
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSkeletal diversity in Pt- and Au-catalyzed annulations of allenedienes: dissecting unconventional mechanistic pathways†
Ronald
Nelson‡
 ab,
Martín
Calvelo‡
a,
Rebeca
García-Fandiño
a,
Agustí
Lledós
c,
Gregori
Ujaque
c,
José L.
Mascareñas
*a and
Fernando
López
*ad
ab,
Martín
Calvelo‡
a,
Rebeca
García-Fandiño
a,
Agustí
Lledós
c,
Gregori
Ujaque
c,
José L.
Mascareñas
*a and
Fernando
López
*ad
aCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Departamento de Química Orgánica, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: joseluis.mascarenas@usc.es
bDepartamento de Química, Universidad Católica del Norte, Av. Angamos 0610, Antofagasta, Chile
cDepartament de Química, Universitat Autònoma de Barcelona, Cerdanyola del Valles, 08193, Catalonia, Spain
dInstituto de Química Orgánica General CSIC, Juan de la Cierva 3, 28006, Madrid, Spain. E-mail: fernando.lopez@csic.es
First published on 27th March 2020
Abstract
We describe the discovery of unprecedented annulation processes of 1,7-allenedienes, promoted by Pt or Au catalysts. These transformations revealed mechanistic pathways that had not been previously observed in reactions involving carbophilic catalysis. In particular, we have found that allenedienes bearing a silyl ether in the carbon tether connecting the diene and the allene divergently afford cyclopropane-embedded tricyclic derivatives, 6,6-fused bicarbocyclic products or 5,6-fused bicarbocyclic systems, depending on the type of Au or Pt catalyst used. We have carried out experimental and computational studies that shed light on the mechanistic reasons behind this rich and unusual skeletal divergence, and provide new lessons on the drastic influence of platinum ancillary ligands on the reaction outcome.
Introduction
Throughout the last two decades, carbophilic catalysis has reshaped the field of organic synthesis by providing an extensive portfolio of methods that allow transformation of simple unsaturated precursors into highly valuable cyclic skeletons.1Our group has contributed to this field with the development of a variety of Pt- and Au-catalyzed formal cycloadditions involving allenes as key reaction partners.2,3 In particular, in 2009, we and the Toste group, independently demonstrated that gold(I) complexes of type LAuCl/AgX could efficiently promote intramolecular cycloadditions of allenedienes of type 1 in a reagent-controlled, divergent manner (Scheme 1a).4–6 Thus, while gold catalysts bearing σ-donating ligands such as IPr (Au1) or JohnPhos (Au2) lead to 5,7-fused bicyclic systems 2 (or 2′), through a formal [4C + 3C] cycloaddition (Scheme 1a, a), catalysts featuring bulky π–acceptor ligands such as Au3 afford [4C + 2C] adducts (3), provided that terminally disubstituted allenes are used (Scheme 1a, b). Theoretical and experimental data confirmed that the reactions involve a common metal carbene intermediate (A), which evolves from 1,2-hydride or 1,2-alkyl migrations to deliver the observed adducts. The divergence of the process is controlled by stereoelectronic factors of the ancillary ligand, the cationic character of the gold carbene, and the substitution pattern at the carbene adjacent positions.4b,7 Therefore, in several cases, mixtures of cycloheptene (2/2′) and cyclohexene (3) adducts are obtained.
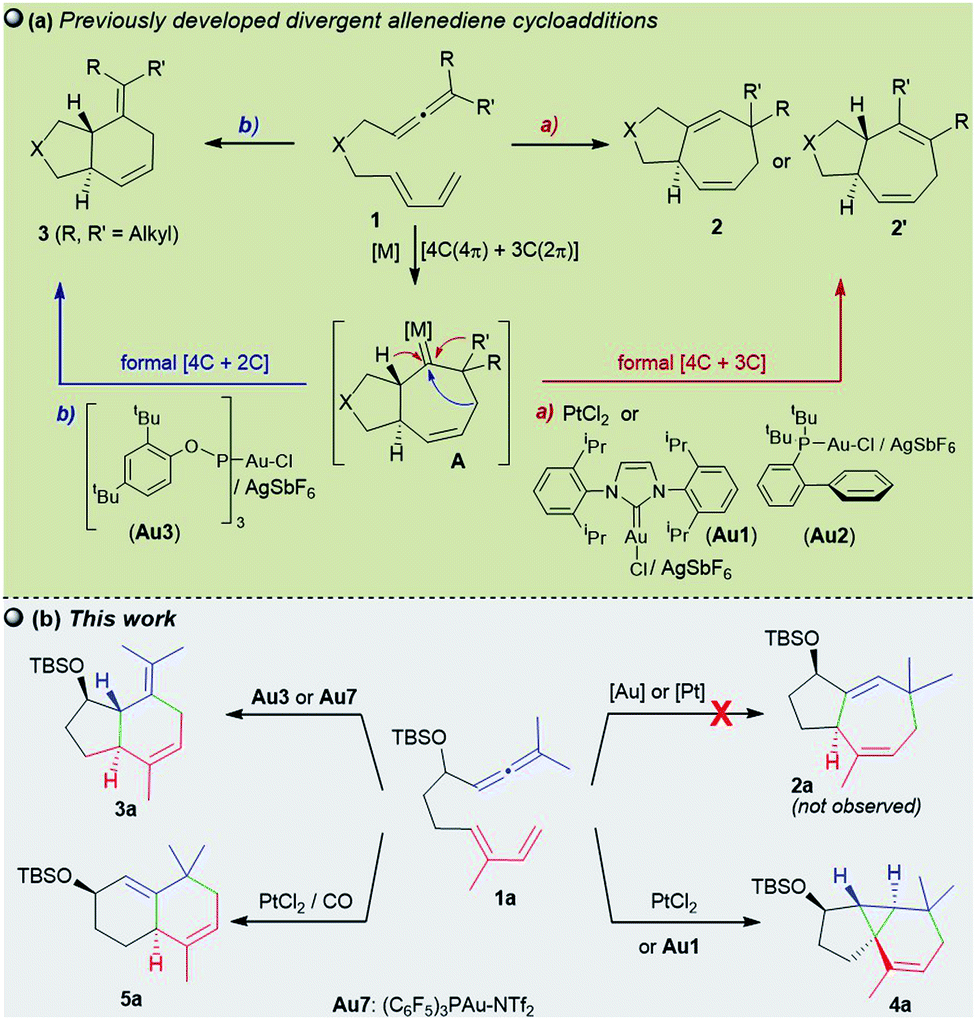 | ||
| Scheme 1 (a) Previous Au(I) and Pt(II)-catalyzed allenediene cycloadditions and (b) current divergent annulations. | ||
Remarkably, the performance of the Au(I) catalysts contrasts with that of their Pt(II) counterparts which, albeit less active, had been previously shown to deliver exclusively cycloheptene products (2/2′).8,9 Indeed, we have exploited this selectivity in short total synthesis of Englerin A, a sesquiterpene natural product with antitumor properties.10
The catalyst-dependent [4C + 3C]/[4C + 2C] annulation of allenedienes was further confirmed by other groups,5,11 and thus it seemed to be a well-established reactivity paradigm. However, we have now found new, surprising annulation pathways that revealed unprecedented ligand-dependent skeletal rearrangements. In particular, we demonstrate herein that 1,7-allenedienes with a silyl ether pendant in the tether (e.g.1a) can divergently afford cyclopropane-embedded tricyclic derivates of type 4, 6,6-fused bicarbocyclic products such as 5, or 5,6-fused bicyclic adducts 3, depending on the Au or Pt catalyst employed (Scheme 1b). The drastic reactivity switch when the PtCl2-catalyzed reactions are carried out in the presence of CO, an effect that is absent in previous Pt-catalysed reactions, is particularly relevant. Curiously, we did not observe the otherwise expected [4 + 3] adducts of type 2 (2a/2a′Scheme 1b).
More importantly, we describe mechanistic studies that shed light on the reasons behind this divergence, and reveal unanticipated pathways that had never been observed in carbophilic metal catalysis. Thus, our work confirms the importance of generally overlooked homo-hyperconjugative interactions in Pt catalysis and shows how subtle conformational and stereoelectronic factors can critically determine the reaction outcome.
Results and discussion
Initial results: discovery of catalyst-dependent multifold paths of allenedienes
We initiated our studies by analyzing the cycloaddition of allenediene 1a, which was designed as a potential precursor of the carbocyclic core of guaiane sesquiterpenes.12 Surprisingly, when allenediene 1a was treated with IPrAuCl/AgSbF6 (Au1) in CH2Cl2, the adduct 2a was not observed. Instead, we obtained a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of the formal [4C + 2C] adduct 3a and the cyclopropyl derivative 4a (isolated in 57% yield), which exhibited an unanticipated 5,3,6-tricyclic skeleton with four consecutive stereocenters (Scheme 2).
:2 mixture of the formal [4C + 2C] adduct 3a and the cyclopropyl derivative 4a (isolated in 57% yield), which exhibited an unanticipated 5,3,6-tricyclic skeleton with four consecutive stereocenters (Scheme 2).
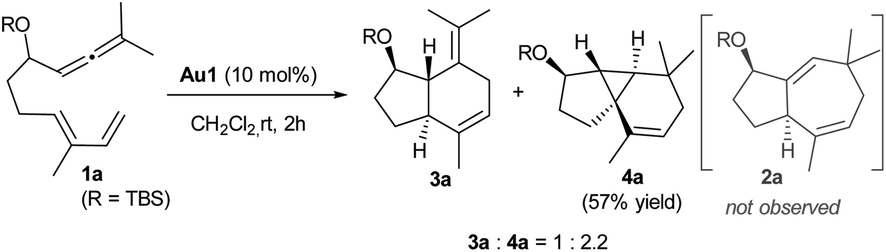 | ||
| Scheme 2 Unexpected reaction outcome of 1a using IPrAuCl/AgSbF6 (Au1). | ||
The structure and relative stereochemistry of both products were determined by NMR, and those of 4a were confirmed by X-ray analysis (Fig. 1).13 This type of tryciclo[4.4.0.01,5]decane skeleton is relevant as it forms the basic core of important natural products like cubebenes, currently used in the fragrance and food industries.14 Notably, while previous routes to these scaffolds involve numerous steps from chiral pool sources,15 the above annulation proposes a straightforward, fully stereoselective entry to their basic cores from readily accessible acyclic precursors.16
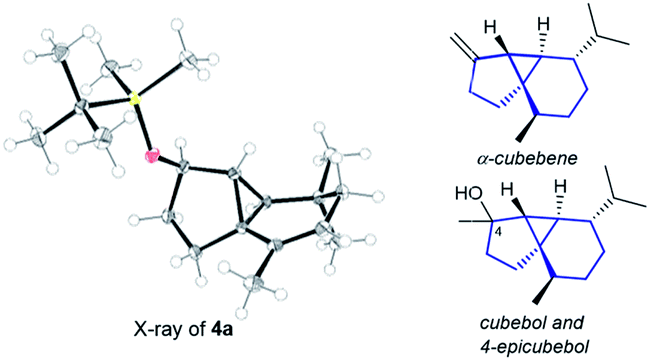 | ||
| Fig. 1 X-ray structure of 4a,13 and the sesquiterpenes cubebols and a cubebene. | ||
The synthetic relevance of the tricyclic product and, specially, the intriguing mechanistic features of the transformation, prompted us to further study the process. The reaction promoted by Au1 can be reproduced in substrate 1b, an allenediene that lacks the methyl group in the diene (Table 1). Again, we obtained a mixture of the tricyclic product 4b and the formal [4C + 2C] adduct 3b, with good selectivity towards the former (4b:3b = 5:1). Accordingly, the cyclopropyl adduct 4b was isolated in a good 70% yield.
|
|
|||||
|---|---|---|---|---|---|
| Entry | [M] | T (°C) | t (h) |
4b:3b ratiob |
Yieldc (%) |
| a Carried out with [M] (10 mol%) in CH2Cl2 (0.05 M), unless otherwise noted. See Tables S1–S2 for the structures of Au complexes and further examples. b Ratio determined by 1H-NMR of the crude mixtures. c Isolated yield. d Complex mixture of products. e Carried out in 1,2-DCE. f Ar = 3,5-(tBu)2C6H3. g Carried out with 5 mol% catalyst. h Carried out in toluene. i Carried out in cyclohexane (with 1 ppm of water). j Carried out in 1,4-dioxane. | |||||
| 1 | IPrAuCl/AgSbF6 (Au1) | rt | 2 | 5:1 |
70 (4b) |
| 2 | JohnPhosAuNTf2 (Au2′) | rt | 12 | — | —d |
| 3 | XPhosAuNTf2 (Au4)e | 80 | 12 | 1.1:1 |
18 (4b) |
| 4 | RuPhosAuNTf2 (Au5) | 40 | 12 | 16:1 |
32 (4b) |
| 5 | IPrMeAuCl/AgNTf2 (Au6) | rt | 2 | 1:0 |
96 (4b) |
| 6 | (ArO)3PAuCl/AgSbF6 (Au3)f | −15 | 1.5 | 1:7 |
66 (3b) |
| 7 | (C6F5)3PAuCl/AgNTf2 (Au7)g | −15 | 1 | 1:12 |
70 (3b) |
| 8 | AuCl3 | rt | 12 | — | —d |
| 9 | AuBr3 | rt | 12 | — | —d |
| 10h | PicAuCl2 | 110 | 8 | 1:0 |
17 (4b) |
| 11h | PtCl2 | 110 | 0.5 | 1:0 |
43 (4b) |
| 12h | [PtCl2(ethene)2]2g | 110 | 1 | 1:0 |
53 (4b) |
| 13h | PtCl2/(C6F5)3P (1:1) |
110 | 1 | 1:0 |
62 (4b) |
| 14h | PtCl2 | 70 | 2 | 1:0 |
81 (4b) |
| 15i | PtCl2 | 50 | 2 | 1:0 |
88 (4b) |
| 16j | PtCl2 | 30 | 2.5 | 1:0 |
99 (4b) |
| 17j | PtCl2 (1 mol%) | 30 | 24 | 1:0 |
92 (4b) |
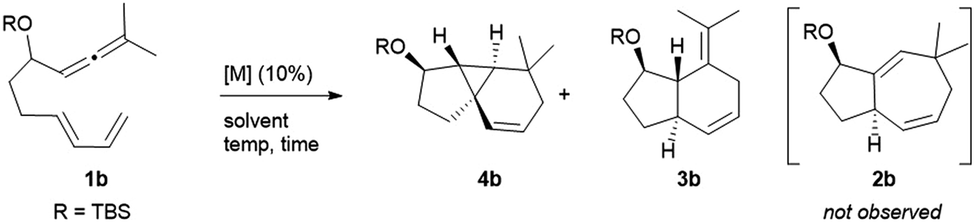
Interestingly, other gold(I) complexes previously reported to promote formal [4C + 3C] cycloadditions of allenedienes, also failed to give the cycloheptenyl adduct 2b. Thus, while the JohnPhos-based catalyst Au2′ provided a complex mixture of products (entry 2), gold(I) complexes with Xphos and Ruphos ligands provided very modest yields of 4b (entries 3 and 4). Noteworthily, the N-heterocyclic carbene gold complex IPrMeAuCl/AgNTf2 (Au6) was completely selective towards the tricyclic product 4b (96% yield at rt, entry 5).
In consonance with previous reports, the formal [4C + 2C] adduct 3b could be obtained in good yields when complexes bearing π–acceptor ligands, such as Au3 or Au7, are used as catalysts (entries 6–7).4b,5 On the other hand, gold(III) complexes such as AuCl3 or AuBr3 provided complex mixtures at rt (entries 8 and 9), whereas the 2-picolinate derivative PicAuCl2 showed some reactivity at high temperature (refluxing toluene), but gave a very modest yield of 4b (17%, after 8 h, entry 10).
Surprisingly, the reaction of 1b with catalytic amounts of PtCl2, in refluxing toluene, also provided the tricyclic product 4b with complete selectivity, but with moderate yield (entry 11). Other catalysts such as [PtCl2(ethene)2]2 or PtCl2/P(C6F5)3 afforded the same product in moderate to good yields, under otherwise identical reaction conditions (entries 12–13). Amongst these catalysts, PtCl2 turned out to be the most efficient one, providing a 81% yield at 70 °C (entry 14). The solvent also played a key role in the rate and efficiency of the process: cyclohexane and 1,4-dioxane proved to be optimal, respectively delivering 4b in 88% yield (2 h at 50 °C, entry 15) and 99% yield (2.5 h at 30 °C, entry 16). Moreover, the reaction could be scaled up (gram scale), and the amount of the catalyst could also be reduced to 1 mol%, without notably affecting the yield and/or selectivity (entry 17).
Surprisingly, when the reaction of 1b with PtCl2 was carried out in the presence of CO (balloon) – a strategy that has been previously employed to accelerate PtCl2 catalyzed processes – we observed a new type of adduct, the biscyclohexenyl product 5b (59% yield), together with minor amounts of 4b (5b:4b ratio = 8:1, Scheme 3). X-ray analysis of a desilylated derivative (5b′) confirmed its 6,6-bicyclic structure.13
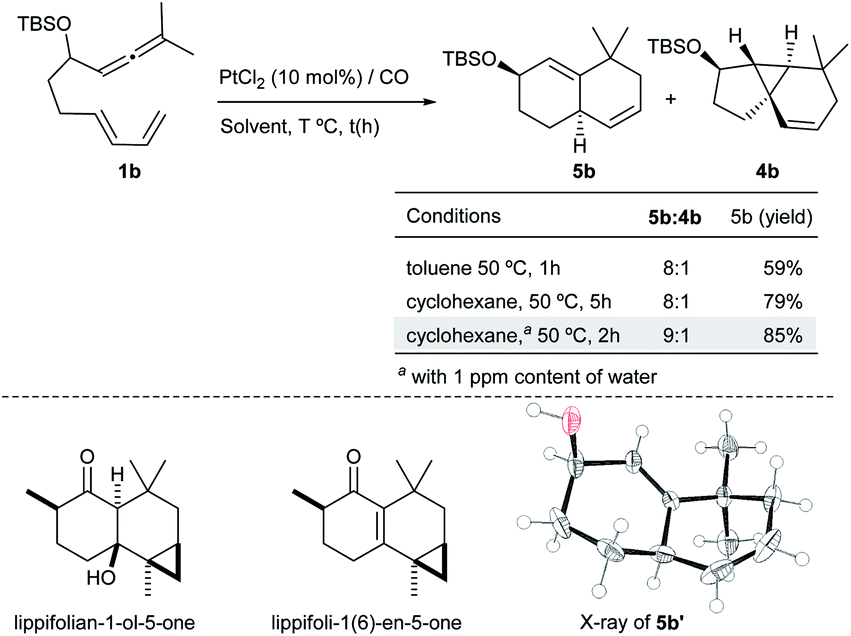 | ||
| Scheme 3 PtCl2-catalyzed reaction of 1b under a CO atmosphere. | ||
This type of bicarbocyclic skeleton is quite common in several sesquiterpenes (Scheme 3, bottom).17 Importantly, carrying out the reaction in cyclohexane (with a 1 ppm content of water) at 50 °C for 2 h resulted in 5b in 85% yield (5b:4b ratio = 9:1). While using completely dry cyclohexane the reaction was slightly less efficient (79% yield, Scheme 3; also see Table S3†).
In analogy with the above results, treatment of the precursor 1a with PtCl2, in cyclohexane at 50 °C, gave the tricyclic adduct 4a (88% yield), whereas performing this reaction under a CO atmosphere produced the bicarbocyclic adduct 5a in 85% yield (Scheme 4). Alternatively, when using Au7 as the catalyst, the same precursor (1a) provides the expected 5,7-fused bicyclic product 3a (54% yield, formal [4 + 2] cycloadduct).13
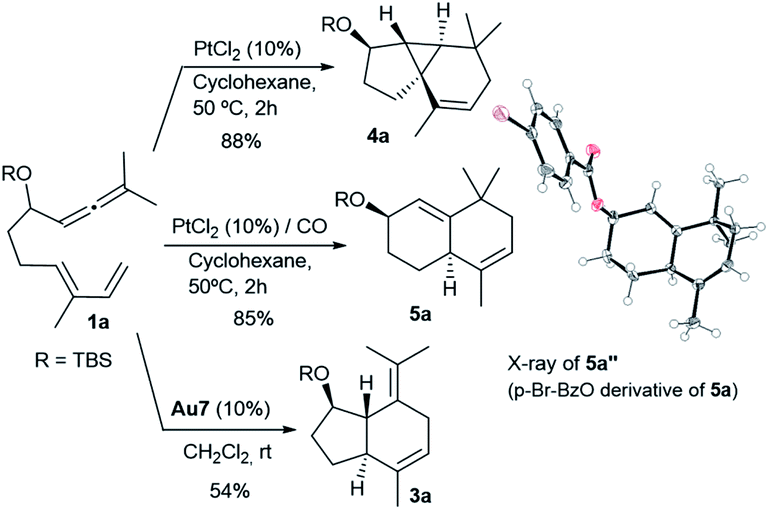 | ||
| Scheme 4 Catalyst-dependent skeletal diversity from 1a. | ||
Overall, these results confirm an interesting case of multifold reactivity, in which the acyclic precursor can be transformed in a catalyst-dependent, divergent manner. Moreover, the above-described examples represent the first demonstration of a drastic change in the outcome of a Pt-catalyzed reaction, when it is carried out in the presence of CO.18
Preliminary exploration of the synthetic potential of the products
A preliminary analysis of the synthetic potential of the tricyclic adducts 4 revealed interesting reactivity profiles (Scheme 5). Therefore, treatment of 4b with H2 (1 atm) over Pd/C (10 mol%) resulted in the hydrogenation of the double bond with concomitant cyclopropane opening, affording the trans-fused bicyclic product 6b in quantitative yield and with complete diastereoselectivity (Scheme 5, eqn (1), conditions a).19 An alternative hydrogenation using Pd(OH)2/C in MeOH also proceeded with equal selectivity, albeit the TBS group was also removed (eqn (1), conditions b). In contrast, the use of PtO2 (10 mol%) under H2 pressure (5 bar) led to the exclusive hydrogenation of the double bond, quantitatively affording the tricyclic system 7b, which presents a carbocyclic core analogous to that of cubeb derivatives (eqn (2)). Finally, the OTBS group of 4b could also be easily converted into its corresponding ketone (8b), paving the way for further variations at this position (eqn (3)).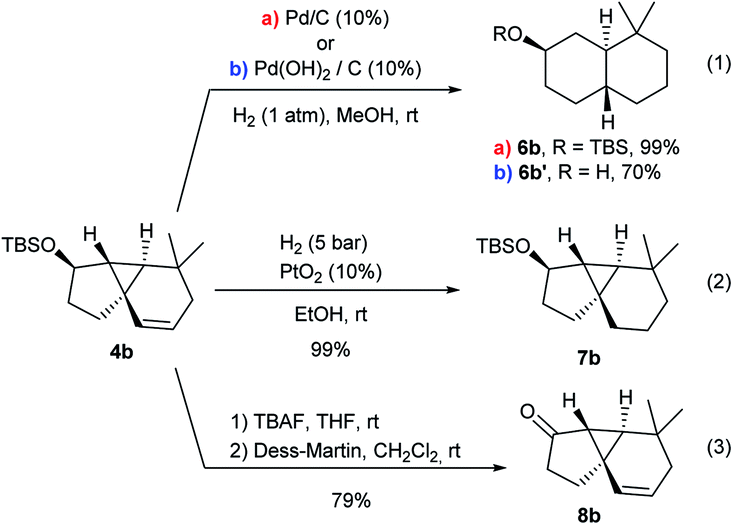 | ||
| Scheme 5 Preliminary exploration of the synthetic potential of adducts 4. | ||
Experimental mechanistic studies
The above annulation reactions are especially intriguing from a mechanistic perspective. In this context, we first questioned whether the 6,6-bicarbocyclic cycloadducts (such as 5b) could be formed by cleavage of the cyclopropyl ring in the adduct 4b. However, treatment of this tricyclic compound with PtCl2/CO, under standard conditions, neither generated 5b nor any other identifiable product (Scheme 6a, eqn (1) and Tables S4–S6†).13 Given that the mechanistic proposals to explain the formation of adducts 2 and 3 invoked the presence of carbene intermediates (A, Scheme 1), and envisioning that the same type of intermediate could also be behind the novel reactivity, we sought to trap the putative carbene species using different oxidants.20 Gratifyingly, after some experimentation (Table S7†), we found that treatment of 1b with IPrAuNTf2 (10 mol%) and 3,5-dichloropyridine oxide (10 equiv) provided, together with the tricyclic adduct 4b (67% yield), a substantial amount of the ketone 9b (≈30% yield, Scheme 6, eqn (2)). This product was identified after reduction of the crude residue with NaBH4, which afforded the alcohols 10b and 10b′ (30% overall yield). Although this result is not fully conclusive, as the pyridine oxide could somehow alter the mechanistic pathway, the formation of cycloheptenone 9b is a priori compatible with a concerted allenediene [4C(4π) + 3C(2π)] cycloaddition that delivers the hypothesized carbene species A′ (Scheme 6a, eqn (2)).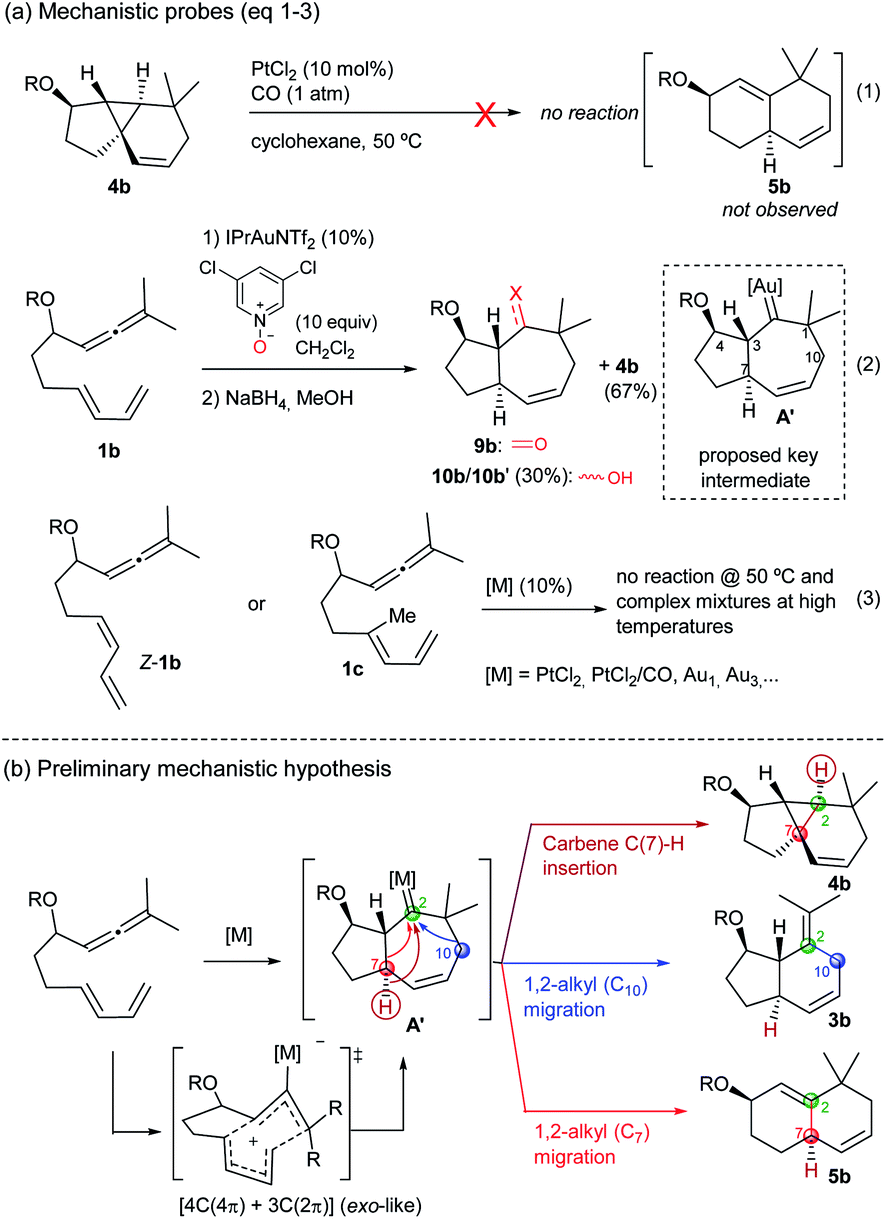 | ||
| Scheme 6 (a) Mechanistic probes (eqn (1)–(3)) and the (b) mechanistic hypothesis. | ||
Importantly, allenedienes Z-1b and 1c, respectively bearing a cis-conjugated diene or a methyl group at the diene internal position, did not provide any product regardless of the type of the catalyst used (Scheme 6a, eqn (3), Tables S8–S9†). These results are consonant with a concerted cycloaddition to provide a metal carbene species like A′ (Scheme 6b), which can evolve in a divergent way to give the observed products. Indeed, we initially envisioned that a putative C(7)–H carbene insertion could provide the tricyclic adduct of type 4,21 whereas alternative 1,2-alkyl migrations of C10 and C7, with concomitant elimination of the catalyst, would produce the adducts 3 (migration of C10) and 5 (migration of C7) (Scheme 6b).
Other allenediene precursors with OTIPS or OTES groups in the tether also provided the same reactivity profile as 1a or 1b (Table S10†). However, treatment of allenediene 1d, equipped with a gem-diester in the tether, with PtCl2 produced the expected seven-membered cycloadduct 2d in quantitative yield (Scheme 7).8 Moreover, when this reaction was carried out under a CO atmosphere, in cyclohexane, we observed the same product (2d) again. Likewise, we also obtained the adduct 2d by using the gold catalyst Au1. Neither the 6,6-fused bicyclic adduct 5d nor the tricyclic product 4d was detected. These results confirm that the structure of the precursor also has a critical influence on the reaction outcome.
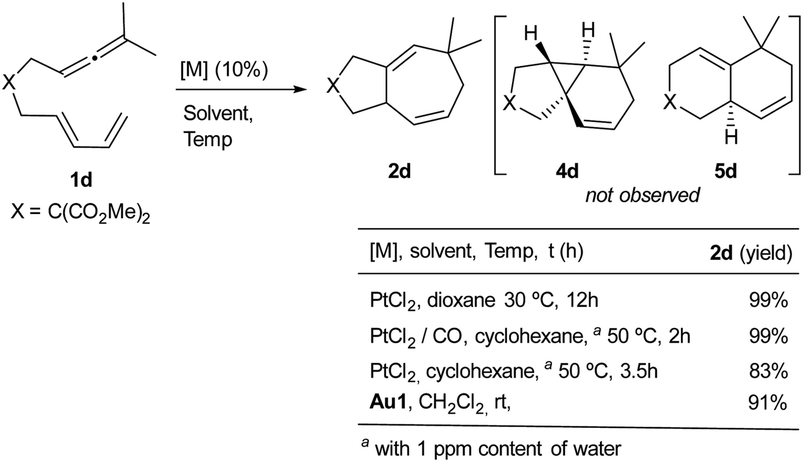 | ||
| Scheme 7 Pt(II)- and Au(I)-catalyzed annulations of 1d. | ||
DFT analysis of the new, divergent processes
The observation that, even in the presence of CO, 1d performs very differently from 1a and 1b, added further interest to the mechanistic study, and to uncover the reasons behind the skeletal divergence. We therefore performed extensive DFT computational studies, using allenedienes 1b and 1d, and different Pt and Au complexes as catalysts.22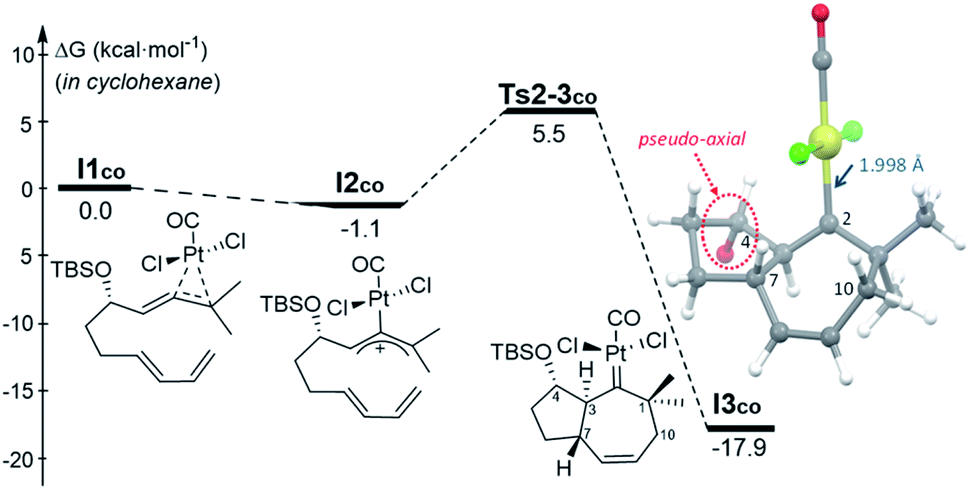 | ||
| Fig. 2 Gibbs energy profile (kcal mol−1) for the activation of 1b by PtCl2(CO) and its subsequent [4C + 3C] cycloaddition in cyclohexane (SMD model). The TBS group is omitted from the 3D representation of I3co for clarity.22 | ||
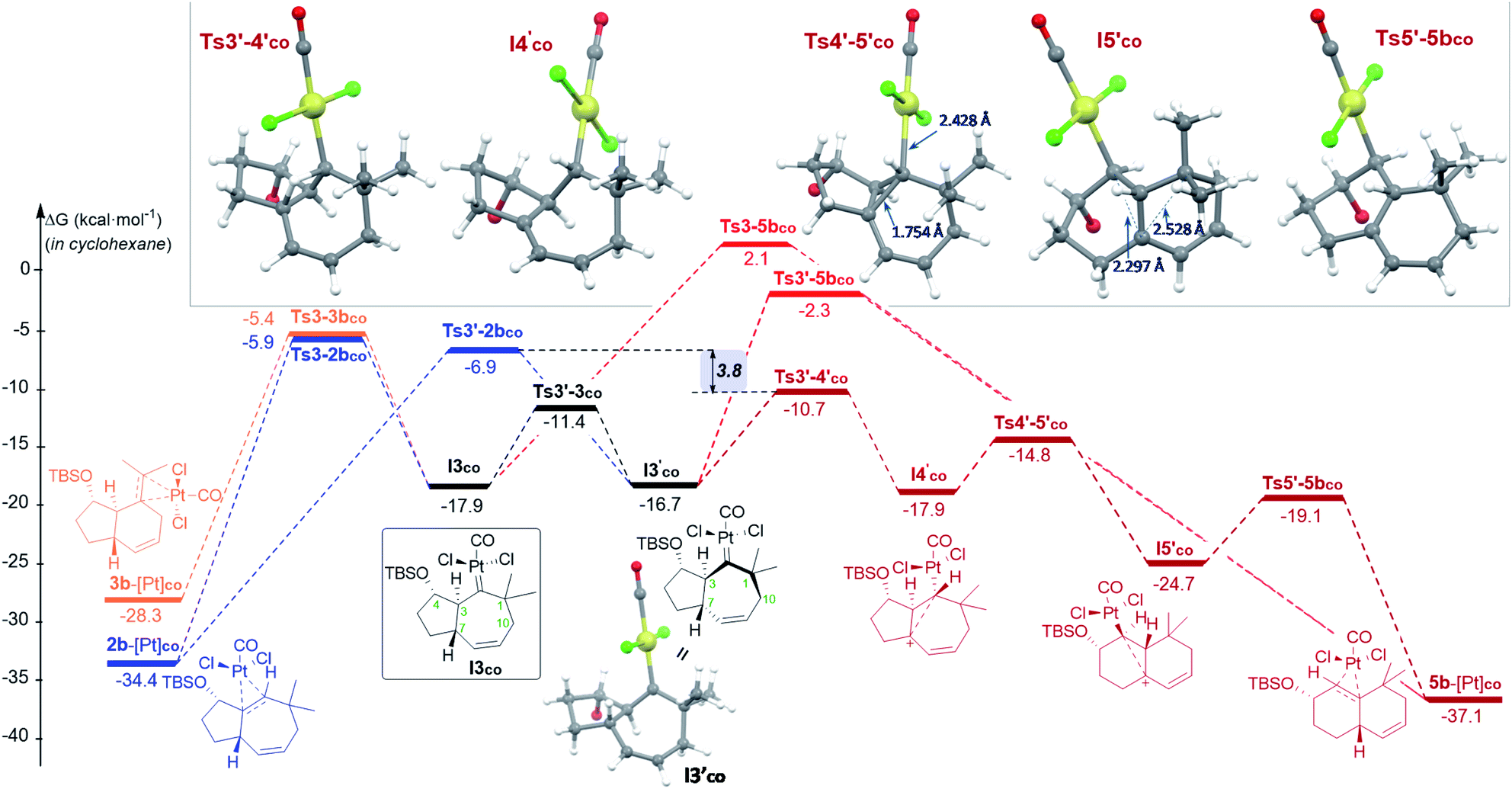 | ||
| Fig. 3 Gibbs energy profile (kcal mol−1) of the reaction of 1b with PtCl2(CO) in cyclohexane: the most relevant divergent pathways from I3co (energy of I1co, shown in Fig. 2, is considered as the reference; see Fig. S8–S9† for a complete profile).22 Optimized structures of selected stationary points (TBS omitted for clarity). | ||
From the cycloheptenyl carbene I3co, we could locate a transition state for the 1,2-alkyl migration of carbon C7 (seven-membered ring contraction, Ts3-5bco), which generates the experimentally observed dicyclohexene product 5b (denoted as 5b-[Pt]co, Fig. 3, red path, Ea: 20 kcal mol−1). However, the 1,2-alkyl (C10) migration leading to the formal [4 + 2] adduct 3b (denoted as 3b-[Pt]co), and the 1,2-hydrogen (H-3) migration leading to cycloheptene 2b (denoted as 2b-[Pt]co) were found to involve significantly lower energy barriers (Fig. 3, blue and orange paths).
Importantly, more detailed DFT scrutiny revealed an unanticipated but energetically more viable alternative, which is initiated by a conformational change in Pt–carbene species I3co to generate I3′co, a conformer in which five out of the seven carbons of the cycloheptene (C3, C7–C10) are almost coplanar (Fig. 3). This species is 1.2 kcal mol−1 less stable than I3co, but accessible through an activation barrier of only 6.5 kcal mol−1 (see Fig. 3 and S8†). Therefore, this easy conformational evolution warrants eventual Curtin–Hammet conditions.
From I3′co we located an unexpected, very favorable migration of the hydrogen at C7 to the Pt–carbene center (C2) to give the carbocationic intermediate I4′co (Fig. 3, magenta path). This is an interesting species which can be viewed as a non-classical carbocation stabilized by homohyperconjugation (also called the γ-effect),25 namely donation of electron density from the back lobe of the Pt–C2 orbital to the empty p orbital of C7. The HOMO−1 molecular orbital of I4′co, obtained by NBO analysis, clearly shows that it is the hyperconjugation, rather than a hypothetical allyl-cation conjugation (with the C8–C9 double bond), what accounts for the relative stability of I4′co (Fig. 4).26
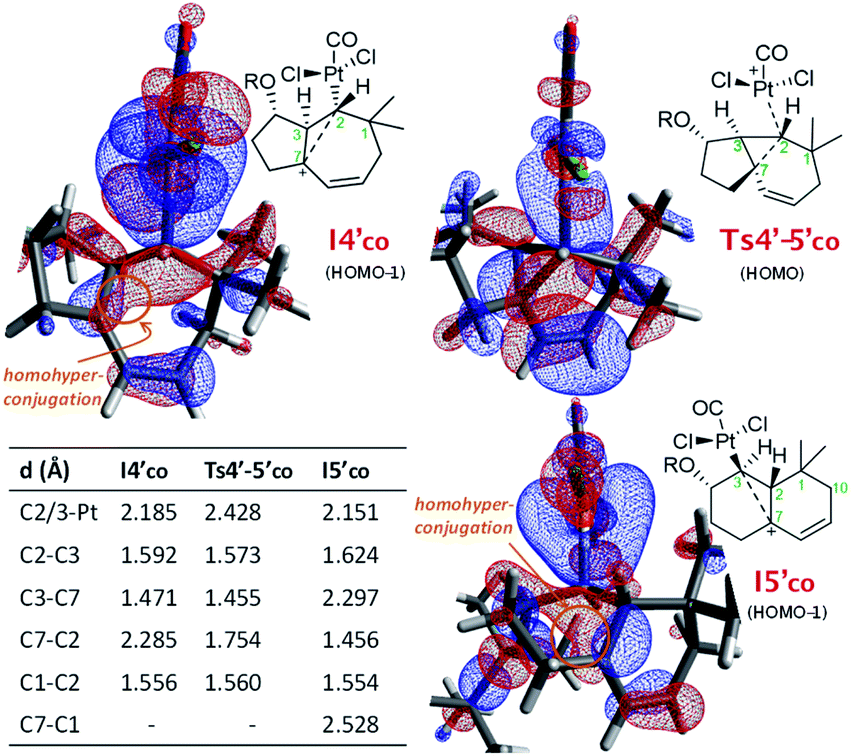 | ||
| Fig. 4 Key distances and NBO representation of relevant HOMO's of I4′co, Ts4′-5′co and I5′co. The TBS groups are truncated for clarity.22 | ||
The atoms in molecules (AIM) topological analysis of I4′co further confirms this bonding pattern, providing for C2 a sum of Wiberg bond indexes (WBIs) of 3.91, a value which is divided into five different bonds. Thus, this analysis provided WBIs of 0.18 and 0.52 for the critical C2–C7 and C2–Pt bonds, whereas those of C2–C3 and C3–C7 bonds are clearly higher, 0.89 and 1.07, respectively (Fig. S10†).27
Importantly, the evolution of I3′co to I4′co is much more favorable than either the 1,2-hydride migration towards 2b (viaTs3′-2bco), or the 1,2-alkyl (C7) migration viaTs3′-5bco [ΔΔG‡ = 3.8 and 8.4 kcal mol−1, respectively; Fig. 3].28
Curiously, DFT calculations indicate that intermediate I4′co could undergo a very easy skeletal rearrangement to the more stable 6,6-bicyclic system I5′co, through an early transition state (Ts4′-5′co, ΔG = 3.1 kcal mol−1), which shows how the C2–C7 bond is being formed with net inversion of configuration at C2 [d(C2–C7) = 1.754 Å] while the Pt complex migrates stereospecifically from C2 to C3. The three different C–C bonds of the cyclopropyl ring generated in this transition state are mostly, but not completely formed (see Fig. 4 for C–C bond distances, and Fig. S10† for WBIs and Laplacian electron density maps). Also, remarkably, in this transition state, the Pt, C2, C3 and C7 atoms, together with the p orbital of C7, adopt a W-shape conformation similar to that found in cyclopropanation reactions of different organometallic species (e.g. Fe, Sn,…), wherein hyperconjugative interactions are also critical.29
The higher stability of I5′co compared to I4′co (6.8 kcal mol−1) is probably the consequence of the release of ring strain and the strengthening of homohyperconjugative interactions. Indeed, the HOMO−1 molecular orbital of I5′co (NBO analysis) shows clear homo-hyperconjugative stabilization by an orbital that extends over C3 and C7 (Fig. 4). Accordingly, the C7–C3 distance in I5′co (2.297 Å) is significantly shorter than that of the C7–C1 bond (2.528 Å, Fig. 3 and 4). Finally, from I5′co, a stereospecific 1,2-migration of the hydrogen at C3 to the carbocation center (C7), with concomitant elimination of PtCl2(CO), delivers the experimentally observed product 5b, which is complexed to the Pt center (denoted as 5b-[Pt]co).
Overall, the above-calculated route towards 5b from I3′co is highly exergonic and proceeds through transition states that exhibit the lowest energy barriers of all the different calculated profiles (ΔG‡ varying from 3.1 to 6.0 kcal mol−1; Fig. 3). Therefore, these data are fully consistent with the experimental observation of adduct 5b as the major product in the reactions promoted by PtCl2 under a CO atmosphere.30
The overall profile for this Pt catalyst is similar to that previously found for PtCl2(CO), but with some key differences (Fig. 5 and S12†). As in the previous case, we found that the conformational change from I3et to I3′et is instrumental in revealing the most favorable energy alternatives (for a complete profile including less favored pathways from I3et, see Fig. S12†). For I3′et, the lowest energy barrier of the processes involves a 1,3-hydrogen migration from C7 to the carbene center (C2), to deliver the carbocationic species I4′et (ΔG‡ = 8.2 kcal mol−1), also stabilized by homohyperconjugation.
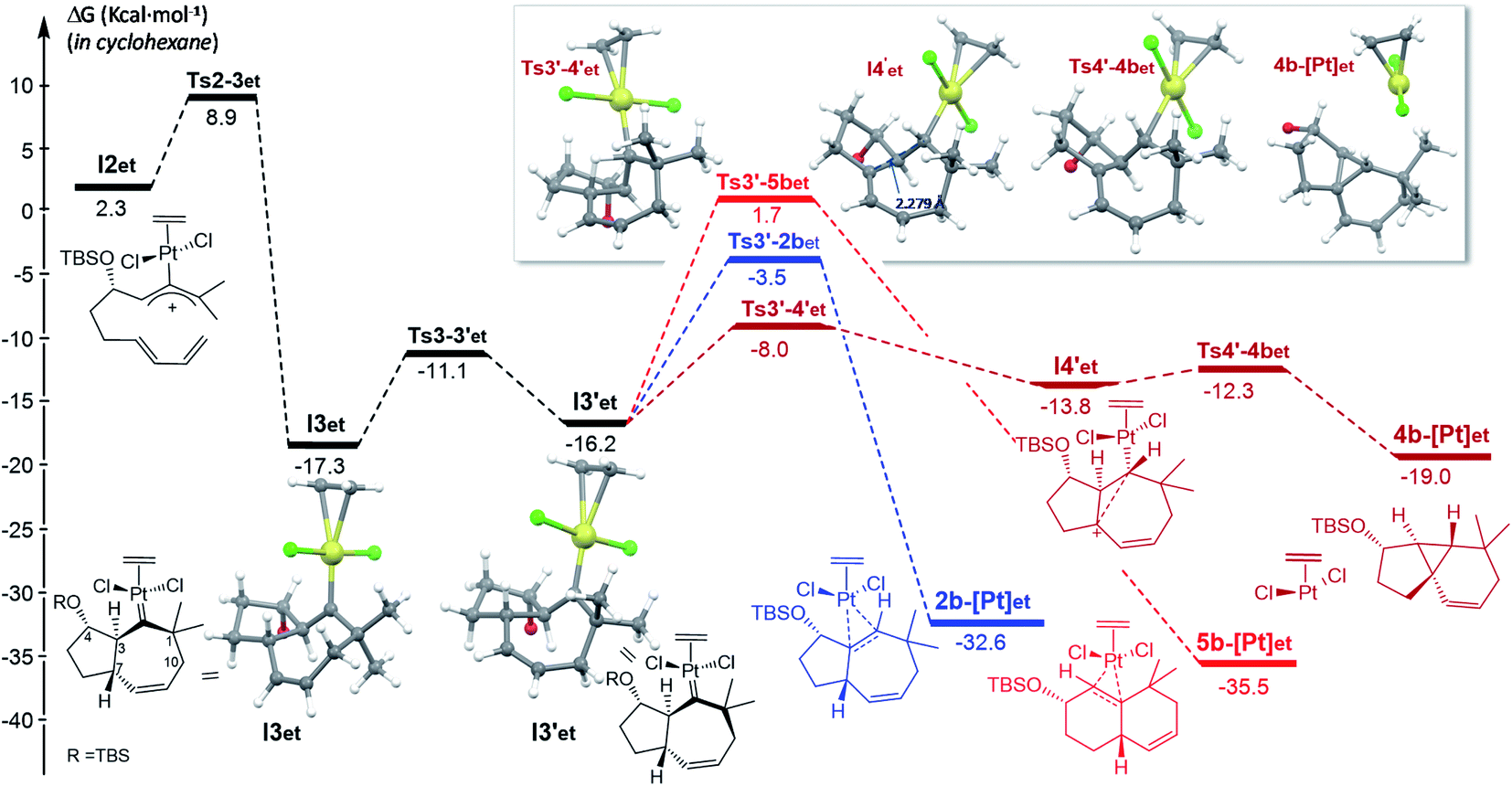 | ||
| Fig. 5 Gibbs energy profile (kcal mol−1) for the reaction of 1b catalyzed by PtCl2(ethene) in cyclohexane; energy of I1et (Fig. S12,† complete profile) is considered as the reference.22 Optimized structures of selected stationary points (TBS omitted for clarity). | ||
Importantly, the presence of ethene at the platinum center, instead of CO, determines a different evolution of this intermediate (vs.I4′co). Indeed, it undergoes a very easy transformation (viaTs4′-4bet, ΔG‡ = 1.5 kcal mol−1) into the cyclopropyl-containing product 4b, with the Pt complex [PtCl2(ethene)] completely de-coordinated from cyclopropane. Overall, the transformation from I3′co into the cyclopropane 4 constitutes a stepwise C(sp3)–H insertion on a Pt-carbene with concomitant cyclopropanation. This is a very uncommon pathway that, to the best of our knowledge, is unprecedented for Pt–carbene species.32
Therefore, the carbocationic species I4′ evolves differently depending on whether the platinum contains a CO ligand or not. The reasons behind this ligand-dependent dichotomy are not fully clear, but they are likely related to the particularly strong σ-donor properties of the CO ligand. Some insights could be obtained from the NBO analysis, which indicates that the carbon attached to the Pt center (C2), has a higher negative charge in I4′co than in I4′et.33 This might favor stronger homohyperconjugation interactions in the former, hampering the decoordination of the Pt–CO complex while favoring the rearrangement and migration of the metal from C2 to C3. Moreover, the Pt atom has a more cationic character in Pt–ethene complexes than in their analogous Pt–CO counterparts, which is in consonance with its tendency to be released (Scheme 8).
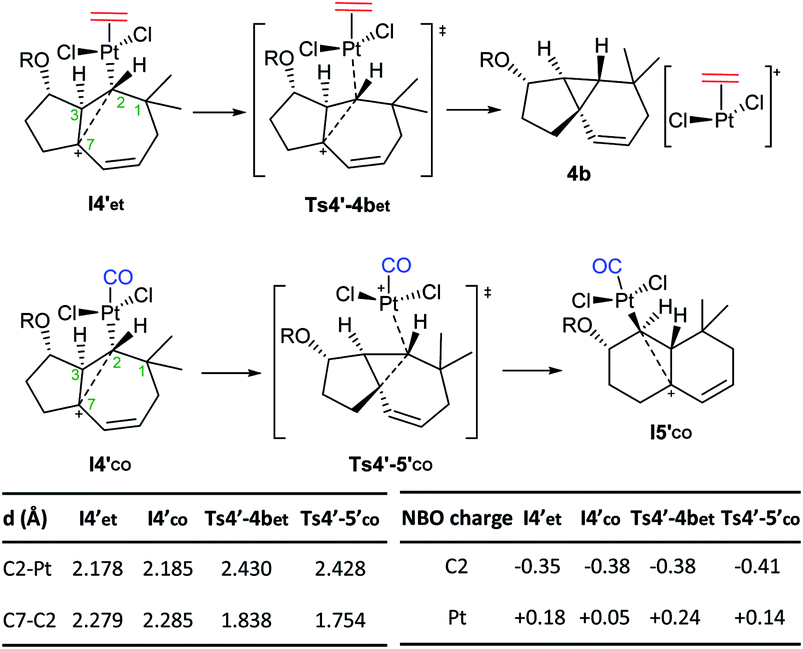 | ||
| Scheme 8 Divergent paths observed with CO and ethene ligands from species I4′. Key distances and NBO charges.22 | ||
Overall, the energy barriers of the pathway leading to cyclopropyl tricycle 4b from I1et are compatible with the thermal requirements of the reaction. The calculations suggest that the initial [4C + 3C] cycloaddition that delivers the carbene I3et is the step with the highest energy barrier (ΔG‡ = 8.9 kcal mol−1). After conformational evolution to I3′et, a 8.2 kcal mol−1 energy barrier connects this carbene species with I4′et, and eventually with the tricyclic product 4b.
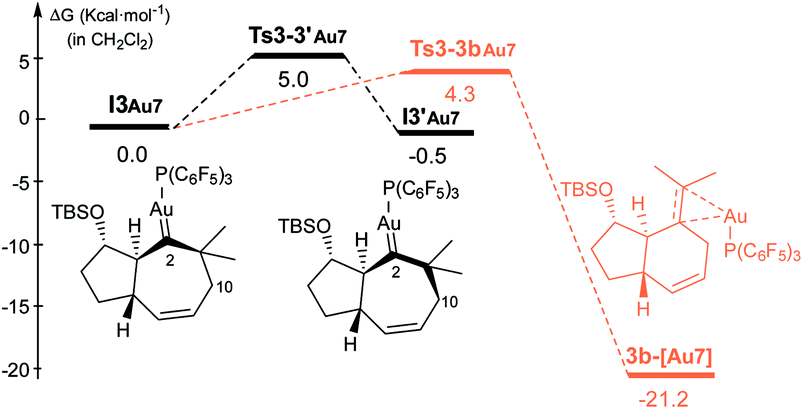 | ||
| Fig. 6 Key steps of the Gibbs energy profile of the reaction of 1b catalyzed by [(F5C6)3P–Au]+, in CH2Cl2.22 | ||
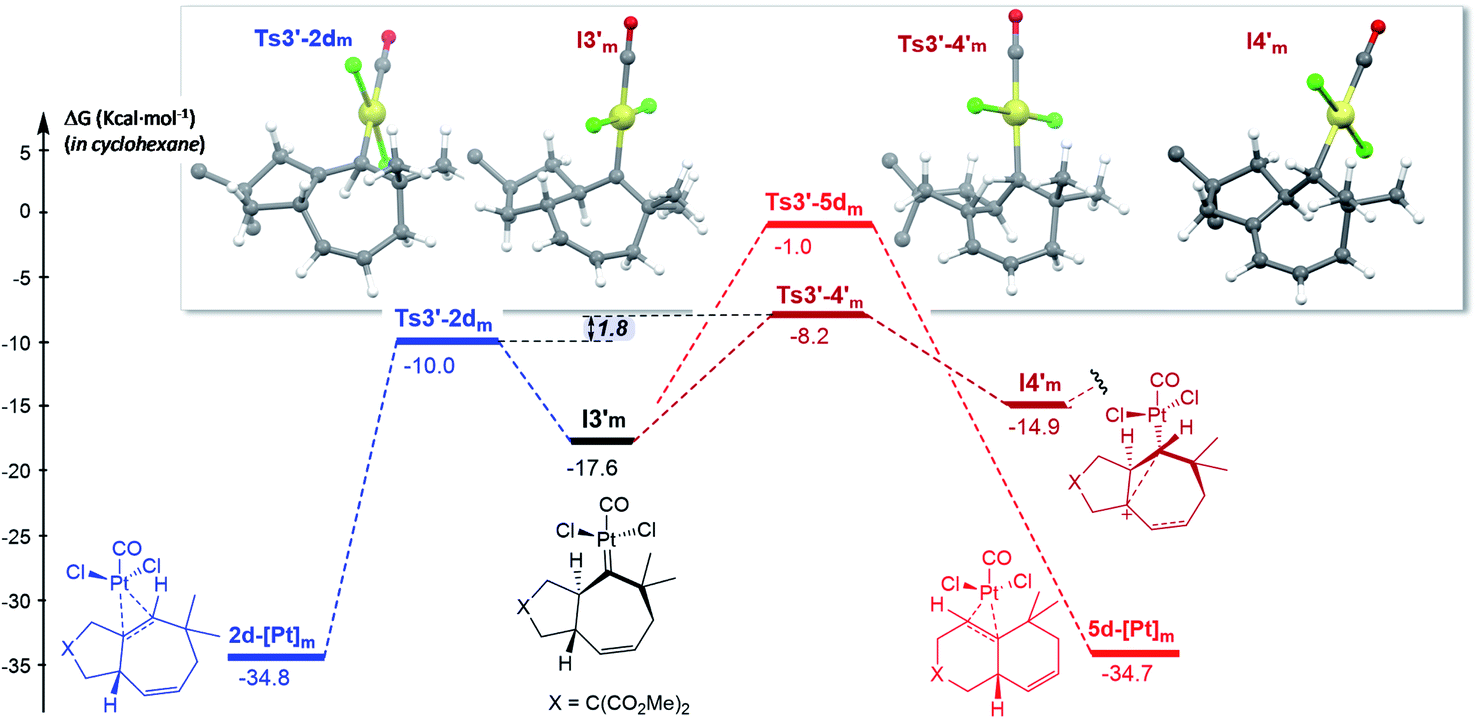 | ||
| Fig. 7 Gibbs energy profile for the reaction of 1d catalyzed by PtCl2(CO) in cyclohexane; energy of I1m (Fig. S15†) is considered as the reference. The gem-diesters [X = C(CO2Me)2] are omitted for clarity. See Fig. S15 and S16† for the entire energy profile.22 | ||
The formation of carbene I3m through a concerted [4C(4π) + 3C(2π)] cycloaddition proceeds with an energy barrier similar to that observed in previous cases, albeit now there are no significant conformational preferences within the ensuing five membered ring (ΔΔG‡ = 0.3 kcal mol−1, Fig. S15†).35 Platinum carbene intermediate, I3m, can also easily evolve to its conformer I3′m, which is almost isoenergetic (Fig. S15†). Remarkably, in contrast to what was observed for 1b, the most energetically favored evolution of I3′m is the 1,2-hydrogen migration leading to the cycloheptene adduct 2d (Ts3′–2dm, ΔG‡ = 7.6 kcal mol−1, Fig. 7).36 The alternative pathway observed for substrate 1b, involving the 1,3-hydrogen migration and generation of carbocationic species I4′m is almost 2 kcal mol−1 more costly (Fig. 7).
A stepwise pathway leading to the decalin 5d from I4′m could not be located, whereas the direct migration of C7, through Ts3′-5d′m presents the highest energetic barrier (>16 kcal mol−1, Fig. 7). Therefore, in consonance with the experimental results the most favored pathway is that leading to the [4C + 3C] adduct 2d.
Comparing the energy profiles of 1d and 1b it can be seen that the barrier associated with the formation of cycloheptene 2d (viaTs3′-2dm) is 2.2 kcal mol−1 lower than that required for obtaining the analog [4C + 3C] adduct 2b from 1b (viaTs3′-2bco, Fig. 8). Moreover, the highest energy barrier in the pathway towards products of type 5 and 4, requiring first the migration of the hydrogen at C7 into the metal carbene center (C2), is considerably higher in the case of the substrate 1d than with 1b [ΔΔG‡ = 3.4 kcal mol−1, Fig. 8].37 Therefore, for substrate 1b, with the OTBS pendant in the connecting tether, there is destabilization of transition state Ts3′-2b′co and stabilization of Ts3′-4′co, and both effects synergistically contribute to the switch of selectivity compared to allenedienes such as 1d.
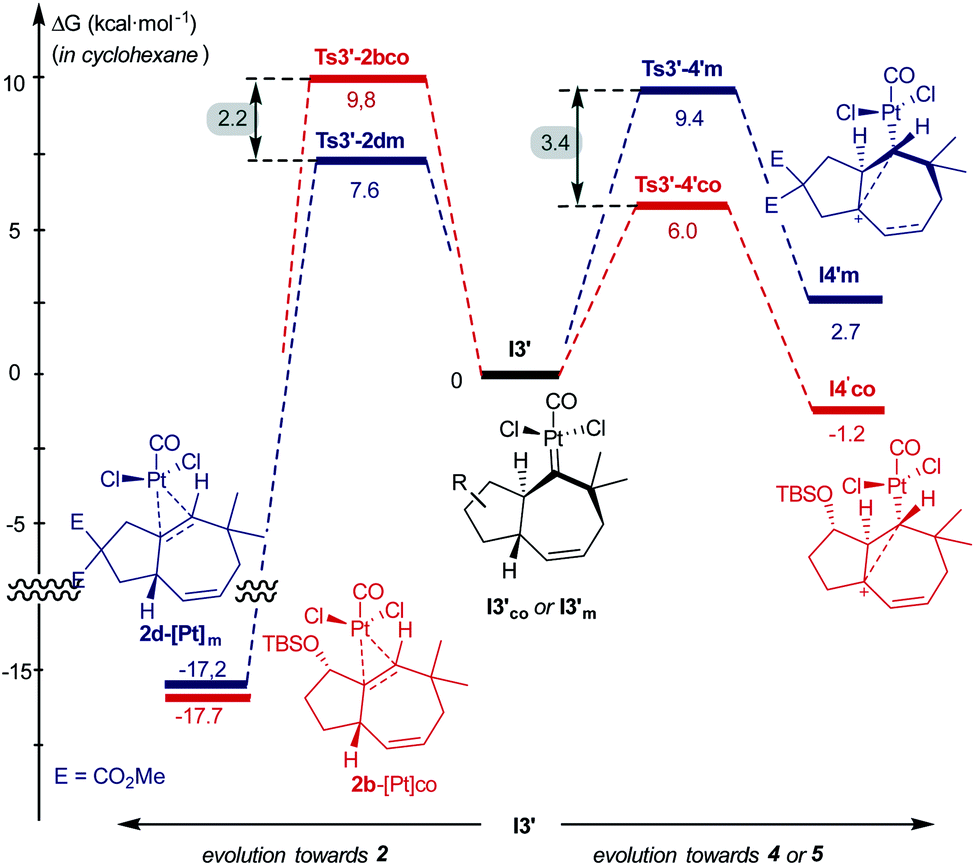 | ||
| Fig. 8 Comparison of energy barriers in pathways to cycloheptene 2 and adducts 4/5, from 1b and 1d; energies of I3′co and I3′m are used as the reference. | ||
But, what are the physical reasons behind these differences? The stabilization of the transition state Ts3′-4′co, corresponding to the migration of the hydrogen atom at C7 to the carbene center, can be qualitatively understood by analyzing the conformations of their immediate precursor I3′co. In particular, the distance between the hydrogen atom that migrates from C7 to C2 is significantly shorter in I3′co than in I3′m [Fig. 9a, d(H7–C2) = 2.455 Å in I3′co and 2.639 Å in I3′m]. These differences are likely associated with a steric clash in I3′co, between one of the methyl groups of the TBS moiety and the chloride atom at the platinum center. This steric congestion forces the Pt center to move far from the methyl, eventually resulting in a higher proximity of the H7 to the empty p-orbital of the carbene at C2 (Fig. 9b). As a result, the orbital contact for the 1,3-hydride migration towards I4′co is facilitated, favoring the overall process towards 5 and 4.
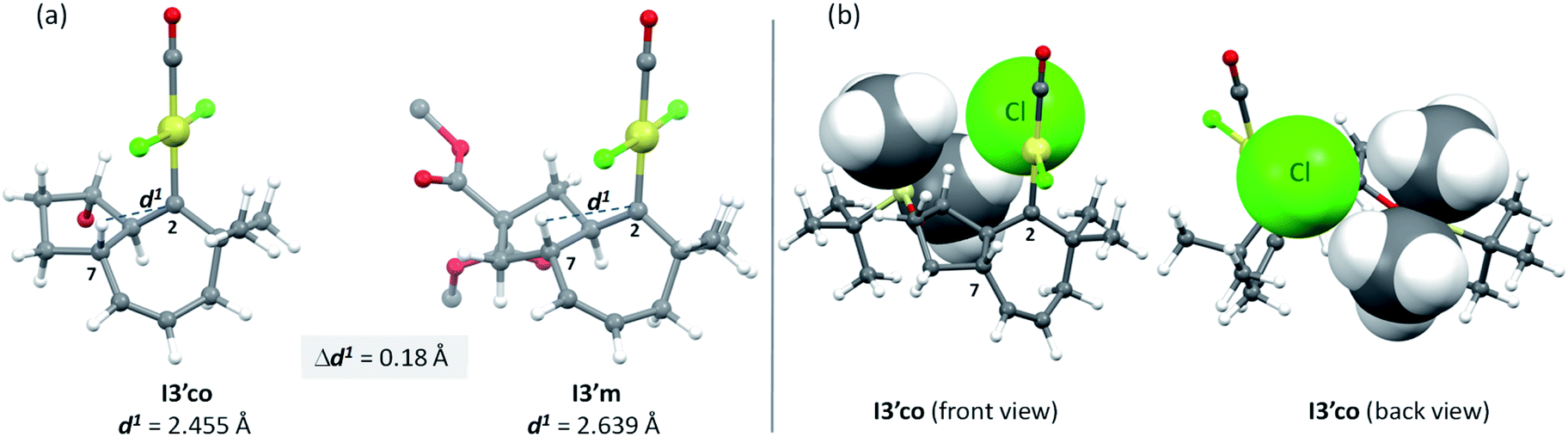 | ||
| Fig. 9 Comparison of H7–C2 distances in I3′co and I3′m (a), and major steric repulsions within I3′co (b); space filling models using Van del Waals radii are used in specific atoms to highlight the steric interactions. | ||
Moreover, the destabilization of the transition state leading to cycloheptene 2b (Ts3′-2bco) can be understood by considering stereoelectronic factors imposed by the OTBS group. During the required 1,2 H migration (from C3 to C2), the TBS group gets quite close to the [PtCl] moiety (Fig. 10a). Moreover, the positive charge generated at the ensuing sp2 carbon, C3, is further intensified by inductive effects generated by the oxygen atom at its β position (see Fig. 10 for atomic charges). In this regard, NBO analysis of this transition state did not show any hyperconjugative interaction, between C3 and the oxygen lone pairs, which could stabilize this positive charge of C3.38 In contrast, in the analog transition state from 1d (Ts3′-2dm, Fig. 10b), there are no such destabilizing interactions, while the positive charge generated at C3 is stabilized by hyperconjugation with its adjacent C4–H bond (NBO analysis, Fig. 10b).
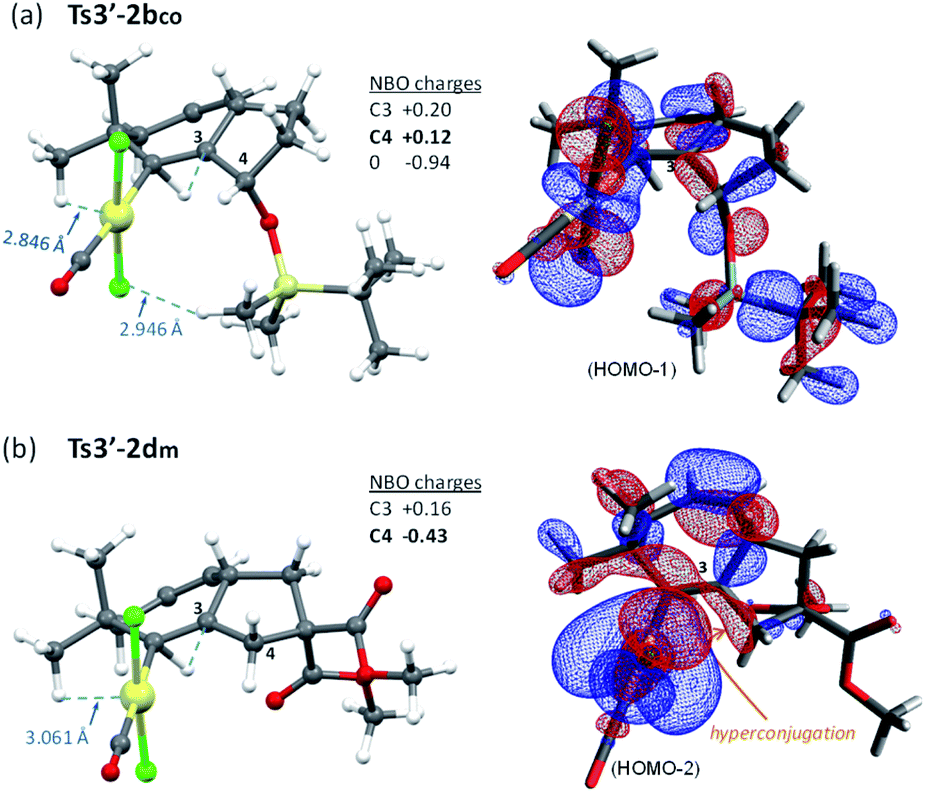 | ||
| Fig. 10 Comparison of steric repulsions (a) and the most relevant HOMO orbitals (b) of Ts3′-2bco and Ts3′-2dm (NBO analysis). | ||
Thus, all these stereoelectronic factors synergistically contribute to determination of the fate of the cycloheptene carbene intermediates.
Importantly, while these results arise from the analysis of specific substrates, the mechanistic findings are relevant to other reactions that occur through similar types of intermediates.
Conclusions
To conclude, the presence of a bulky silyl ether substituent in the connecting tether of allenedienes of type 1, allowed the exploration of novel, intramolecular annulations promoted by carbophilic catalysts. Remarkably, the different pathways can be finely tuned by choosing appropriate ligands at the metal center, which helped to obtain a variety of carbocyclic skeletons in a divergent and highly stereoselective manner.The unexpected rearrangements revealed a number of previously unidentified, mechanistic features that are related to the whole field of synthetic and organometallic chemistry. The effect of a CO ligand in the evolution of platinum intermediates formed after the initial [4C + 3C] cycloaddition step is specifically significant. The presence of the CO ligand hampers the release of the platinum complex from key intermediates, and favors rearrangement processes involving non-classical cationic intermediates which are stabilized by homo-hyperconjugation. These types of interactions, almost ignored in Pt catalysis, should be taken into account when designing other skeletal rearrangements. Finally, we also demonstrate how the obtained annulation products can be selectively manipulated, paving the way for the synthetic applications of this methodology.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received financial support from the Spanish MINECO (SAF2016-76689-R, CTQ2016-77047-P, CTQ2017-84767-P, CTQ2017-87889-P, RYC-2016-20335 and RTI2018-098795-A-I00), the Xunta de Galicia (ED431C 2017/19, 2015-CP082, Centro Singular de Investigación de Galicia accreditation 2019–2022, ED431G 2019/03 and predoctoral fellowship ED481A-2017/068 to M. C.), the ERDF, ERC (Adv. Grant No. 340055), the Orfeo-Cinqa network (CTQ2016-81797-REDC) and CONICYT (grant to RN: PFCHA/72130218). Facilities provided by the Galician Supercomputing Centre (CESGA) are acknowledged. Dr. Isaac Alonso, Ivan Varela and. Almudena Couce-Rios are acknowledged for preliminary work.Notes and references
- (a) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed; (b) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395–403 CrossRef CAS; (c) A. Fürstner, Acc. Chem. Res., 2014, 47, 925–938 CrossRef PubMed; (d) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed; (e) D. Pflästerer and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 1331–1367 RSC; (f) Z. Zheng, Z. Wang, Y. Wang and L. Zhang, Chem. Soc. Rev., 2016, 45, 4448–4458 RSC.
- (a) J. L. Mascareñas, I. Varela and F. López, Acc. Chem. Res., 2019, 52, 465–479 CrossRef PubMed; (b) F. López and J. L. Mascareñas, Beilstein J. Org. Chem., 2013, 9, 2250–2264 CrossRef PubMed. see also: (c) D. C. Marcote, I. Varela, J. Fernández-Casado, J. L. Mascareñas and F. López, J. Am. Chem. Soc., 2018, 140, 16821–16833 CrossRef CAS PubMed.
- For the first Pt-catalyzed reaction of allenes, see: (a) C. Stephan, C. Munz and H. Tom Dieck, J. Organomet. Chem., 1994, 468, 273–278 CrossRef CAS; see also: (b) T. Ishiyama, T. Kitano and N. Miyaura, Tetrahedron Lett., 1998, 39, 2357–2360 CrossRef CAS; (c) Y. Yasunori, F. Ryou, Y. Akihiko and M. Norio, Chem. Lett., 1999, 28, 1069–1070 CrossRef; For the first Au-catalyzed reactions of allenes, see: (d) A. S. K. Hashmi, L. Schwarz, J.-H. Choi and T. M. Frost, Angew. Chem., Int. Ed., 2000, 39, 2285–2288 CrossRef CAS; (e) A. S. K. Hashmi, T. M. Frost and J. W. Bats, J. Am. Chem. Soc., 2000, 122, 11553–11554 CrossRef CAS; (f) A. Hoffmann-Röder and N. Krause, Org. Lett., 2001, 3, 2537–2538 CrossRef PubMed; (g) N. Krause, A. Hoffmann-Röder and J. Canisius, Synthesis, 2002, 2002, 1759–1774 CrossRef.
- (a) B. Trillo, F. López, S. Montserrat, G. Ujaque, L. Castedo, A. Lledós and J. L. Mascareñas, Chem. – Eur. J., 2009, 15, 3336–3339 CrossRef CAS PubMed; (b) I. Alonso, B. Trillo, F. López, S. Montserrat, G. Ujaque, L. Castedo, A. Lledós and J. L. Mascareñas, J. Am. Chem. Soc., 2009, 131, 13020–13030 CrossRef CAS; see also: (c) I. Alonso, H. Faustino, F. López and J. L. Mascareñas, Angew. Chem., Int. Ed., 2011, 50, 11496–11500 CrossRef CAS PubMed.
- P. Mauleón, R. M. Zeldin, A. Z. González and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 6348–6349 CrossRef PubMed.
- For an interesting gold-catalyzed cycloaddition of alkyne-1,3-dienes, see: A. Fürstner and C. C. Stimson, Angew. Chem., Int. Ed., 2007, 46, 8845–8849 CrossRef PubMed.
- (a) D. Benitez, E. Tkatchouk, A. Z. Gonzalez, W. A. Goddard and F. Dean Toste, Org. Lett., 2009, 11, 4798–4801 CrossRef CAS PubMed; (b) S. Montserrat, I. Alonso, F. López, J. L. Mascareñas, A. Lledós and G. Ujaque, Dalton Trans., 2011, 40, 110095–111105 RSC; (c) A. H. Christian, Z. L. Niemeyer, M. S. Sigman and F. D. Toste, ACS Catal., 2017, 7, 3973–3978 CrossRef CAS PubMed; For studies describing particular characteristics of gold-carbenes, see: (d) L. Nunes dos Santos Comprido, J. E. M. N. Klein, G. Knizia, J. Kästner and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2015, 54, 10336–10340 CrossRef CAS PubMed; (e) L. Nunes dos Santos Comprido, J. E. M. N. Klein, G. Knizia, J. Kästner and A. S. K. Hashmi, Chem. – Eur. J., 2016, 22, 2892–2895 CrossRef CAS.
- B. Trillo, F. López, M. Gulías, L. Castedo and J. L. Mascareñas, Angew. Chem., Int. Ed., 2008, 47, 951–954 CrossRef CAS.
- For other reports comparing Au and Pt catalytic profiles, see: (a) M. Pernpointner and A. S. K. Hashmi, J. Chem. Theory Comput., 2009, 5, 2717–2725 CrossRef CAS PubMed; (b) A. Leyva-Pérez and A. Corma, Angew. Chem., Int. Ed., 2012, 51, 614–635 CrossRef; (c) A. S. K. Hashmi, E. Kurpejovic, W. Frey and J. W. Bats, Tetrahedron, 2007, 63, 5879–5885 CrossRef CAS.
- R. Nelson, M. Gulías, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2016, 55, 14359–14363 CrossRef CAS.
- (a) M. Alcarazo, T. Stork, A. Anoop, W. Thiel and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 2542–2546 CrossRef CAS; see also: (b) B. W. Gung, D. T. Craft, L. N. Bailey and K. Kirschbaum, Chem. – Eur. J., 2010, 16, 639–644 CrossRef CAS.
- See for instance: G. Liu and D. Romo, Angew. Chem., Int. Ed., 2011, 50, 7537–7540 CrossRef CAS.
- See the ESI† for further details.
- See for instance: M. I. Velazco, L. Wuensche and P. Deladoey, Use of Cubebol as a Flavoring Ingredient, EP1040765, Firmenich, S.A., 2000 Search PubMed.
- (a) A. Fürstner and P. Hannen, Chem. – Eur. J., 2006, 12, 3006–3019 CrossRef; (b) C. Fehr and J. Galindo, Angew. Chem., Int. Ed., 2006, 45, 2901–2904 CrossRef CAS; (c) D. M. Hodgson, S. Salik and D. J. Fox, J. Org. Chem., 2010, 75, 2157–2168 CrossRef CAS PubMed; (d) S. M. Kim, J. H. Park, S. Y. Choi and Y. K. Chung, Angew. Chem., Int. Ed., 2007, 46, 6172–6175 CrossRef CAS PubMed; (e) C. Fehr, I. Magpantay, J. Arpagaus, X. Marquet and M. Vuagnoux, Angew. Chem., Int. Ed., 2009, 48, 7221–7223 CrossRef CAS PubMed.
- Allenedienes of type 1 can be synthesized in an asymmetric fashion using a Ru-catalyzed asymmetric transfer hydrogenation (see ref. 10), which paves the way for an enantioselective entry to these scaffolds.
- C. M. Cerda-García-Rojas, A. D. C. Coronel, M. E. P. De Lampasona, C. A. N. Catalán and P. Joseph-Nathan, J. Nat. Prod., 2005, 68, 659–665 CrossRef.
- The precedents on the effect of CO in the catalytic activity of PtCl2 basically deal with changes in reaction rates and/or yields, see: (a) A. Fürstner, P. W. Davies and T. Gress, J. Am. Chem. Soc., 2005, 127, 8244–8245 CrossRef PubMed; (b) Y. Gimbert, L. Fensterbank, V. Gandon, J. P. Goddard and D. Lesage, Organometallics, 2013, 32, 374–376 CrossRef CAS; for an isolated example wherein the use of CO modifies the endo vs. exo selectivity of a cycloisomerization, see: (c) A. Fuente-Hernández, P. Costes, P. Kalck, U. Jáuregui-Haza, O. Dechy-Cabaret and M. Urrutigoïty, Appl. Organomet. Chem., 2011, 25, 815–819 CrossRef.
- The structure of 6b could be determined by X-ray analysis of a derivative. See the ESI† for details.
- Although the oxidative trapping of monosubstituted Pt- and Au-carbenes is known, that of disubstituted carbenes is almost unprecedented, due to their lower reactivity; see reference 4a and: S. Montserrat, H. Faustino, A. Lledòs, J. L. Mascareñas, F. López and G. Ujaque, Chem. – Eur. J., 2013, 19, 15248–15260 CrossRef CAS.
- Very few, isolated examples of C–H insertions with concomitant cyclopropanation have been reported with Pt or Au-carbene species. For Au, see: (a) Y. Horino, T. Yamamoto, K. Ueda, S. Kuroda and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 2809–2811 CrossRef CAS PubMed; (b) G. Lemière, V. Gandon, K. Cariou, A. Hours, T. Fukuyama, A. L. Dhimane, L. Fensterbank and M. Malacria, J. Am. Chem. Soc., 2009, 131, 2993–3006 CrossRef; (c) A. Escribano-Cuesta, V. López-Carrillo, D. Janssen and A. M. Echavarren, Chem. – Eur. J., 2009, 15, 5646–5650 CrossRef CAS. For Pt, see: (d) H. Funami, H. Kusama and N. Iwasawa, Angew. Chem., Int. Ed., 2007, 46, 909–911 CrossRef CAS; (e) C. H. Oh, J. H. Lee, S. M. Lee, H. J. Yi and C. S. Hong, Chem. – Eur. J., 2009, 15, 71–74 CrossRef CAS PubMed.
- (a) DFT calculations have been carried out with Gaussian 09. The geometries of all species were optimized using the B3LYP hybrid functional together with the 6-31G(d) basis set for C, H, O, P, Si and Cl and the LANL2DZ basis set for Pt and Au. Single-point calculations of the optimized systems were carried out using hybrid functional M06 together with the 6-3111++g(d,p) basis set for C, H, O, P, Si and Cl, and the Stuttgart-Dresden (SDD) ECP for Pt and Au, either in cyclohexane (for Pt catalysis) or dichloromethane (for gold catalysis) as solvents (using the SMD model). See the ESI† for further details and references; (b) For clarity reasons, we depicted the enantiomeric series.
- According to previous reports, we considered PtCl2(CO) as the active catalyst generated from PtCl2/CO; see ref. 18b and (a) H. Alper, Y. J. Huang, D. B. Dellamico, F. Calderazzo, N. Pasqualetti and C. A. Veracini, Organometallics, 1991, 10, 1665–1671 CrossRef CAS; (b) D. B. Dell'Amico, R. Bini, F. Calderazzo, L. Carbonaro, L. Labella and A. Vitullo, Organometallics, 2005, 24, 4427–4431 CrossRef; (c) D. B. Dell'Amico, R. Bini, F. Calderazzo, L. Carbonaro, L. Labella and A. Vitullo, Organometallics, 2006, 25, 4913–4916 CrossRef; Moreover, according to the literature, the reduction of Pt(II) to Pt(0) under these conditions seems very unlikely, see: (d) D. A. Evans, M. F. Hallam, D. M. P. Mingos and R. W. M. Wardle, J. Chem. Soc., Dalton Trans., 1987, 1889–1895 RSC; (e) See also the ESI for control experiments with Pt(0) complexes.
- A complete computational analysis from the pseudo-equatorial conformer (I3coeq) was also carried out but we could not locate more favored pathways towards any of the products. Indeed, the lowest energy pathway involves a transition state in which the OTBS group is relocated in a pseudo-axial disposition (Fig. S9†)..
- (a) I. V. Alabugin, G. dos Passos Gomes and M. A. Abdo, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1389 Search PubMed; (b) Z. Alamiddine and S. Humbel, Front. Chem., 2014, 1, 1–9 RSC.
- (a) NBO calculations did not provide any HOMO or HOMO-n molecular orbital featuring an allyl cation bonding interaction between C7, C8 and C9; (b) The orbital HOMO−1 is considered instead of the HOMO since the latter exclusively involves the [PtCl2] moiety. The energy difference between them is 0.34 eV.
- Related non-classical Pt-stabilized carbocationic intermediates were only proposed by Gagné in the context of cycloisomerization of 1,6-dienes, see: F. Bell, J. Holland, J. C. Green and M. R. Gagné, Organometallics, 2009, 28, 2038–2045 CrossRef CAS.
- From intermediate I3′co, we could not locate a 1,2-alkyl (C10) migration leading to the formal [4 + 2] adduct 3b, likely due to an unfavorable conformation.
- (a) M. Brookhart and Y. Liu, J. Am. Chem. Soc., 1991, 113, 939–944 CrossRef CAS; (b) C. P. Casey and N. A. Strotman, J. Am. Chem. Soc., 2004, 126, 1699–1704 CrossRef CAS PubMed; (c) J. B. Lambert, L. A. Salvador and J. H. So, Organometallics, 1993, 12, 697–703 CrossRef CAS.
- We also located an analogous route towards 5b from the initial conformer I3co, through related species I4co and I5co (Fig. S8B†). Nonetheless, this route is significantly less favored than that from I3′co.
- Additionally, we theoretically analyzed the reaction of 1b with the catalyst resulting from PtCl2/P(C6F5)3 (Table 1, entry 13). The energy profile (Fig. S13†) is not significantly different from that found for PtCl2(ethene).
- For an isolated case with a Pt-vinylidene, see: (a) Z. F. Li, Y. Fan, N. J. Deyonker, X. Zhang, C. Y. Su, H. Xu, X. Xu and C. Zhao, J. Org. Chem., 2012, 77, 6076–6083 CrossRef CAS PubMed; In general, stepwise C(sp3)–H insertions on metal carbenes are rare. A handful of feasible cases involving Rh and Au carbenes have been proposed when stable carbocations are generated from the initial hydride migration, see: (b) J. Hansen, J. Autschbach and H. M. L. Davies, J. Org. Chem., 2009, 74, 6555–6563 CrossRef CAS PubMed; (c) S. Bhunia and R.-S. Liu, J. Am. Chem. Soc., 2008, 130, 16488–16489 CrossRef CAS; (d) Y. Wang, Z. Zheng and L. Zhang, J. Am. Chem. Soc., 2015, 137, 5316–5319 CrossRef CAS.
- This trend is observed in all stationary points throughout the energy profiles with PtCl2(CO) and PtCl2(ethene).
- For a related preliminary analysis with a [IPrAu]+ model catalyst related to Au1, also consonant with the experimental results, see Fig. S14.†.
- Both conformations of the ensuing five-membered ring were analyzed and the energy difference between them was negligible (see Fig. S15†). Nonetheless, the most favored one, analogous to the previously denoted as pseudo-equatorial, has been considered for Fig. 7 (see the full profile in Fig. S16†).
- Moreover, the next favoured process is the homologous 1,2-H migration from the conformer I3m (viaTs3-2dm, ΔG‡ = 8.1 kcal·mol−1, Fig. S16†).
- Similar trends are also observed when analyzing the homologous pathways from the conformers I3m and I3co (Fig. S17†).
- J. S. Kudavalli and R. A. More O'Ferrall, Beilstein J. Org. Chem., 2010, 6, 1035–1042 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental procedures, optimization of the catalyst, characterization of all new compounds (including NMR spectra), computational details (including Fig. S8–S18), and Cartesian coordinates are provided. CCDC 1964184–1964188. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00650e |
| ‡ R. N. and M. C. contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |