
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyto-friendly polymerization at cell surfaces modulates cell fate by clustering cell-surface receptors†
Jing
Qi‡
a,
Weishuo
Li‡
ac,
Xiaoling
Xu
a,
Feiyang
Jin
a,
Di
Liu
a,
Yan
Du
a,
Jun
Wang
a,
Xiaoying
Ying
a,
Jian
You
a,
Yongzhong
Du
 *a and
Jiansong
Ji
*b
*a and
Jiansong
Ji
*b
aInstitute of Pharmaceutics, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, 310058, China. E-mail: duyongzhong@zju.edu.cn
bKey Laboratory of Imaging Diagnosis and Minimally Invasive Intervention Research, Lishui Hospital of Zhejiang University, Lishui, 323000, China. E-mail: lschrjjs@163.com
cState Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, Center of Advanced Pharmaceuticals and Biomaterials, China Pharmaceutical University, Nanjing, 210009, China
First published on 25th March 2020
Abstract
Lots of strategies, e.g. using multivalent synthetic polymers, have been developed to control the spatial distribution of cell-surface receptors, thus modulating the cell function and fate in a custom-tailored manner. However, clustering cell-surface receptors via multivalent synthetic polymers is highly dependent on the structure as well as the ligand-density of the polymers, which may impose difficulties on the synthesis of polymers with a high density of ligands. Here, we pioneered the utilization of a cyto-friendly polymerization at the cell surface to cluster cell-surface receptors. As a proof of concept, an anti-CD20 aptamer conjugated macromer was initially synthesized, which was then efficiently and stably introduced onto the Raji cell surface via ligand–receptor interaction. With the assistance of an initiator, i.e. ammonium peroxysulfate (APS), the macromer bound onto the Raji cell surface polymerized, inducing the clustering of CD20 receptors, and thereby triggering cell apoptosis. This cell-surface polymerization induced cell-surface receptor crosslinking could alternatively be applied in modulating the fates and functions of other cells, especially those mediated by the spatial distribution of cell-surface receptors, such as T cell activation. Our work opens new possibilities in the area of chemical biology to some extent.
Introduction
The spatial orientations of cell surface receptors are of critical importance in controlling cellular signaling cascades.1 Lots of cell behaviors, including growth factor signaling,2 immune system function,3 cell communication,4 cell adhesion,5 cell migration,5 cell activation6 and cell apoptosis,7 are believed to be regulated by the clustering of cell-surface receptors. Therefore, it is highly convincing that, by reprogramming the receptor–ligand interactions, we could manipulate cell fate and function in a custom-tailored manner. Many strategies,8 such as those involving molecular nanopatterns,9 magnetic actuation,10 polymers11 and DNA nanostructures,12 have been developed to control the cluster of the cell-surface receptors, thus modulating the cell fate. Among all the techniques applied, multivalent synthetic polymers have attracted lots of interest. For instance, Kiessling's group has demonstrated the capacity of neoglycopolymers bearing L-selectin-binding carbohydrates for promoting L-selectin clustering and subsequent leukocyte shedding.4 Kopeček and colleagues have synthesized an antibody-conjugated, multivalent N-(2-hydroxypropyl)methacrylamide (HPMA) polymer, which is bound to cell-surface receptors (i.e. CD20) to initiate downstream cellular events (i.e. apoptosis) through multivalent clustering of CD20 receptors.13 It is worth noting that the clustering of cell-surface receptors in aforementioned scenarios is highly dependent on the structure as well as the ligand-density of polymers; however, the synthesis of polymers with a high density of ligands may be relatively difficult.Conducting polymerization at the live cell surface, a novel technology in polymer chemistry, has emerged recently. For example, Berron et al. introduced the photoinitiator onto the cell surface by antigen–antibody recognition, and the photoinitiator-primed cells were then exposed to monomer solution and light to produce a nanothin polymeric shell around them;14 Choi and his colleagues developed a cyto-compatible atom transfer radical polymerization method for grafting polymers from living cells with the use of polydopamine priming;15 Pioneering work from Hawker's group introduced polymerization initiating chain-transfer-agent groups onto the cell surface and then well-defined grafting chains could be directly initiated from the cell surface, thus achieving a high polymer-grafting efficiency.16 In the aforementioned scenarios, there are two typical applications of conducting polymerization at cell surface. One is to modulate cell–surface properties (i.e. charges and recognition capability) as well as cellular activities (i.e. cell division and cell aggregation); the other is to encapsulate live cells for cellular protection.13–15 However, to the best of our knowledge, clustering cell-surface receptors via polymerization at the cell surface has not been reported.
Herein, we hypothesize that polymerization at the cell surface could serve as an alternative strategy to induce cell-surface receptor clustering. As a proof of concept, we chose the well-established CD20 clustering-induced apoptosis in B-cell cancer cells as a model system. CD20 is one of the most reliable biomarkers for B-cell non-Hodgkin lymphoma (NHL).17 It is a non- or slow-internalizing antigen that remains on the cell surface when bound to a complementary antibody (Ab).18 Moreover, the clustering of CD20-bound antibodies with a secondary antibody results in apoptosis.7 This system is composed of an anti-CD20 aptamer-modified macromer and an initiator, ammonium persulfate (APS). An anti-CD20 aptamer-modified macromer could bind to a non-Hodgkin's lymphoma Raji cell surface via the interaction between CD20 receptors and the aptamer,19 and then the subsequent application of APS would trigger the polymerization of the macromer binding to the Raji cell surface, which would induce apoptosis.
Results and discussion
The synthesis of the macromers involved the reaction between a thiol-derivatized macromer and a maleimide-functionalized anti-CD20 aptamer, as illustrated in Fig. 1A. pHEMA, 1, was first synthesized by the reversible addition–fragmentation chain transfer (RAFT) polymerization of 2-hydroxyethyl methacrylate (HEMA), followed by acetylation to give double-band-decorated macromer 2. The structures of 1 and macromer 2 were confirmed by proton NMR (Fig. S1 and S2†). By changing the feed ratio of methacryloyl chloride, macromer 2 with various double-band contents from 4% to 32% were obtained (Table 1 and Fig. S3†). The thiol ester in macromer 2 was then reduced to afford the thiol-derivatized macromer 3 (Mn = 5.1 kDa and double-bond content = 4%, Table 1 and Fig. S4†). Then macromer 3 was conjugated to a maleimide-functionalized anti-CD20 aptamer to give macromer 4, with a Mn of 53.4 kDa and a double-bond content of 4% (Table 1). The SEC traces of macromers 3 and 4 and the aptamer are shown in Fig. 1B. A significant shift of signal is observed, which demonstrates the successful conjugation of macromer 3 to the aptamer. Furthermore, the polyacrylamide gel electrophoresis (SDS-PAGE) was conducted by labeling the macromers with FITC. Fig. 1C suggests a much slower moving speed of macromer 4 in comparison with macromer 3, as a result of the increase of molecular weight.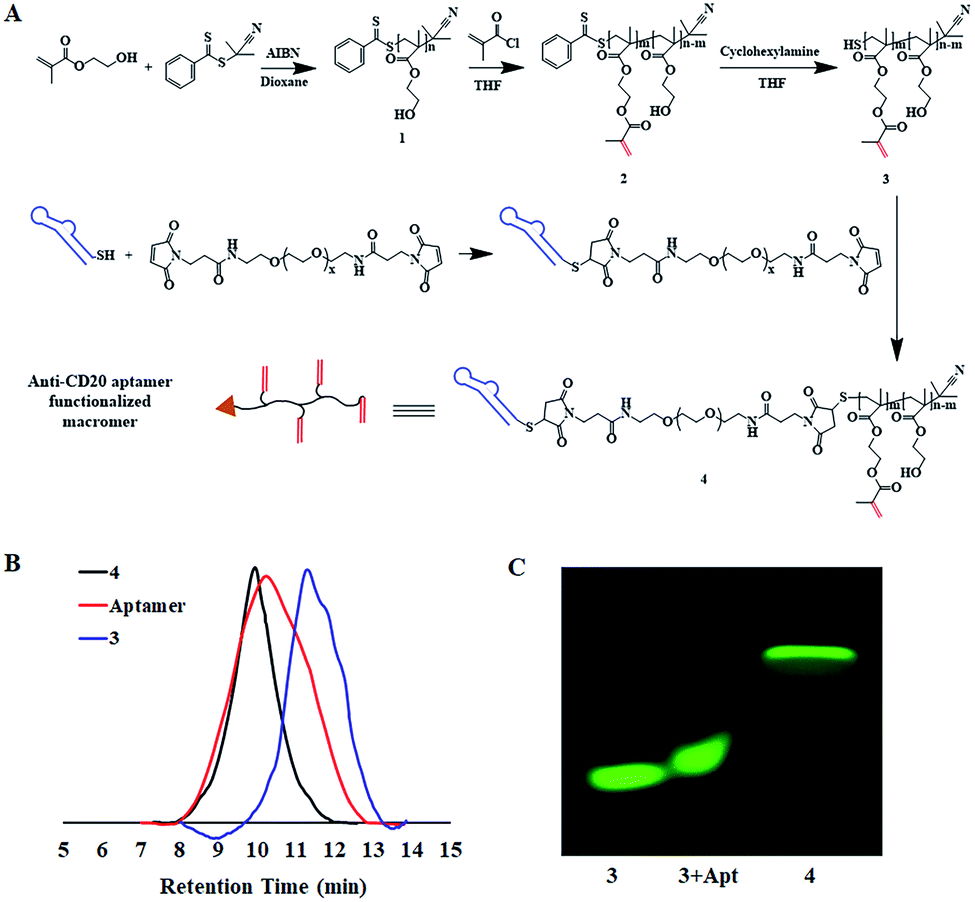 | ||
| Fig. 1 Synthesis and characterization of macromers. (A) Synthetic scheme of macromers; (B) the SEC traces of macromers and aptamer; (C) the SDS-PAGE for FITC-tagged macromers and the mixture of FITC-3 with aptamer. | ||
The key step of polymerization-induced receptor-crosslinking is based on the binding of macromer 4 onto the surface of Raji cells. So the expression of the CD20 receptor on Burkitt's lymphoma Raji cells was first studied. As presented in Fig. 2A, Raji cells are CD20-overexpressed when compared to other cancer cell lines (e.g. hepatocellular carcinoma BEL 7402, human non-small cell lung cancer A549, gastric cancer SCG, as well as human acute T-lymphocytic leukemia cells Jurkat). Then, the binding efficiency of macromer 4 onto Raji cells was investigated by flow cytometry (FACS) and confocal fluorescence microscopy. The effective selectivity of macromer 4 for the Raji cells in a mixed cell population is demonstrated in Fig. S5,† indicating the high interaction between aptamer and CD20 receptor. As shown in Fig. 2B, the binding of macromer 4 onto Raji cells is in a concentration-dependent manner: with the increase of macromere 4's concentration from 0 to 3 μM, the mean fluorescence intensity of Raji cell increases from 0 to 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. Then, the impact of incubation-time on binding was studied. As shown in Fig. 2C and 2D, with incubation-time extending from 1 to 8 h, the fluorescent intensity of macromer 4 on the Raji cell surface does not show any significant increase, which suggests a short time, such as 1 h, is sufficient enough for the binding. Besides, the green fluorescence of macromer 4 co-localizes well with the red fluorescence of the Raji cell membrane, which indicates that macromer 4 can effectively and stably bind onto the surface of Raji cells without detectable internalization within 8 h. It is worth noting that the aptamer displays almost the same binding pattern as that of macromer 4 (Fig. S6†), which suggests macromer 4 entirely inherits the binding capacity of aptamer. To further quantify the amount of macromer 4 bound onto the surface of Raji cells, ferrocene-tagged macromer 4 was synthesized (Fig. 2E and S7†). The successful tag of ferrocene was verified by the change of the visual color of macromer 4 from white to gray. In addition, the tag efficiency was measured by inductively coupled plasma mass spectrometry (ICP-MS) and determined at 3.5 mol iron per mol macromer 4. And, consistent with the results obtained from FITC-tagged macromer 4, the amount of macromer 4 bound onto the cell surface grows with the increase of the feed concentration of macromer 4, up to 2.0 × 10−6 nmol macromer 4 per cell (Fig. 2F).
000. Then, the impact of incubation-time on binding was studied. As shown in Fig. 2C and 2D, with incubation-time extending from 1 to 8 h, the fluorescent intensity of macromer 4 on the Raji cell surface does not show any significant increase, which suggests a short time, such as 1 h, is sufficient enough for the binding. Besides, the green fluorescence of macromer 4 co-localizes well with the red fluorescence of the Raji cell membrane, which indicates that macromer 4 can effectively and stably bind onto the surface of Raji cells without detectable internalization within 8 h. It is worth noting that the aptamer displays almost the same binding pattern as that of macromer 4 (Fig. S6†), which suggests macromer 4 entirely inherits the binding capacity of aptamer. To further quantify the amount of macromer 4 bound onto the surface of Raji cells, ferrocene-tagged macromer 4 was synthesized (Fig. 2E and S7†). The successful tag of ferrocene was verified by the change of the visual color of macromer 4 from white to gray. In addition, the tag efficiency was measured by inductively coupled plasma mass spectrometry (ICP-MS) and determined at 3.5 mol iron per mol macromer 4. And, consistent with the results obtained from FITC-tagged macromer 4, the amount of macromer 4 bound onto the cell surface grows with the increase of the feed concentration of macromer 4, up to 2.0 × 10−6 nmol macromer 4 per cell (Fig. 2F).
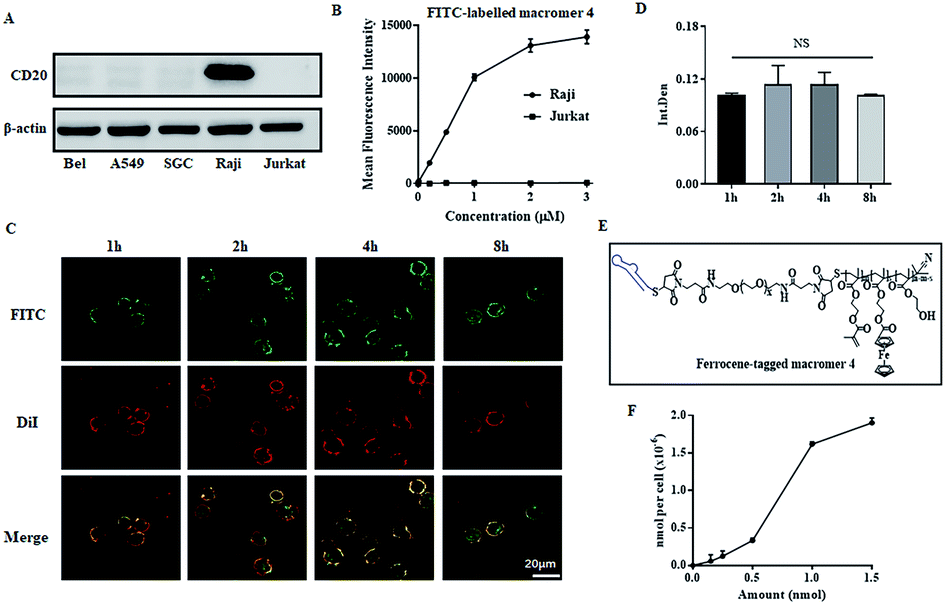 | ||
| Fig. 2 Macromer 4 binding onto cell surface. (A) Expression of CD20 receptors on various cancer cells; (B) FACS analysis of Raji cells incubated with FITC-labeled macromer 4 at different concentrations; fluorescent images (C) and semi-quantitative results (D) of Raji cells exposed to FITC-labeled macromer 4 for various time periods, the cell membrane was labeled by DiI; (E) the structure of ferrocene-tagged macromer 4; (F) the amount of macromer 4 bound onto the Raji cells, measured by ICP-MS based on ferrocene-tagged macromer 4. The results are expressed as mean and SD. NS means no significant difference, n = 3 (Student's t test). | ||
To conduct polymerization at the cell surface, the toxicities of initiator and macromer 4 were first measured by CCK8. As shown in Fig. 3A, macromer 4 is cyto-compatible with a cell viability higher than 80%, when its concentration is under 2 μM. The cell viabilities are all around 100% when exposed to APS at various concentrations from 0.5 to 5 mM, indicating a negligible cytotoxicity of the initiator APS (Fig. 3B). Then, the cell viability incubated with APS for different time-periods was studied. As presented in Fig. 3C, when the incubation-time is shorter than 30 min, the presence of APS is cyto-friendly without significant loss of cell viability. Taken together, for all the following cell-surface polymerizations, unless it is specifically stated, the concentration of macromer and initiator are set at 2 μM and 5 mM, respectively, while the initiation duration is fixed at 30 min. To verify the polymerization of macromer 4 at the cell surface, as presented in Fig. 3D, SEM images of Raji cells with or without polymerization were acquired. Without polymerization, Raji cells show a rough cell surface with the presence of lots of microvilli. In contrast, the conduction of polymerization results in a smoother cell surface, which could be due to the shielding effect of an in situ generated crosslinked polymer (Fig. 3E). To further visualize the polymerization at the cell surface, ferrocene-tagged macromer 4 was applied. The ferrocene-tagged macromer 4 shows a random and irregular morphology in solution, but after polymerization, the ferrocene-tagged macromer 4 assembled into a defined morphology with a round shape (Fig. 3F). Therefore, it is believable that the defined assemblies on the TEM images of Raji cells originate from the polymerization of ferrocene-tagged macromer 4 bound on the cell surface. To extract the polymerized crosslinked polymer from the cellular milieu, biotin-tagged macromer 4 was synthesized (Fig. S8 and S9†). Macromer 4 extracted from both cellular milieu and the solution was re-dispersed into water, and the images are displayed in Fig. 3G. The polymerized, crosslinked 4 does not dissolve well in water compared to unpolymerized 4, and large particles are present in both polymerized cases, which was further verified by the dynamic light scan (Fig. 3H).
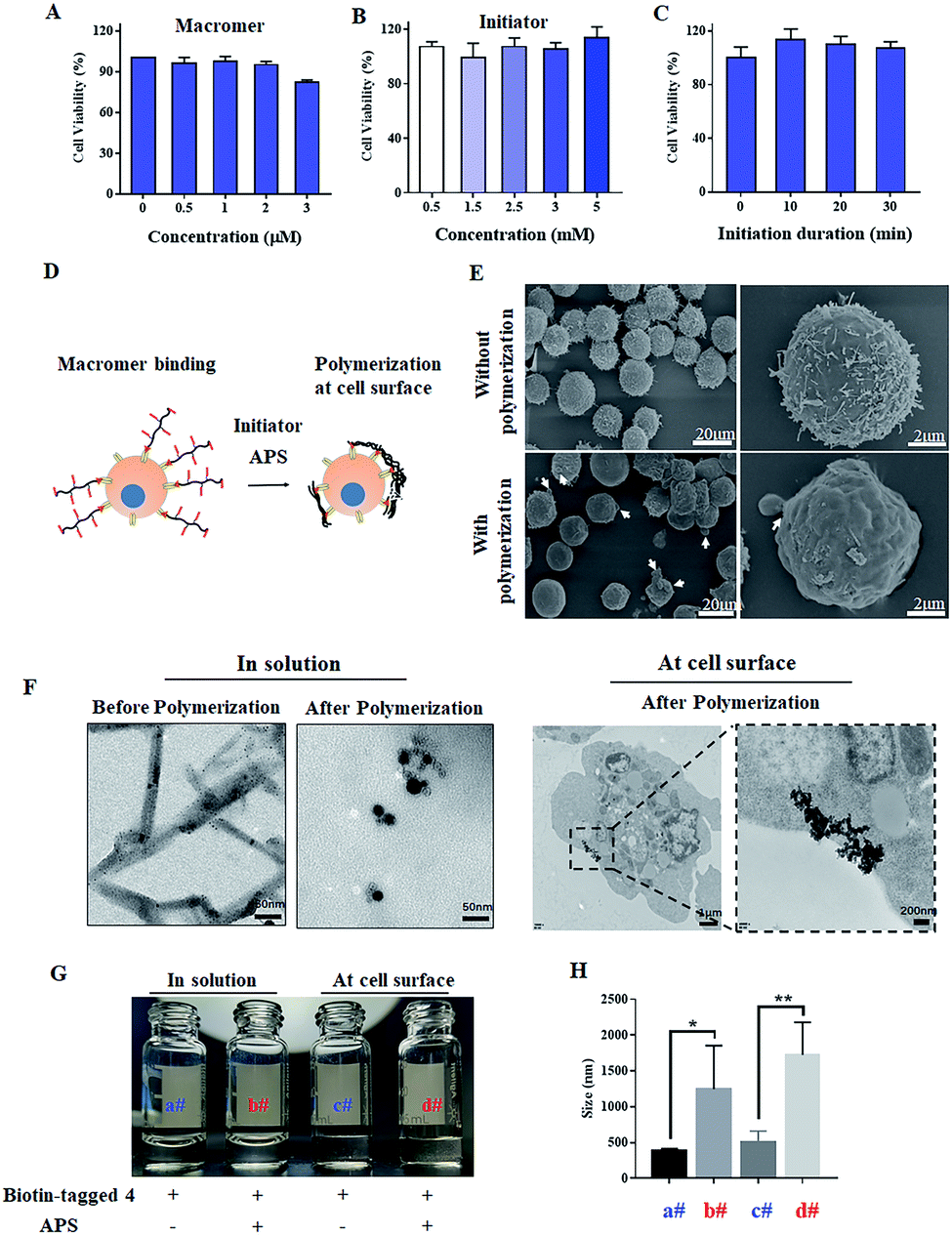 | ||
| Fig. 3 Polymerization at cell surface. Cytotoxicity of macromer (A), initiator (B) and initiation duration (C); (D) schematic illustration of polymerization at the cell surface; (E) the SEM images of Raji cells with and without polymerization; (F) the TEM images of ferrocene-tagged macromer 4 with and without polymerization in solution or at the cell surface. The images (G) and hydrodynamic diameter (H) of the extracted unpolymerized and polymerized biotin-tagged macromer 4 in water. The results are expressed as mean and SD. *p < 0.05, **p < 0.01, n = 3 (Student's t test). | ||
Encouraged by the successful polymerization of macromer 4 at the cell surface, the polymerization-induced receptor-clustering was studied by monitoring the fate of the Raji cell, which could be modulated by the spatial distribution of CD20 receptors-, as illustrated in Fig. 4A. It should be pointed out that apoptotic bodies (Fig. 3E, white arrows) as well as fragments of the Raji cells (Fig. 3F) appeared after polymerization, which implies the killing potential of the cell-surface polymerization. Further on, as presented in Fig. 4B, macromer 4 did not cause any significant toxicities. Comparably, the following cell-surface polymerization initiated by the addition of the initiator, APS, leads to a high cytotoxicity, killing nearly 80% of the Raji cells, when the concentration of macromer 4 is at 2 μM. This cell-surface polymerization mediated cell-killing behavior is believed to be induced by CD20-receptor-clustering, other than the harmful polymerization environment. To illustrate this assumption, macromer, namely maleimide-functionalized macromer 3, was introduced onto the cell surface by chemical modification, instead of ligand–receptor interaction. The chemical introduction of maleimide-functionalized macromer 3 onto the Raji cells was confirmed by confocal fluorescent images, presented in Fig. S10,† in which the fluorescence of FITC-labeled macromer 3 was well co-localized with the fluorescence of the cell membrane. And then the polymerization was conducted. No significant toxicity towards Raji cells was observed at all the tested concentrations (Fig. 4C), indicating that the cell-surface polymerization itself, a process characterized by large amounts of free radicals, does not harm Raji cells obviously.
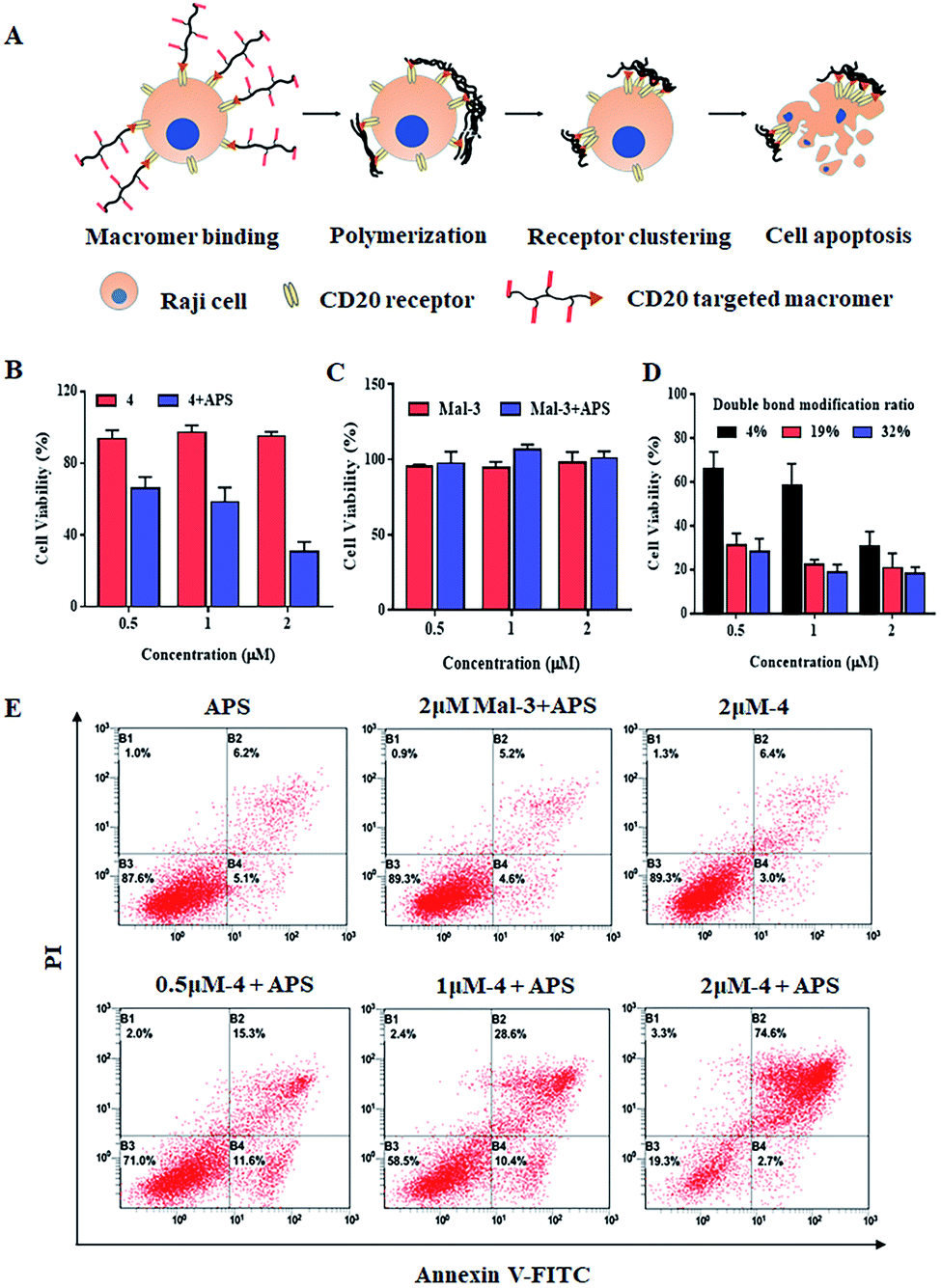 | ||
| Fig. 4 Polymerization at the cell surface inducing CD20-receptor clustering. (A) Schematic illustration of cell-surface polymerization induced CD20 receptor clustering, thus causing the apoptosis of Raji cells; (B–D) Raji cell's viability with or without cell-surface polymerization of different macromers measured by CCK-8. (E) The apoptotic-inducing efficiency of cell-surface polymerization of different macromers monitored by FACS. | ||
Moreover, when the concentration of macromer 4 is low, i.e. 0.5 and 1 μM, the cell-killing capacity decreases to 40% and 50%, respectively. This could be due to the fact that different concentrates of macromer 4 induce various degrees of CD20-receptor clustering, thereby resulting in different levels of cell apoptosis. To verify this assumption, a series of macromer 4 with various modification ratios of the double-bond were applied. A much more toxic behavior of the cell-surface polymerization of macromer 4 with a higher modification ratio of the double-bond (e.g. 19% and 32%) is achieved: 0.5 μM of macromer 4 kills nearly 70% of the Raji cells, which is twice as high as that of macromer 4 (content of double-bond = 4%) (Fig. 4D). Additionally, the cell-killing property of cell-surface polymerization was evaluated by the Annexin V-FITC-PI Apoptosis Detection Kit. Fig. 4E suggests the cytotoxicity of cell-surface polymerization was apoptosis-based. Cell-surface polymerization of 2 μM of macromer 4 leads to an apoptosis ratio of 74.6%, compared to 28.6% and 15.3% for 1 and 0.5 μM, respectively. The potentially harmful polymerization environment does not cause significant cell apoptosis with an apoptosis ratio of 5.2%, which is in accordance with the results acquired from CCK8 assay.
Afterwards, the apoptosis signals triggered by CD20-receptor clustering were elucidated. As previously reported, CD20 is constitutively associates with lipid rafts.20 As illustrated in Fig. S11A,† clustering of the CD20-receptor leads to the aggregation of lipid rafts, followed by the activation of Src-family protein tyrosine kinases (Src-PTKs), influx of calcium ion, destruction of mitochondrial membrane potential, activation of caspase 3, and initiation of apoptosis.21 So, we investigated the effect of cell-surface polymerization on the induction of calcium-associated apoptosis. After cell-surface polymerization of 2 μM of macromer 4, a significant increase of the intracellular calcium ion concentration (Fig. S11B and D†) and decrease of the mitochondrial membrane potential (Fig. S11C and E†) are observed. In addition, an obvious increase in caspase 3 activity is observed (Fig. S11F†), thereby increasing the apoptosis proportion (Fig. S11G†). However, when the cells were treated with β-CD to extract cholesterol, a component of lipid rafts, from the cell membrane, CD20-receptor clustering cannot lead to the aggregation of lipid rafts and the downstream signaling events cannot be triggered. Besides this, when cells were treated with PP2, an inhibitor of Src-PTKs, the calcium-associated apoptosis induced by CD20-receptor clustering was almost completely reversed, indicating that Src-PTKs act proximally to the CD20-triggered changes in the cytoplasmic calcium ion. Together with the experiments using extracellular calcium chelator, EGTA, our findings verified that the apoptosis of the CD20-receptor clustering model is calcium associated, and the calcium-associated apoptosis is dependent on the aggregation of lipid rafts and the following activation of Src-PTKs.
Conclusions
In summary, macromers introduced onto a cell surface via aptamer-CD20 receptor interaction polymerized at the cell surface with the assistance of an initiator, APS. And the cell-surface polymerization induced CD20-receptor clustering, which effectively initiates the apoptotic signals in the cells. To a broader extent, this cell-surface polymerization could be applied in modulating the fates and functions of other cells, especially those mediated by spatial distribution of cell-surface receptors, such as T cell activation. Our work opens up new possibilities in the area of chemical biology to some extent.Experimental section
Please refer to the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the New Century 151 Talent Project of Zhejiang Province; the Natural Science Foundation of Jiangsu Province (BK20180552); the National Natural Science Foundation of China (81803465).Notes and references
- T. Iskratsch, H. Wolfenson and M. P. Sheetz, Nat. Rev. Mol. Cell Biol., 2014, 15, 825 CrossRef CAS.
- C. H. Heldin, Cell, 1995, 80, 213 CrossRef CAS.
- R. N. Germain and I. Stefanova, Annu. Rev. Immunol., 1999, 17, 467 CrossRef CAS.
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Curr. Opin. Chem. Biol., 2000, 4, 696 CrossRef CAS.
- J. D. Humphrey, E. R. Dufresne and M. A. Schwartz, Nat. Rev. Mol. Cell Biol., 2014, 15, 802 CrossRef CAS.
- M. Fourcin, S. Chevalier, C. Guillet, O. Robledo, J. Froger, A. Pouplard-Barthelaix and H. Gascan, J. Biol. Chem., 1996, 271, 11756 CrossRef CAS.
- D. Shan, J. A. Ledbetter and O. W. Press, Blood, 1998, 91, 1644 CrossRef CAS.
- K. Zhang, H. Gao, R. Deng and J. Li, Angew. Chem., Int. Ed., 2019, 58, 4790 CrossRef CAS.
- V. Hortiguela, E. Larranaga, F. Cutrale, A. Seriola, M. Garcia-Diaz, A. Lagunas, J. Andilla, P. Loza-Alvarez, J. Samitier, S. Ojosnegros and E. Martinez, Nano Lett., 2018, 18, 629 CrossRef CAS.
- R. J. Mannix, S. Kumar, F. Cassiola, M. Montoya-Zavala, E. Feinstein, M. Prentiss and D. E. Ingber, Nat. Nanotechnol., 2008, 3, 36 CrossRef CAS PubMed.
- Z. Liu, Y. Liu, Y. Chang, H. R. Seyf, A. Henry, A. L. Mattheyses, K. Yehl, Y. Zhang, Z. Huang and K. Salaita, Nat. Methods, 2016, 13, 143 CrossRef CAS.
- A. Shaw, V. Lundin, E. Petrova, F. Fordos, E. Benson, A. Al-Amin, A. Herland, A. Blokzijl, B. Hogberg and A. I. Teixeira, Nat. Methods, 2014, 11, 841 CrossRef CAS.
- T. W. Chu, J. Y. Yang and J. Kopeček, Biomaterials, 2012, 33, 7174 CrossRef CAS.
- (a) J. L. Lilly, G. Romero, W. Xu, H. Y. Shin and B. J. Berron, Biomacromolecules, 2015, 16, 541 CrossRef CAS; (b) C. S. Bahney, T. J. Lujan, C. W. Hsu, M. Bottlang, J. L. West and B. Johnstone, Eur. Cells Mater., 2011, 22, 43 CrossRef CAS PubMed ; discussion 55.
- J. Y. Kim, B. S. Lee, J. Choi, B. J. Kim, J. Y. Choi, S. M. Kang, S. H. Yang and I. S. Choi, Angew. Chem., Int. Ed., 2016, 55, 15306 CrossRef CAS PubMed.
- J. Niu, D. J. Lunn, A. Pusuluri, J. I. Yoo, M. A. O'Malley, S. Mitragotri, H. T. Soh and C. J. Hawker, Nat. Chem., 2017, 9, 537 CrossRef CAS.
- P. Stashenko, L. M. Nadler, R. Hardy and S. F. Schlossman, J. Immunol., 1980, 125, 1678 CAS.
- O. W. Press, A. G. Farr, K. I. Borroz, S. K. Anderson and P. J. Martin, Cancer Res., 1989, 49, 4906 CAS.
- L. Zhang, Y. Hu, B. Cao, J. Duan, Z. Liu and X. Yang, Basic Res. Clin. Med., 2014, 34, 628 Search PubMed (in Chinese with English Abstract).
- (a) J. P. Deans, H. D. Li and M. J. Polyak, Immunology, 2002, 107, 176 CrossRef CAS PubMed; (b) J. M. Hartley, T. W. Chu, E. M. Peterson, R. Zhang, J. Y. Yang, J. Harris and J. Kopeček, Chembiochem, 2015, 16, 1725 CrossRef CAS PubMed.
- (a) L. Li, J. Y. Yang, J. W. Wang and J. Kopeček, Macromol. Biosci., 2018, 18, 1700196 CrossRef PubMed; (b) J. K. Hofmeister, D. Cooney and K. M. Coggeshall, Blood Cells, Mol., Dis., 2000, 26, 133 CrossRef CAS PubMed; (c) T. L. Unruh, H. Li, C. M. Mutch, N. Shariat, L. Grigoriou, R. Sanyal, C. B. Brown and J. P. Deans, Immunology, 2005, 116, 223 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06385d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |