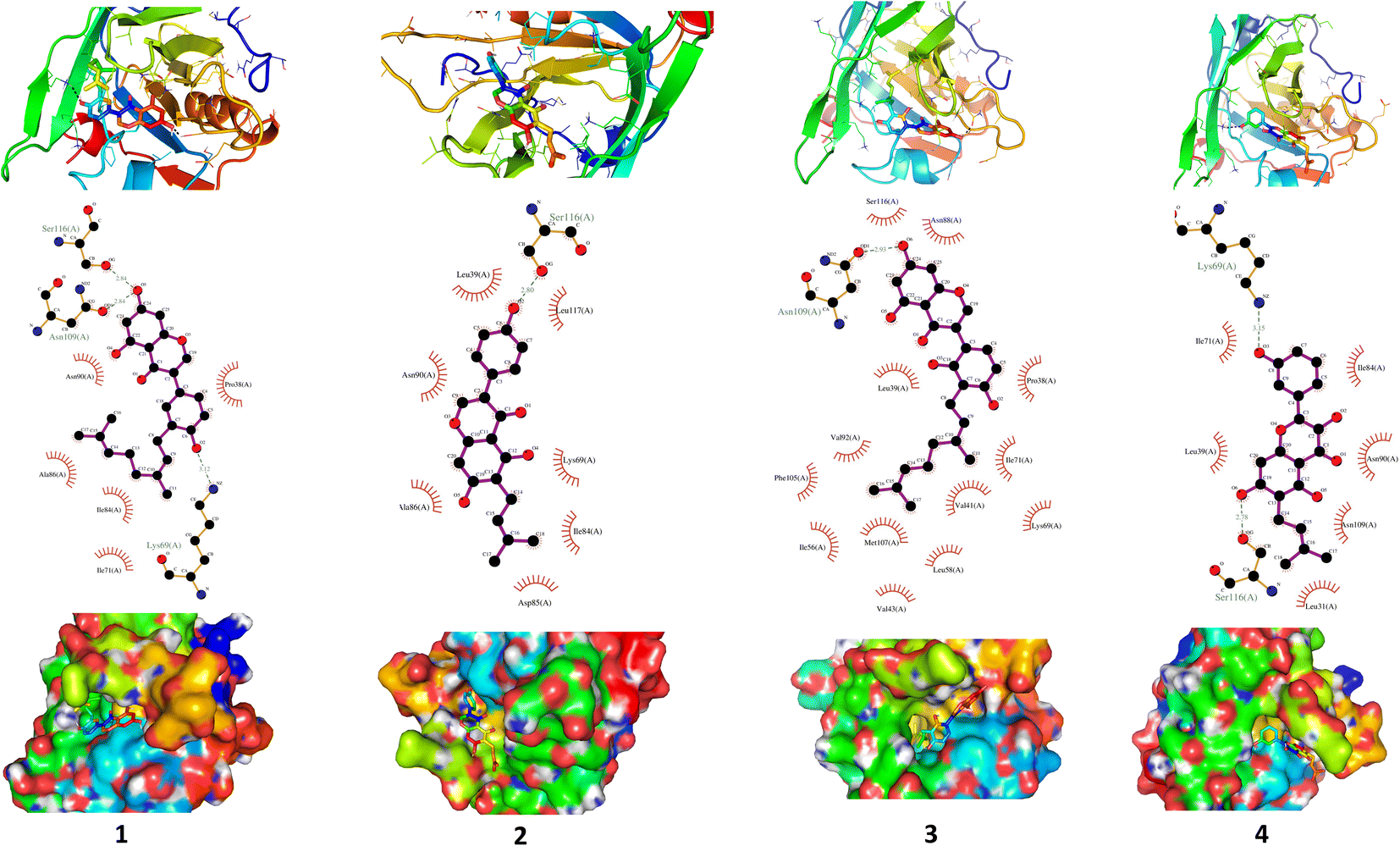
| 5 | −7.5 | Asn90 and Asn109 | Asn88, Glu108 and Ser116 |
| 6 | −7.5 | Lys69, Asn90, Asn109 and Ser116 | Pro38, Leu39, Ile84 and Glu108 |
| 7 | −7.6 | Lys69, Asn90, Asn109 and Ser116 | Leu39, Ile71, Ile84, Asn88, Met107 and Glu108 |
| 8 | −7.1 | Lys69 and Asn88 | Pro38, Leu39, Ile71, Asn90, Met107, Asn109 and Ser116 |
 | ||
| Fig. 2 Binding interactions of compounds (1–4) with BLG. The tube model shows the active site of the drug–protein complex, and the interacting residues are represented as stick model (up). Surface representation of the binding pocket occupied by drugs under investigation (down). The active site interacting polar and hydrophobic residues are shown in the middle. | ||
 | ||
| Fig. 3 Binding profile of compounds (5–8) with BLG. The drug–protein complex binding sites are introduced as tube model, while residues are depicted as stick model (up). Surface representation of BLG binding pocket occupied by drugs under assessment (down). The residues exerting polar and hydrophobic interactions with the drugs are represented in the middle. | ||
The stability of geometrical conformation of biological macromolecules is believed to be dependent on ligand-protein polar and hydrophobic interactions. Polar bonding represents the main route for the binding of drugs at the binding cavity of a specific protein. Consequently, these hydrogen bonds effectively participate in ligand's alliance and correlation to the target active site.65 The contribution of these polar interactions to the binding affinity depends mainly on desolvation power and the newly generated hydrogen bonds. The hydrophobic interactions significantly affect the binding affinities of drug's lipophilic surfaces to the binding pocket hydrophobic areas. Thus, an adequate geometrical correlation of the ligand to protein active site is crucial for a thermodynamically favorable drug–target interaction.
The output of our docking studies revealed the affinity of the isolated phytochemicals towards BLG, and thus reflected a high probability of these compounds to from stable drug–protein complexes. This inference could be attributed to the resulting low binding energies of these flavonoids, ranging from −6.9 to −7.8 kcal mol−1. These low binding interaction energies reflected a high probability of employing these flavonoids as controlling factor for BLG transport function to biological active sites. All isolated phytochemicals occupied the same active site with common amino acids residues. The dense network of polar bonds for all flavonoids confirms their compatibility to BLG active site. Previously reported studies on BLG revealed the availability of three active sites with the central cavity as the essential binding pocket for hydrophobic drugs, and this cavity represents the favorable binding site for flavonoids. Our docking results manifested the binding of isolated flavonoids to the entrance of the internal cavity with both polar and hydrophobic interactions present with considerable existence for the hydrophobic bonding. In general, the molecular modelling analysis of the isolated flavonoid-BLG pattern could provide trustworthy foretelling of the conceivable routes for the production of BLG binding inhibitors and activators.
3.3. MD simulations studies
The interaction of BLG with the isolated flavonoids was evaluated by carrying out MD simulations for BLG and BLG-flavonoids complexes with extensive analysis of MD trajectories along with the hydrogen bonding profile. The determined parameters included root mean square deviations (RMSD), radius of gyration (Rg), solvent accessible surface area (SASA), and root mean square fluctuation (RMSF). The interaction of the isolated compounds with residues in the binding cavity of BLG was monitored along the 10 ns MD simulation run through the assessment of the hydrogen bonding patterns of various drug–protein interaction (Fig. 4). The polar bonding profile of compounds 1, 3, 4 and 6 displayed two hydrogen bonds with different interacting residues in the BLG binding site, one hydrogen bond is significantly stable and the other is relatively weak during the course of 10 ns MD simulations run. Three hydrogen bonds were detected in compounds 2 and 7 complexes, among them two are strong for compound 2 and the third is weak while in compound 7 only one hydrogen bond is stronger. Compounds 5 and 8 form four hydrogen bonds, two are strong and the remaining are weak.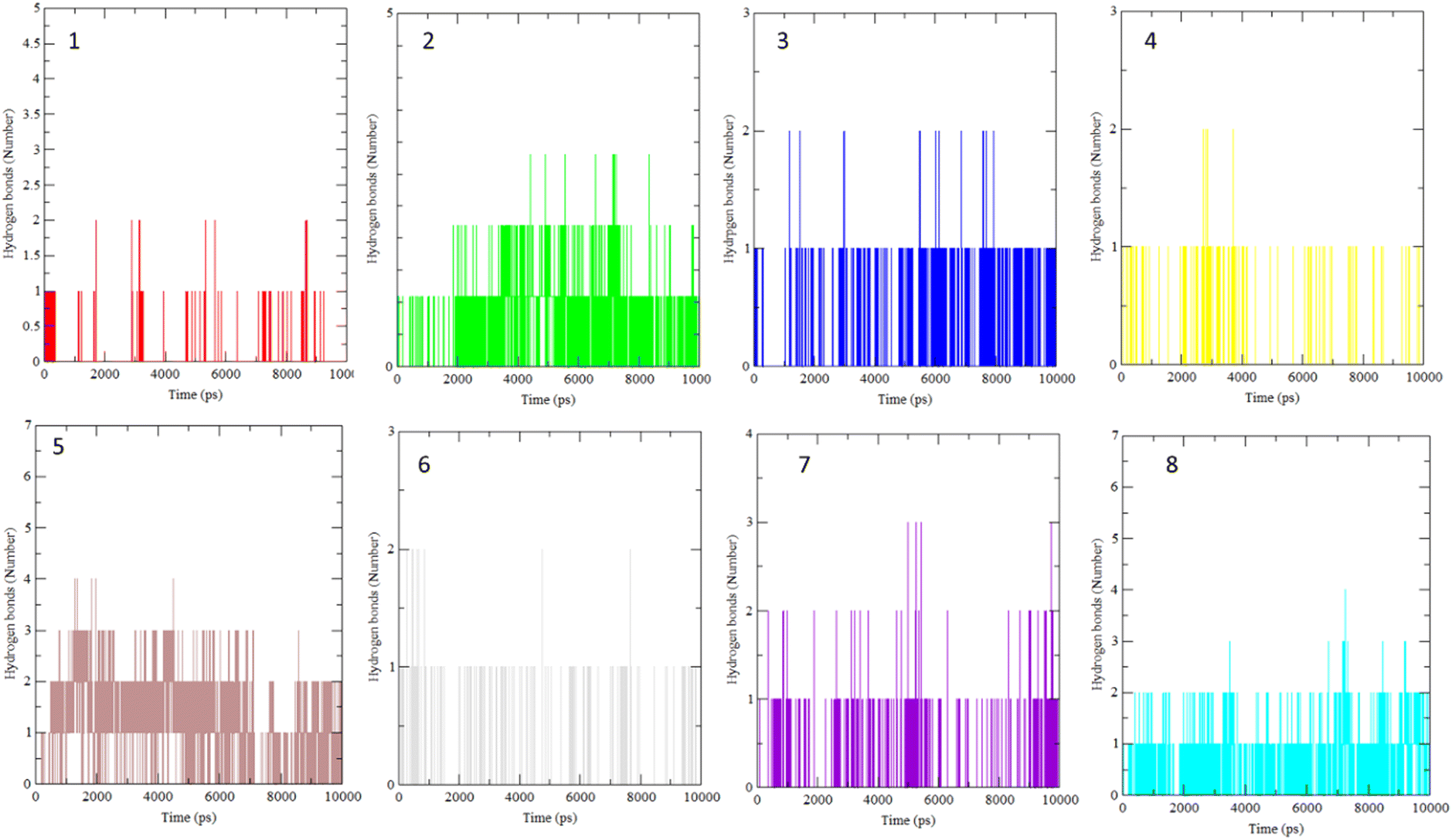 | ||
| Fig. 4 Overall intensity of hydrogen bonds after 10 ns MD simulations for the BLG-flavonoids complexes (1–8). | ||
The stability and integrity of trajectories through the whole 10 ns MD simulations were emphasized by studying the RMSD of backbone atoms of BLG against time for the target protein as standard and for various drug–protein complexes (1–8), as represented in Fig. 5A. From this plot, RMSD of BLG and BLG-flavonoid complexes achieved equilibrium and oscillated all over the median value after nearly 2.5 ns and kept with minimum energy till the end of the MD simulation run. Thus, the protein backbone atoms RMSD demonstrate comparable patterns of all structures, reflecting their stability and equilibrium for most of the time (between 2.5 and 10 ns). Reduced values of RMSD of BLG-flavonoid complexes indicate that the bonding between flavonoids and BLG minimized the degrees of freedom of the target protein dynamic. In addition, the obtained values of radius of gyration (Rg) of the standard BLG and BLG–flavonoid complexes were plotted as a function of time, as shown in Fig. 5B. The tightness of the protein skeleton along the folding and unfolding capacities during the 10 ns MD run were estimated from the obtained thermodynamically based Rg values. The Rg values attained a stabilized pattern at approximately 2500 ps in different systems, suggesting that the simulation reached equilibrium after this time. As shown in Fig. 5B, the compactness of the protein skeleton along the simulation was estimated because of the decrease in Rg values of the backbone atoms. The initial Rg value of BLG and BLG-flavonoid complexes was close to 1.44 nm, which agrees with the previously published experimental data. Furthermore, Fig. 5B demonstrates that the Rg estimation of BLG is not affected by the forming complexes with flavonoids. This implied that the nature of BLG was not altered during binding to ligands.
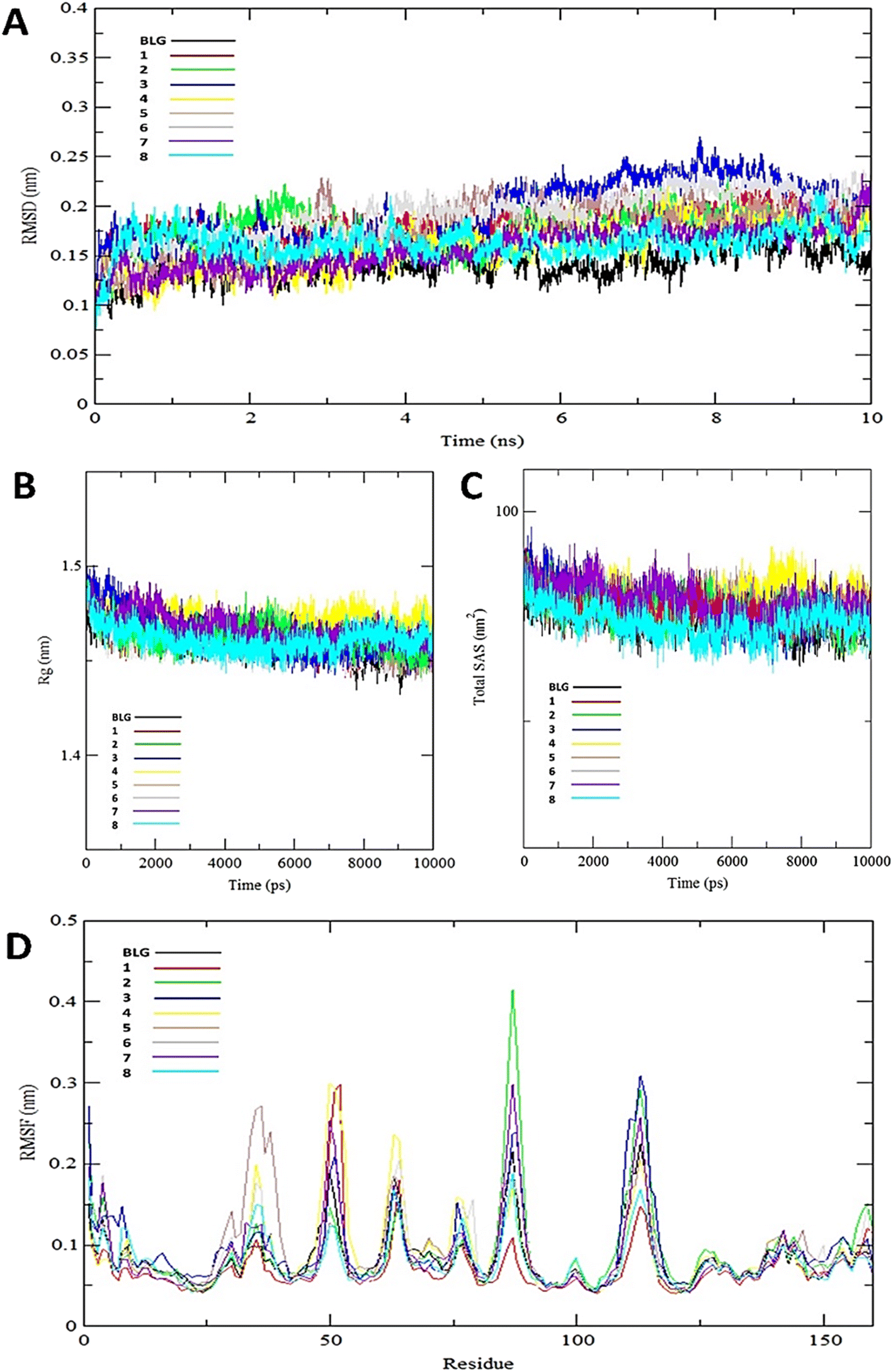 | ||
| Fig. 5 Various backbone RMSD patterns of BLG in free form and BLG-flavonoids complexes during 10 ns MD simulation (A), radius of gyration (Rg) of the backbone of BLG and BLG-flavonoids complexes (B), total solvent accessible surface (SAS) of BLG and BLG-flavonoids complexes (C), and root mean square fluctuation (RMSF) values of BLG and BLG-flavonoids complexes (D). | ||
The total solvent accessible surface areas (SASA) were studied for the protein in free and liganded form (1–8) during the 10 ns MD simulations as shown in Fig. 5C. Interestingly, the divergence behaviors of SASA in free BLG and BLG-liganded complexes (1–8) resemble the alteration in Rg values. This variation confirmed the accuracy and precision of the outcomes of our MD calculations.
A detailed analysis of the RMSF profile of free BLG and BLG-flavonoid complexes (1–8) was performed, and the assessed findings manifested the fluctuations of interacting residues in the free and liganded forms (Fig. 5D). The obtained plot was developed as a function of the number of interacting residues according to the 10 ns trajectory. Fluctuations at the binding site of BLG were detected in the range of 0.04 to 0.05 nm in the obtained RMSF profile. Contrary to BLG in its free form, no considerable fluctuations were detected at the drug's binding cavity in various complexes (Fig. 5D). Consequently, the resulted data is obviously pointed out to the fact that amino acids at the active site in the main central channel show minimized fluctuations for each flavonoid. Thus, the active site of isolated drugs architecture is mainly exhibiting an almost rigid behavior along the MD simulation run. The interaction between the target BLG and isolated flavonoids is represented in Fig. 6.
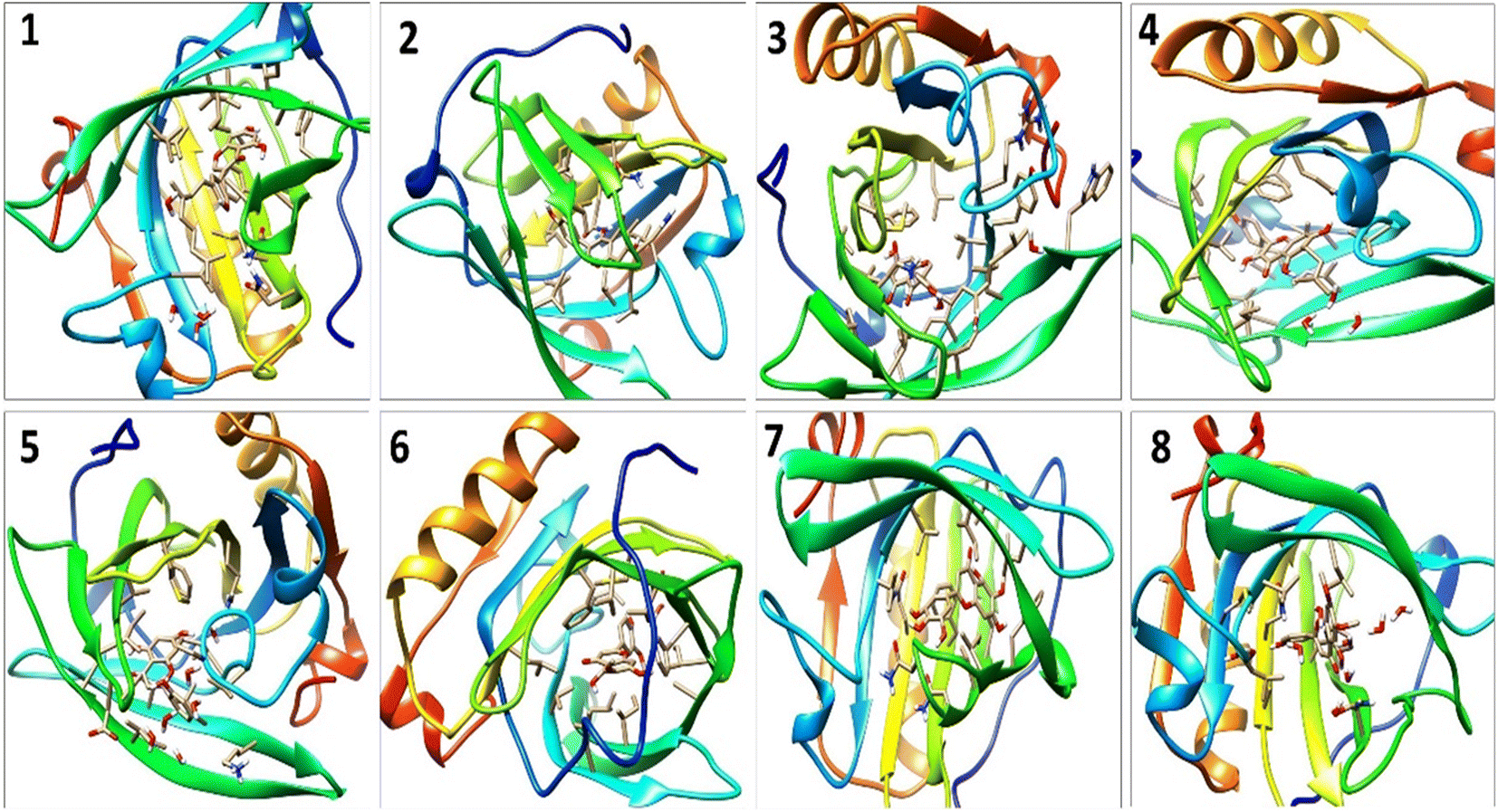 | ||
| Fig. 6 The binding modes of isolated flavonoids in the active site of BLG as estimated by MD simulations. | ||
3.4. DFT studies on the structure–antioxidant activity relationship
Free radicals are oxidant species with well-acknowledged deleterious effects on the cells. The adverse influence, particularly the cellular damage, caused by free radicals on the human body is alleviated by the antioxidant defense system. Incompetence of the antioxidant defense system is compensated by outside source antioxidants. Natural antioxidant can be found in food, and the vast majority exist in the form of flavonoids.66 Flavonoids are naturally occurring antioxidant phenolics with a potent ability to contend free radicals via chelating, scavenging, deactivating prooxidant enzymes, and activating detoxification enzymes.67 It has been depicted that the ability of flavonoids to act as antioxidants is determined by its activity toward scavenging free radicals.68 The ease of H-abstraction is agreed to be the leading factor controlling the antioxidant activity of flavonoids.23 Therefore, three conceivable mechanisms (HAT, SET-PT and SPLET) are proposed for the radical scavenging activities of the isolated flavonoids (1–8). Thus, the O–H bond dissociation may proceed via any of the three mechanisms. However, the three free radical scavenging mechanisms can coexist depending on the thermodynamic equilibrium existing through their different steps.25The isolated flavonoids, including isoflavones and flavones, are featured by the common carbon skeleton of flavonoids of rings A, B and C. Even though the initial geometries of the optimized structures of the isolated phytochemicals were estimated according to stereochemical data obtained from optical rotation and spectroscopic data, a large number of conformations for all isolated flavonoids was examined by random alteration of the dihedral angle C2′–C1′–C2–C3 and that of the geranyl and prenyl groups to detect the most stable conformation for each compound. Results of these calculations revealed that variation of C2′–C1′–C2–C3 angles in some of the isolated flavonoids resulted in quite meager influence on the final optimized geometries of our compounds. All isolated compounds are characterized by OH groups at positions 4'-, 5- and 7- except for compounds 5 and 8, where the OH group at positions 4′ is absent and OH at position 2′ is present instead in compound 5. Meanwhile in compound 8 a methoxy group is located at position 7- instead of the OH group. The OH group at position 3- appears only in compound 4, while position 3′ is occupied by OH in compound 7. Three free radical intermediates are resulted from the H-abstraction process from each hydroxyl in compounds 1, 2, 5 and 6, while in compounds 3, 4 and 7 four radical species are produced and only two radicals are resulted from the H-abstraction in compound 8.
The output of DFT calculations of the BDE and reaction enthalpies (kcal mol−1 at 298.15 K) for each active OH group (minimum BDE) for eight flavonoids from C. scoparia in gas and aqueous phases are represented in Tables 2–4, respectively. Interestingly, the radicals obtained by H-abstraction from position 4ꞌ are the most stable and represent the active site in all isolated flavonoids except compounds 3 and 5 in gas phase and compounds 3, 4 and 5 in water solution. The variation of the active site in compound 4 between positions 4′ in gas phase and 3 in water is obviously due to hydrogen bonding. In water, the hydrogen bonding between OH at position 3 and the carbonyl oxygen is attenuated by the hydrogen bonding with water molecules. Consequently, the ease of H-abstraction from position 3 is increased in water. The majority of 4′-Flav-O˙ possessing stabilities are higher than their rivals because of the hydrogen bonding with the neighboring oxo moiety in most cases. The data shown in Table 2 revealed that the homolytic hydroxyl bond dissociation is the thermodynamically prevailing mechanism for the radical scavenging activity. Compounds 7 and 6 showed the lowest BDE values and consequently they are considered the most active antioxidant agents among all isolated flavonoids. This notion was supported by the coincided results obtained by the DPPH assay in which these compounds possessed the lowest IC50 values. Contrary to the small discrimination in BDE values of the isolated flavonoids, there is a great variation in IC50 values which might be attributed to the environment that plays a crucial role in the antioxidant potential of the compounds under investigation. By checking the results of the calculations in water (Table 4) of the reaction enthalpies of the eight compounds, the homolytic bond dissociation mechanism seemed the thermodynamically less favorable one when compared with the SPLET Pathway. Generally, the energy requirements of the initial step in SPLET route (PA) are lower than that needed for the homolytic bond dissociation, and consequently the SPLET route is the predominant pathway for the radical scavenging activities of isolated flavonoids in water solution. Furthermore, a plot of the IC50 values of the isolated compounds versus overall energy demand of the SPLET mechanism afforded a linear relationship with a square correlation value of 0.962 (Fig. 7A).
| Flavonoid | Phase | 4′-OH | 5-OH | 7-OH | 3-OH | 3′-OH | 2′-OH |
|---|---|---|---|---|---|---|---|
| 1 | Gas | 79.63 | 98.52 | 88.02 | — | — | — |
| Water | 79.21 | 92.71 | 87.29 | — | — | — | |
| 2 | Gas | 81.42 | 95.89 | 85.47 | — | — | — |
| Water | 80.17 | 90.88 | 85.03 | — | — | — | |
| 3 | Gas | 78.37 | 97.93 | 89.19 | — | — | 76.52 |
| Water | 78.32 | 92.11 | 88.67 | — | — | 76.78 | |
| 4 | Gas | 80.06 | 91.36 | 83.74 | 80.23 | — | — |
| Water | 79.19 | 86.35 | 82.58 | 75.89 | — | — | |
| 5 | Gas | — | 105.92 | 87.51 | — | — | 83.3 |
| Water | — | 104.13 | 87.35 | — | — | 82.69 | |
| 6 | Gas | 68.74 | 78.75 | 70.94 | — | — | — |
| Water | 70.32 | 77.94 | 74.26 | — | — | — | |
| 7 | Gas | 60.66 | 78.12 | 80.59 | — | 70.11 | — |
| Water | 63.95 | 77.33 | 81.23 | — | 70.94 | — | |
| 8 | Gas | 82.64 | 91.01 | — | — | — | — |
| Water | 82.54 | 86.28 | — | — | — | — |
| Flavonoid | Active site | SPLET | SET-PT | BDE | RSA | ||||
|---|---|---|---|---|---|---|---|---|---|
| PA | ETE | PA + ETE | IP | PDE | IP + PDE | IC50 | |||
| a SPLET, sequential proton loss electron transfer; SET-PT, single electron transfer followed by proton transfer; BDE, bond dissociation enthalpy; RSA, radical scavenging activity; PA, proton affinity; ETE, electron transfer enthalpy; IP, ionization potential; PDE, proton dissociation enthalpy. | |||||||||
| 1 | 4′-OH | 333.93 | 61.56 | 395.49 | 164.07 | 231.42 | 395.49 | 79.63 | 19.54 |
| 2 | 4′-OH | 337.21 | 60.07 | 397.28 | 167.36 | 229.92 | 397.28 | 81.42 | 22.38 |
| 3 | 2′-OH | 328.15 | 64.82 | 392.97 | 162.91 | 230.06 | 392.97 | 76.52 | 15.83 |
| 4 | 4′-OH | 326.20 | 70.32 | 396.52 | 165.13 | 231.39 | 396.52 | 80.06 | 12.27 |
| 5 | 2′-OH | 327.09 | 72.07 | 399.16 | 178.21 | 220.95 | 399.16 | 83.30 | 25.93 |
| 6 | 4′-OH | 313.35 | 71.25 | 384.60 | 153.72 | 230.88 | 384.60 | 68.74 | 5.42 |
| 7 | 4′-OH | 259.38 | 117.73 | 377.11 | 169.13 | 207.98 | 377.11 | 60.66 | 3.73 |
| 8 | 4′-OH | 274.38 | 124.12 | 398.50 | 156.70 | 241.80 | 398.50 | 82.64 | 24.11 |
| Flavonoid | Active site | SPLET | SET-PT | BDE | RSA | ||||
|---|---|---|---|---|---|---|---|---|---|
| PA | ETE | PA + ETE | IP | PDE | IP + PDE | IC50 | |||
| a SPLET, sequential proton loss electron transfer; SET-PT, single electron transfer followed by proton transfer; BDE, bond dissociation enthalpy; RSA, radical scavenging activity; PA, proton affinity; ETE, electron transfer enthalpy; IP, ionization potential; PDE, proton dissociation enthalpy. | |||||||||
| 1 | 4′-OH | 32.57 | 45.79 | 78.36 | 79.89 | −1.53 | 78.36 | 79.21 | 19.54 |
| 2 | 4′-OH | 31.31 | 48.00 | 79.31 | 80.74 | −1.43 | 79.31 | 80.17 | 22.38 |
| 3 | 2′-OH | 29.08 | 47.43 | 76.51 | 78.11 | −1.60 | 76.51 | 76.78 | 15.83 |
| 4 | 3-OH | 29.07 | 46.55 | 75.62 | 77.71 | −2.09 | 75.62 | 75.89 | 12.27 |
| 5 | 2′-OH | 26.56 | 55.27 | 81.83 | 89.20 | −7.37 | 81.83 | 82.69 | 25.93 |
| 6 | 4′-OH | 16.83 | 52.64 | 69.47 | 70.80 | −1.33 | 69.47 | 70.32 | 5.42 |
| 7 | 4′-OH | 12.36 | 51.32 | 63.68 | 81.57 | −17.89 | 63.68 | 63.95 | 3.73 |
| 8 | 4′-OH | 24.40 | 57.28 | 81.68 | 72.76 | 8.92 | 81.68 | 82.54 | 24.11 |
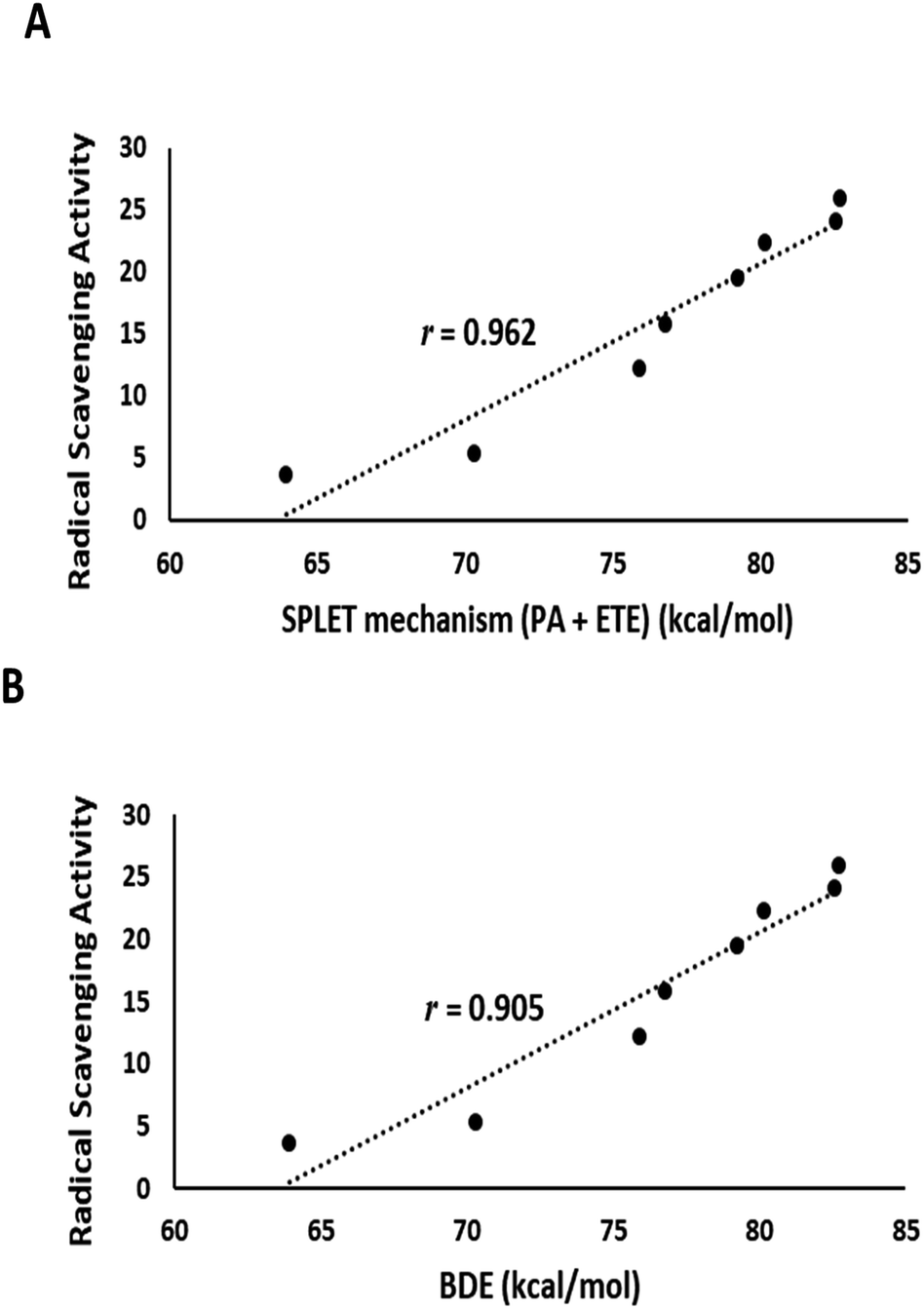 | ||
| Fig. 7 (A) Plot of the experimental DPPH assay values against total energy demand for the SPLET mechanism in water solution. (B) A plot of the experimental DPPH assay values against gas phase BDE. | ||
Data in Table 2 represents the estimated BDE (in kcal mol−1 at 298.15 K) in gas and aqueous phases for various O–H groups in the isolated compounds (1–8). The arrangement of BDE values of OH substituents in gas phase is similar to that calculated in water solution. Because water system is more imitative to the bulk polarity of the cell environments and gas phase is generally seen as an ideal state, our findings targeted the mechanisms of antioxidant activities in water solution. Interestingly, the order of experimental antioxidant efficacy of different drugs is correlated with the theoretically predicted arrangement, emphasizing that H-abstraction from OH substituents is likely to be the optimum evidence for the radical scavenging activity of phenolics (Fig. 7B).
The spin density is a rationally trustworthy parameter that offers representation of stabilities of various radical species.21 The minimal BDE and the ease of radical formation are mainly attributed to high delocalization of the radical spin density.69 The influence of H-abstraction on the electron delocalization of various radical species was undertaken by evaluating local spin densities distributions at the depletion site (Fig. S19–S26†). Interestingly, the spin densities of various flavonoids radicals are more likely to be delocalized for radicals resulting from B ring than those originating from A ring (5-OH and 7-OH). This inference is confirmed by the fact that all active sites (minimum BDE values) in studied phytochemicals are located in B ring (4′-OH and 2′-OH). For instance, in compound 1 the Mulliken spin population is 0.357 on the oxygen atom at position 4′ of the radical and 0.431 on position 7 of the same compound. Consequently, this effect results in lowering the BDE values of OH groups in B rings than the A ring counterparts. Compound 7 showed the highest antioxidant potential and minimum BDE value (4′-OH) with two radical species originating from B ring and two from A ring. This compound showed the lowest spin density (0.307) at position 4′ among all radical species. On the other hand, compounds 2 and 5 showed the lowest antioxidant activities with the highest BDE values of the active sites. The spin densities of the active sites of these compounds (4′-OH and 2′-OH, respectively) were 0.382 and 4, respectively. The assessment of spin populations of isolated flavonoid radicals revealed carbon atom at position 1′ is the most likely radical center followed by oxygen atom from which hydrogen is removed. These outputs lead to the conclusion that spin densities distributions play a crucial role in predicting stabilities of radical species.
Generally, the activity of flavonoids as antioxidant agents is strongly influenced by variation of hydroxyl substitution on both rings A and B and conjugation extent between B and C rings.70 The results of our calculation led to the inference that substitution at B ring offers a relatively stable phenoxy radical upon H-abstraction. Also, the existence of both 3-OH and 5-OH groups might result in improving the antioxidant activity as seen in compound 4. Another leading factor for maximizing stability of the phenoxy radical in compound 7 (minimum BDE, highest RSA) is the o-dihydroxylation at the B ring (4'-, 5′-OH), this structural feature offers extra stability and better radical scavenging activity by hydrogen bonding or expanded electron delocalization.
3.5. XO inhibitory activity of C. scoparia flavonoids
XO is a molybdenum-containing enzyme that generates reactive oxygen species (ROS) and catalyzes the oxidation of hypoxanthine and the production of uric acid. Activation of XO and its mediated ROS production and oxidative stress are implicated in several diseases.71 Besides the radical scavenging activity of the isolated flavonoids, we investigated their XO inhibitory activity using in vitro assay and molecular docking simulations. The results represented in Fig. 8 show the inhibitory effect of C. scoparia flavonoids and allopurinol (AL). Compounds 1, 2, 4 and 5 showed lower IC50 values when compared with the other compounds. The output of molecular docking revealed the activity of isolated phytochemicals against XO as represented in Table 5 and, Fig. 9 and supplementary Fig. S17 and S18.† All isolated flavonoids were shown to occupy the main central cavity of the enzyme. A relatively low binding energies (−7.2 to −9.0 kcal mol−1) were obtained with compounds 1 and 4 showed the lowest binding energy. The estimated low binding energy indicated the activity of isolated compounds against XO. All ligands were shown to exhibit polar interactions with the binding cavity of the enzyme. A large number of hydrophobic interactions were detected for compounds 4 and 5. In addition, many commonly known significant amino acid residues were included in the binding interactions of these complexes. These findings highlighted the XO inhibitory activity of C. scoparia flavonoids.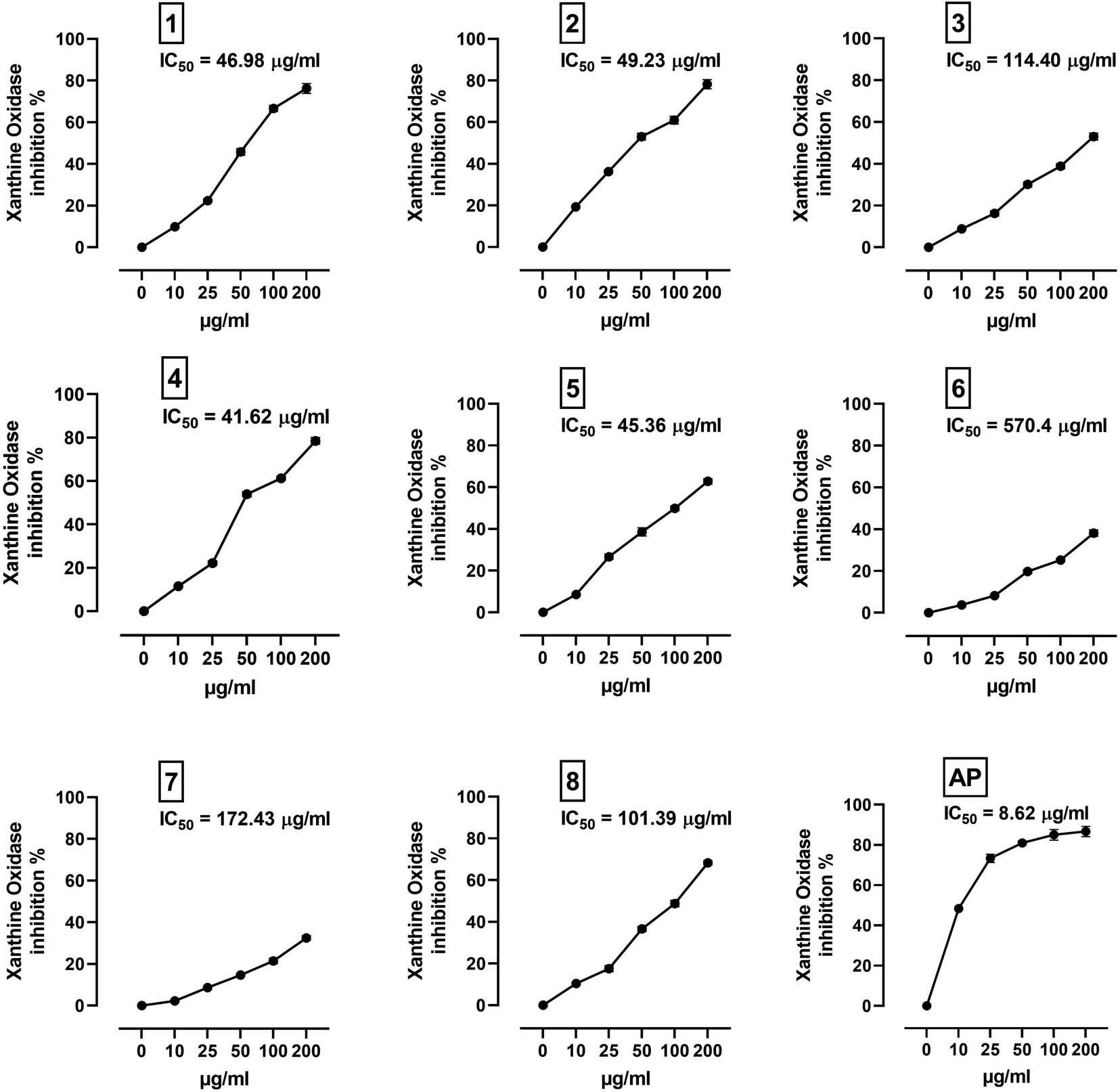 | ||
| Fig. 8 XO inhibitory activity of C. scoparia flavonoids and allopurinol. Data are mean ± SEM, (N = 3). | ||
| Flavonoid | Binding affinity (kcal mol−1) | Polar bonds | Hydrophobic interactions |
|---|---|---|---|
| 1 | −9 | Gln585 and Arg912 | Try592, Gly795, Gly796, Phe798, Met1038, Gly1039, Gln1040, Gln1194 and Glu1261 |
| 2 | −8 | Ile698 and Glu733 | Thr697, Glu699, Tyr735, Leu948, Asn866, Leu905, Tyr1213, Ser1214 and Arg1295 |
| 3 | −7.6 | Glu699, Glu733, Leu1211, His1212 and Arg1295 | Ile696, Thr697, Tyr735 Leu843, Asn866 and Try1213 |
| 4 | −8.2 | Ser1082 | Gln767, Phe798, Gly799, Glu802, Phe911, Arg912, Phe914, Gln1040, Lys1045, Thr1077, Ala1078, Ser1080, Thr1083, Gln1194, Ala1258, Val1259 and Gly1260 |
| 5 | −7.8 | Tyr135 and Lys1304 | Ile696, Thr697, Gly737, Gly738, Pro841, Asn866, Leu905, Glu1210, Leu1211, Tyr1213 and Pro1299 |
| 6 | −7.3 | Tyr592, Arg912 and Gln1194 | Gln585, Leu744, Gly795, Phe798, Met1038, Gly1039 and Gln1040 |
| 7 | −7.3 | Tyr592, Leu744, Arg912 and Gln1194 | Gln585, Gly795, Phe798, Met1038, Gly1039, Gln1040 and Gly1197 |
| 8 | −7.2 | Asp740, His741 and Glu1209 | Phe742, Gly1197, Val1200, Gln1201, Ile1229, Pro1230, Ala1231 and Phe1232 |
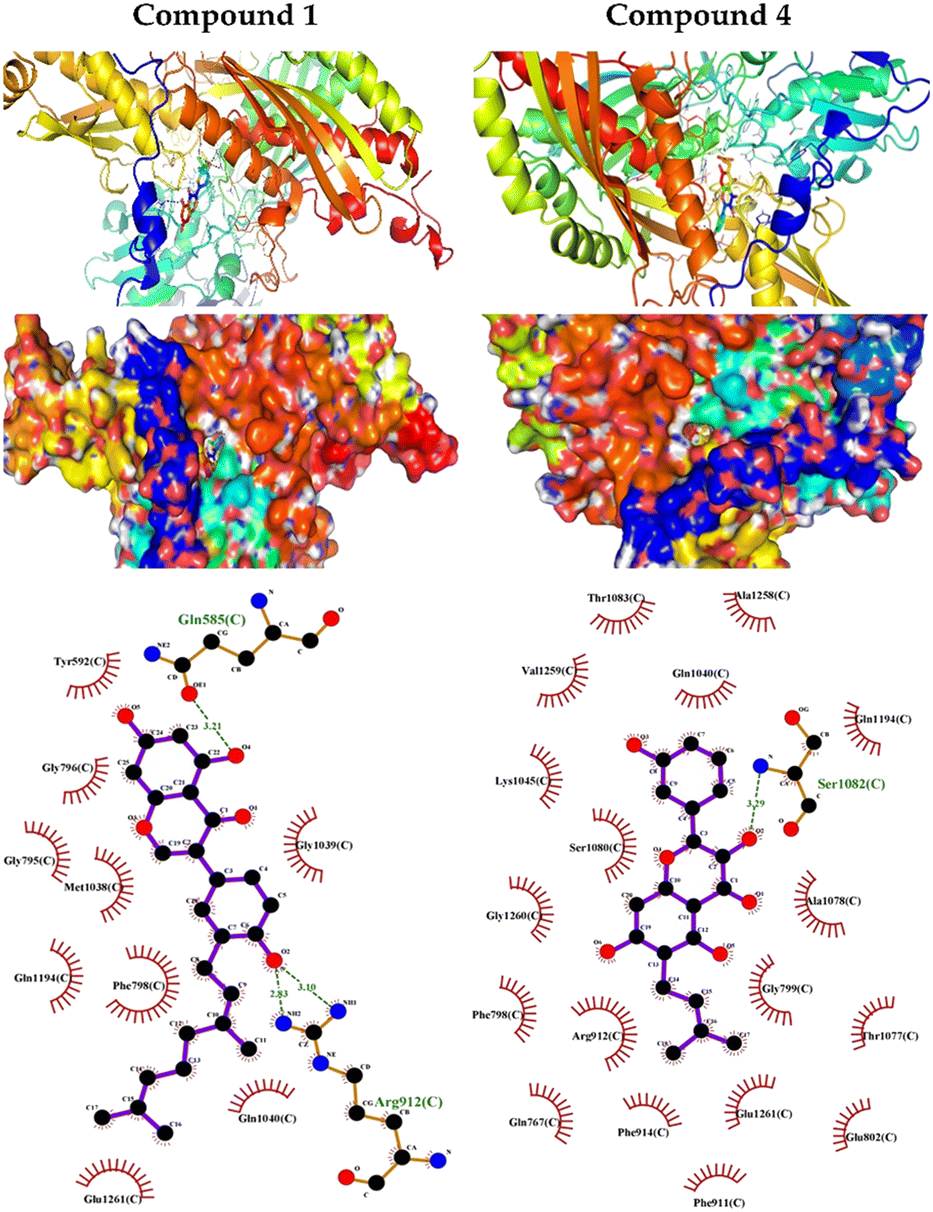 | ||
| Fig. 9 Molecular docking showing the binding modes of compounds 1 and 4 with XO. | ||
4. Conclusions
The outcomes of our DFT calculations indicated that the antioxidant activity mechanism of the isolated flavonoids might be operated by SPLET pathway in water dissolution and by HAT mechanism in the gas phase. The compounds showed in vitro radical scavenging and XO inhibitory activities. The most potent antioxidants are compounds 6 and 7 that possess the lowest ionization potential and BDE values. The SET-PT route is a thermodynamically less favorable mechanism for all flavonoids. The nature of hydrogen atom transfer and neighboring group effect in flavonoids would have a great effect on the thermodynamic features of these flavonoids. Based on docking outcomes, the isolated flavonoids can bind to the appropriate binding site on BLG structure exhibiting polar and hydrophobic interactions, and the obtained low binding affinities suggested the formation of stable drug–protein complexes. The MD simulation investigations produced significant contributions to figure out the impact of the ligand interaction on BLG configurational modification and the stability of BLG-flavonoid complexes in the water phase. Our MD calculations revealed the relative energy minimization of the target protein and flavonoid complexes around 2500 ps. Besides, the resemblance of BLG and BLG-flavonoid complexes atomic fluctuations pattern proposed the rigidity of the ligand-binding pocket structure along the course of 10 ns MD simulations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors extend their appreciation to Princess Nourah bint Abdulrahman University researcher supporting project number (PNURSP2023R342), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors extend their appreciation to the Researchers Supporting Project number (RSPD2023R628), King Saud University, Riyadh, Saudi Arabia. The allocation of computing time at the Centro de Computación Científica of the Universidad Autónoma de Madrid is also acknowledged.References
- S. M. Emery and K. L. Gross, J. Appl. Ecol., 2005, 42, 60–69 CrossRef.
- Y. Kumarasamy, L. Nahar, P. J. Cox, L. N. Dinan, C. A. Ferguson, D. A. Finnie, M. Jaspars and S. D. Sarker, Pharm. Biol., 2003, 41, 203–206 CrossRef CAS.
- C. Panagouleas, H. Skaltsa, D. Lazari, A.-L. Skaltsounis and M. Sokovic, Pharm. Biol., 2003, 41, 266–270 CrossRef CAS.
- M. Grieve, F. Leyel and M. Manya, A Modern Herbal : The Medicinal Culinary Cosmetic and Economic Properties Cultivation and Folk-Lore of Herbs Grasses Fungi Shrubs & Trees with All Their Modern Scientific Uses, Dover Publications, New York, 1982 Search PubMed.
- S. A. Ahmed and E. M. Kamel, Sci. World J., 2014, 2014, 274207 Search PubMed.
- A. N. Khan, I. Fatima, U. A. Khaliq, A. Malik, G. A. Miana, Z.-u.-R. Qureshi and H. Rasheed, Molecules, 2011, 16, 2053–2064 CrossRef CAS PubMed.
- J. Masso, M. Bertran and T. Adzet, Plantes Med. Phytotherap., 1979, 13, 41–45 CAS.
- M. Kaij-a-Kamb, M. Amoros and L. Girre, Pharm. Acta Helv., 1992, 67, 178–188 CAS.
- I. Fernández, J. Pedro and E. Polo, Phytochemistry, 1995, 38, 655–657 CrossRef.
- V. Tickholm, Student's flora of Egypt, Cairo University Publishing, Beirut, 2nd edn, 1974 Search PubMed.
- D. Youssef and A. W. Frahm, Planta Med., 1995, 61, 570–573 CrossRef CAS PubMed.
- D. Youssef and A. W. Frahm, Planta Med., 1994, 60, 572–575 CrossRef CAS PubMed.
- D. Youssef and A. W. Frahm, Planta Med., 1994, 60, 267–271 CrossRef CAS PubMed.
- D. T. Youssef, Phytochemistry, 1998, 49, 1733–1737 CrossRef CAS PubMed.
- M. Bruno, S. Bancheva, S. Rosselli and A. Maggio, Phytochemistry, 2013, 95, 19–93 CrossRef CAS PubMed.
- S. H. Thilakarathna and H. P. Rupasinghe, Nutrients, 2013, 5, 3367–3387 CrossRef PubMed.
- S.-Y. Wu, M. D. Pérez, P. Puyol and L. Sawyer, J. Biol. Chem., 1999, 274, 170–174 CrossRef CAS PubMed.
- G. Kontopidis, C. Holt and L. Sawyer, J. Mol. Biol., 2002, 318, 1043–1055 CrossRef CAS PubMed.
- T. Sarwar, M. A. Husain, S. U. Rehman, H. M. Ishqi and M. Tabish, Mol. Biosyst., 2015, 11, 522–531 RSC.
- M. M. Silva, M. R. Santos, G. Caroço, R. Rocha, G. Justino and L. Mira, Free Radic. Res., 2002, 36, 1219–1227 CrossRef CAS PubMed.
- P. Trouillas, P. Marsal, D. Siri, R. Lazzaroni and J.-L. Duroux, Food Chem., 2006, 97, 679–688 CrossRef CAS.
- M. Leopoldini, T. Marino, N. Russo and M. Toscano, J. Phys. Chem. A, 2004, 108, 4916–4922 CrossRef CAS.
- E. M. Kamel, A. M. Mahmoud, S. A. Ahmed and A. M. Lamsabhi, Food Funct., 2016, 7, 2094–2106 RSC.
- D. Amic, D. Davidovic-Amic, D. Beslo, V. Rastija, B. Lucic and N. Trinajstic, Curr. Med. Chem., 2007, 14, 827–845 CrossRef CAS PubMed.
- D. Amić, V. Stepanić, B. Lučić, Z. Marković and J. M. D. Marković, J. Mol. Model., 2013, 19, 2593–2603 CrossRef PubMed.
- H.-Y. Zhang and H.-F. Ji, New J. Chem., 2006, 30, 503–504 RSC.
- N. Cotelle, J.-L. Bernier, J.-P. Catteau, J. Pommery, J.-C. Wallet and E. M. Gaydou, Free Radic. Biol. Med., 1996, 20, 35–43 CrossRef CAS PubMed.
- J. Cheel, C. Theoduloz, J. A. Rodríguez, P. D. Caligari and G. Schmeda-Hirschmann, Food Chem., 2007, 102, 36–44 CrossRef CAS.
- M. Ozyürek, B. Bektaşoğlu, K. Güçlü and R. Apak, Anal. Chim. Acta, 2009, 636, 42–50 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed.
- K. B. Wiberg, J. Comput. Chem., 1986, 7, 379 CrossRef.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- J. Rimarčík, V. Lukeš, E. Klein and M. Ilčin, Theochem, 2010, 952, 25–30 CrossRef.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab initio molecular orbital theory, John Wiley, New York, 1986 Search PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- E. M. Kamel and A. M. Lamsabhi, Org. Biomol. Chem., 2020, 18, 3334–3345 RSC.
- H. J. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- E. Lindahl, B. Hess and D. Van Der Spoel, J. Mol. Model., 2001, 7, 306–317 CrossRef CAS.
- W. F. van Gunsteren, S. Billeter, A. Eising, P. Hünenberger, P. Krüger, A. Mark, W. Scott and I. Tironi, Biomolecular simulation: the GROMOS96 manual and user guide, Vdf Hochschulverlag AG an der ETH Zürich, Zürich, 1996 Search PubMed.
- A. W. Schüttelkopf and D. M. Van Aalten, Acta Crystallogr. Sect. D Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef PubMed.
- H. Berendsen, J. Postma, W. Van Gunsteren and J. Hermans, in Intermolecular Forces, ed. B. Pullman, Springer, Dordrecht, 1981 Search PubMed.
- B. Hess, C. Kutzner, D. Van Der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- L. Kang, J. X. Zhou and Z. W. Shen, Chin. J. Chem., 2007, 25, 1323–1325 CrossRef CAS.
- Z. F. Shi, C. Lei, B. W. Yu, H. Y. Wang and A. J. Hou, Chem. Biodivers., 2016, 13, 445–450 CrossRef CAS PubMed.
- J. Lingham, N. T. Keen and T. Hymowitz, Phytochemistry, 1977, 16, 1943–1946 CrossRef.
- S. d. O. Oyama, L. A. d. Souza, D. C. Baldoqui, M. H. Sarragiotto and A. A. Silva, Quim. Nova, 2013, 36, 800–802 CrossRef CAS.
- X. Tan, Y. H. Song, C. Park, K.-W. Lee, J. Y. Kim, D. W. Kim, K. D. Kim, K. W. Lee, M. J. Curtis-Long and K. H. Park, Bioorg. Med. Chem., 2016, 24, 153–159 CrossRef CAS PubMed.
- T. Saitoh, T. Kinoshita and S. Shibata, Chem. Pharm. Bull., 1976, 24, 1242–1245 CrossRef CAS.
- S. Mouffok, H. Haba, C. Lavaud, C. Long and M. Benkhaled, Rec. Nat. Prod., 2012, 6, 292–295 CAS.
- Y. Miyaichi, E. Hanamitsu, H. Kizu and T. Tomimori, Chem. Pharm. Bull., 2006, 54, 435–441 CrossRef CAS PubMed.
- R. N. Yadava and K. Saurabh, J. Asian Nat. Prod. Res., 1998, 1, 147–152 CrossRef CAS PubMed.
- Z. Hajdú, A. Martins, O. Orbán-Gyapai, P. Forgo, N. Jedlinszki, I. Máthé and J. Hohmann, Rec. Nat. Prod., 2014, 8, 299–302 Search PubMed.
- M. A. A. Alwahsh, M. Khairuddean and W. K. Chong, Rec. Nat. Prod., 2015, 9, 159–163 Search PubMed.
- M. H. Abukhalil, O. E. Hussein, M. Bin-Jumah, S. A. M. Saghir, M. O. Germoush, H. A. Elgebaly, N. M. Mosa, I. Hamad, M. M. Qarmush, E. M. Hassanein, E. M. Kamel, R. Hernandez-Bautista and A. M. Mahmoud, Environ. Sci. Pollut. Res., 2020, 27, 30118–30132 CrossRef CAS PubMed.
- S. B. Kedare and R. Singh, J. Food Sci. Technol., 2011, 48, 412–422 CrossRef CAS PubMed.
- O. Dangles, Curr. Org. Chem., 2012, 16, 692–714 CrossRef CAS.
- N. Nenadis and M. Z. Tsimidou, Food Res. Int., 2012, 48, 538–543 CrossRef CAS.
- C. J. Parkinson and P. M. Mayer, J. Chem. Soc., Perkin Trans. 2, 1999, 2305–2313 RSC.
- Z. Marković, D. Milenković, J. Đorović, J. M. Dimitrić Marković, V. Stepanić, B. Lučić and D. Amić, Food Chem., 2012, 134, 1754–1760 CrossRef PubMed.
- N. Malik, P. Dhiman, E. Sobarzo-Sanchez and A. Khatkar, Curr. Top. Med. Chem., 2018, 18, 2154–2164 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01661g |
| This journal is © The Royal Society of Chemistry 2023 |