
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
meta-Selective olefination of fluoroarenes with alkynes using CO2 as a traceless directing group†
Andrew R. A.
Spencer
,
Rishi
Korde
,
Marc
Font
and
Igor
Larrosa
 *
*
School of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: igor.larrosa@manchester.ac.uk
First published on 31st March 2020
Abstract
Over the last few decades C–H olefination has received significant interest, due to the importance and usefulness of aryl olefins both as synthetic targets and intermediates. While a wide range of ortho-olefination protocols have been developed, only a small number of meta-olefinations are currently available. Importantly, the most common approach to meta-olefination, using a large meta-directing template, is not suitable for substrates such as fluorobenzenes, which cannot be derivatised. We report that the meta-selective olefination of fluoroarenes can be achieved via the use of CO2 as a traceless directing group, which can be easily installed and removed in a one-pot process. Furthermore, this approach avoids the use of stoichiometric Ag(I)-salts, commonly used in C–H olefinations, and affords complete meta- over ortho/para-regioselectivity.
Introduction
Over the last decades, direct C–H bond activation has emerged as a powerful tool providing a wide variety of novel disconnections simplifying access to, and accelerating the synthesis of complex molecules.1 Aryl olefins are synthetically important motifs, as useful intermediates in synthesis,2 and also due to their widespread presence in bioactive molecules and pharmaceuticals.3 Consequently, large efforts have been devoted to the development of C–H olefination methodologies.4 While a large number of strategies have been developed for the olefination of C–H bonds ortho to a directing group,5 comparatively few exist for those in meta positions.6 To date, the only available ‘direct’ strategy for meta-olefination, involves the use of the U-shaped directing groups pioneered by the group of Yu (Scheme 1a).7 This approach has been used to perform meta-olefinations on derivatives of benzyl alcohols,8N-methyl anilines,9 phenyl acetic acids,10 benzyl sulfonyl esters,11 benzoic acids,12 aromatic carbonyls13 and aryl boronic acids,14 using alkenes as coupling partners. The major drawback of this approach arises from the need to install the large U-shaped directing group, covalently bound, and its subsequent removal after the C–H olefination, as separate synthetic steps. In addition, stoichiometric toxic Ag(I)-salts are required as terminal oxidants in these oxidative couplings. A recent report has expanded the applicability of this strategy to the Rh(III)-catalysed meta-olefination of hydrocinnamic acid derivatives using alkynes as coupling partners (Scheme 1b).15 However, despite it being a redox neutral process, it still requires over three equivalents of Ag(I)-salts as an additive. Furthermore, in addition to the main meta-olefination product, 5–10% of the sometimes difficult to separate ortho and para olefination products are typically obtained. Additionally, the U-shaped directing group strategy is only applicable to aromatics containing a group that can be easily derivatised.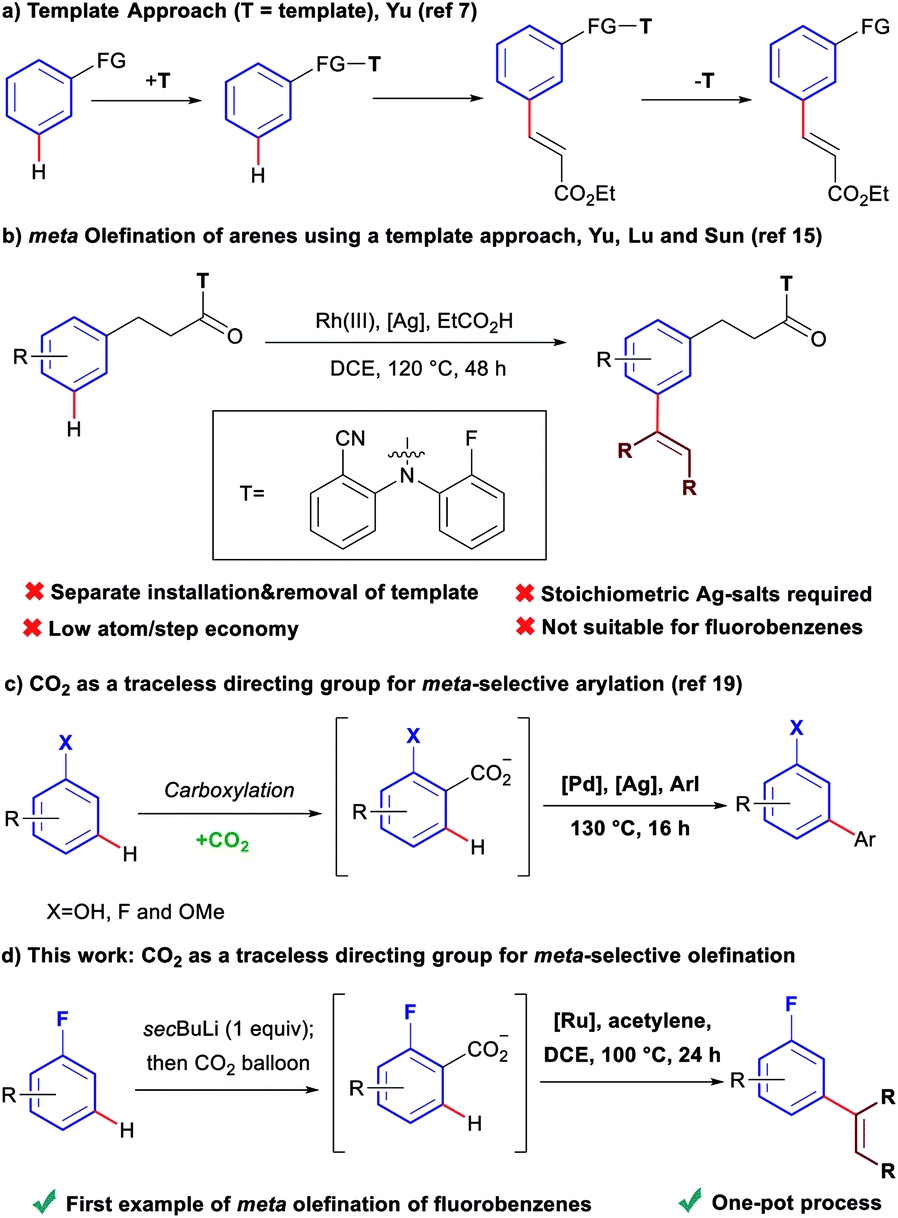 | ||
| Scheme 1 Comparison of the template approach and the traceless directing group relay strategy for the meta olefination of arenes. | ||
Fluoroarenes are recurring structural motifs in pharmaceuticals, agrochemicals, organic materials and other biologically relevant compounds.16 Approximately 30% of pharmaceuticals and 40% of agrochemicals currently contain at least one fluorine atom, usually at the aromatic ring.16 Thus, the direct C–H functionalisation of monofluorobenzenes can provide straightforward access to valuable materials (Fig. 1).3b–d While monofluoroarenes generally present low reactivity towards direct C–H olefination, a number of examples have been reported that use the arene as cosolvent to achieve ortho, para-selective olefination.17 Some pioneering methods for direct olefination using the fluoroarene as the limiting reagent have been reported by Yu et al., but mixtures of ortho, meta and para substitution are obtained.18 However, meta-selective olefination has never been achieved. Importantly, the U-shaped directing group strategy cannot be applied to this class of substrates. We have previously shown that CO2 can be used as a traceless directing group for the meta-selective arylation of phenols, fluorobenzenes and anisoles (Scheme 1c).19 The process relies on the easy carboxylation of these aromatic substrates, to install a temporary carboxylate directing group. The carboxylate can then direct the arylation before it is cleaved, in a tandem process, thus allowing a one-pot meta-arylation to proceed. However, the CO2 traceless directing group approach has never been demonstrated on any other type of functionalisation. Herein we report the first example of a meta-olefination of fluorobenzenes (Scheme 1d). This ruthenium-catalysed process involves the in situ installation and removal of a carboxylate, from CO2, uses alkynes as coupling partners and avoids the need for stoichiometric use of Ag(I)-salts.
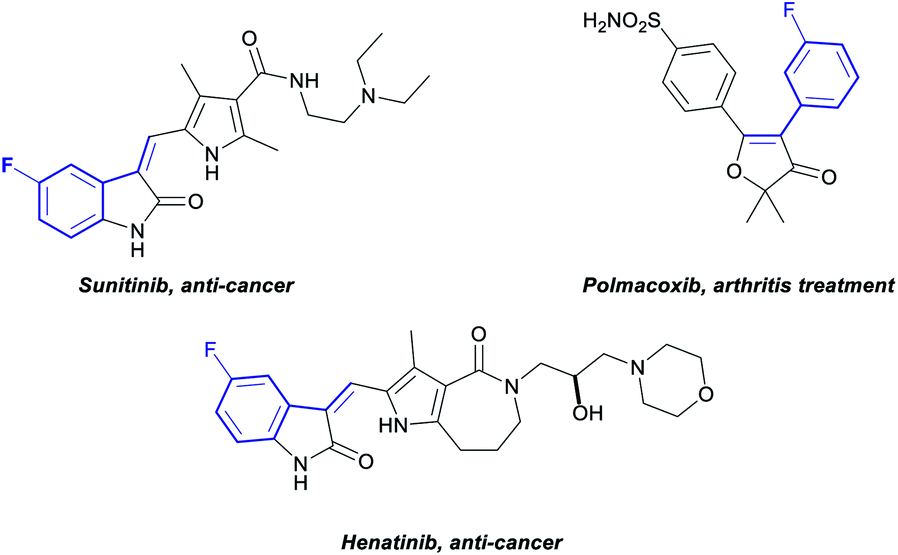 | ||
| Fig. 1 Commercially available pharmaceuticals containing a meta alkenyl fluoroarene. | ||
Results and discussion
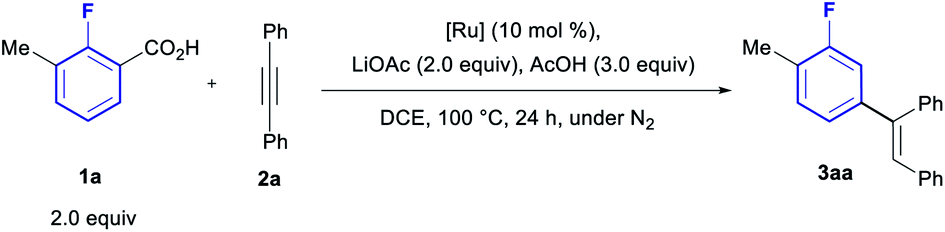
We have previously developed an optimised protocol for the lithiation/carboxylation of fluoroarenes suitable for combination in a one-pot process with a Pd-catalysed tandem arylation/protodecarboxylation, leading to the meta-arylation of fluoroarenes.19c In 2016, three methods for the Ru-catalysed tandem ortho-olefination/protodecarboxylation of benzoic acids by hydroarylation of alkynes were reported by Hartwig and Zhao,20 Ackermann21 and Gooβen.22 Miura and co-workers have also reported numerous methods for the ortho-olefination/protodecarboxylation of benzoic acids using acrylates and styrenes as coupling partners.23 We envisaged these methods could be ideally adapted to operate in combination with the directed ortho-metalation/carboxylation approach to furnish the desired meta-olefination of fluorarenes.We started our investigation by probing the decarboxylative olefination of the fluorotoluic acid 1a with diphenyl acetylene (2a), using Ackermann's protocol (Table 1, entry 1).21 To evaluate the effect of the installation of the carboxylic acid using ortho-lithiation during the desired one-pot process we tested the addition of 2 equiv. of LiOAc (entry 2), revealing a significant negative effect in reactivity. Gratifyingly, addition of 3 equiv. of AcOH efficiently reversed the effect of the presence of the Li-salt (entry 3), providing a method to ensure compatibility of the protocol with the carboxylation step. In previous work on Ru-catalysed ortho-arylation of polyfluorobenzenes we observed an inhibitory effect of coordinated p-cymene, leading to the development of the arene-free Ru-precatalyst [Ru(tBuCN)6][BF4]2.24 However, the use of this catalyst in the olefination reaction led to no product formation (entry 4), suggesting the η6-coordinated arene is essential for reactivity towards olefination. Accordingly, the weaker coordinating benzene-complex led to poor reactivity (entry 5), whereas the highly coordinating C6Me6-bearing Ru complex gave an improved yield, an effect which has previously been observed by Gooβen.25
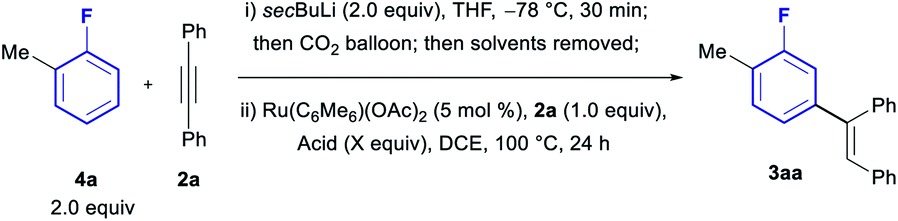
We then moved to optimize the full one-pot protocol, starting from ortho-fluorotoluene (4a, Table 2). Carboxylation of the fluoroarene was observed to occur in nearly quantitative conversion using secBuLi at −78 °C for 30 min, followed by quenching with CO2. Subsequent addition to the same flask of AcOH (3 equiv.), alkyne 2a and 5 mol% Ru(C6Me6)(OAc)2 in DCE led to the formation of the meta-olefinated product 3aa in an excellent 85% yield (entry 1). The use of 4 equiv. or 5 equiv. of AcOH led to reduced yields (entries 2 and 3). Examination of other organic acids also led to lower yields (entries 4–6), revealing AcOH as the optimal acid to facilitate this one-pot process.
|
|
||
|---|---|---|
| Entry | Acid (equiv.) | 4aa (yield%) |
| 1 | AcOH (3) | 85 |
| 2 | AcOH (4) | 75 |
| 3 | AcOH (5) | 72 |
| 4 | PivOH (3) | 1 |
| 5 | iBuCOOH (3) | 71 |
| 6 | TFA (3) | 5 |
With the optimised conditions in hand, we investigated the generality of the process with regards to the fluoroarene core (Scheme 2). Substitution patterns in ortho, meta or para positions were all tolerated, albeit only the relatively small F-atom was compatible in para (3ad). When larger groups were installed in the para position such as Me and Cl, no reactivity could be observed (3al and 3am). Furthermore, the reaction is in all cases completely selective towards mono-olefination and towards the meta position, with no traces of neither bisolefination nor other regioisomers observed by NMR and GCMS analysis of the reactions, even for simple fluorobenzene (3ab). Both electron withdrawing groups (3ac, 3ad, 3ag and 3ah) and donating groups (3aa, 3ae, 3af and 3aj) were compatible with the procedure. Chloroarenes (3ah) were also tolerated with no traces of de-halogenated products. Biaryl and naphthyl-based aromatic systems were also suitable substrates (3ai and 3ak).
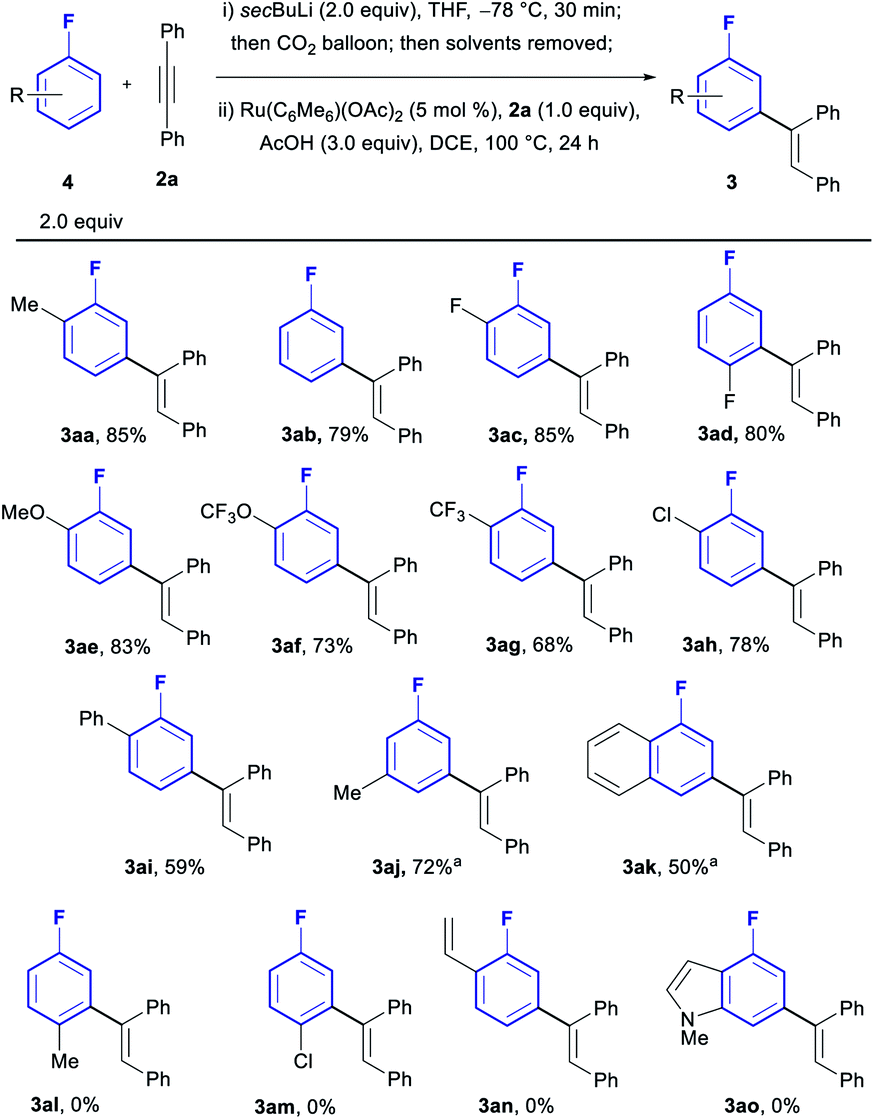 | ||
| Scheme 2 Scope in fluoroarene core. a10 mol% catalyst used. | ||
Subsequently we investigated the scope with respect to the alkyne coupling partner (Scheme 3). Both electron donating (3ba and 3ca) and electron withdrawing groups (3da, 3ea and 3fa) were reactive giving excellent yields. Heterocyclic moieties were also tolerated (3ga). While bisalkyl acetylenes were incompatible with the procedure (3la), unsymmetrical alkyl, aryl-acetylenes led to completely regioselective addition at the carbon adjacent to the alkyl group (3ha and 3ia). Diesters and unsymmetrical ester, aryl-acetylenes were also tolerated offering a handle for further functionalisation (3ja and 3ka) with ethyl phenyl propiolate preferentially forming the α-aryl ester (3ja). No product was observed when terminal acetylenes were used (3ma).
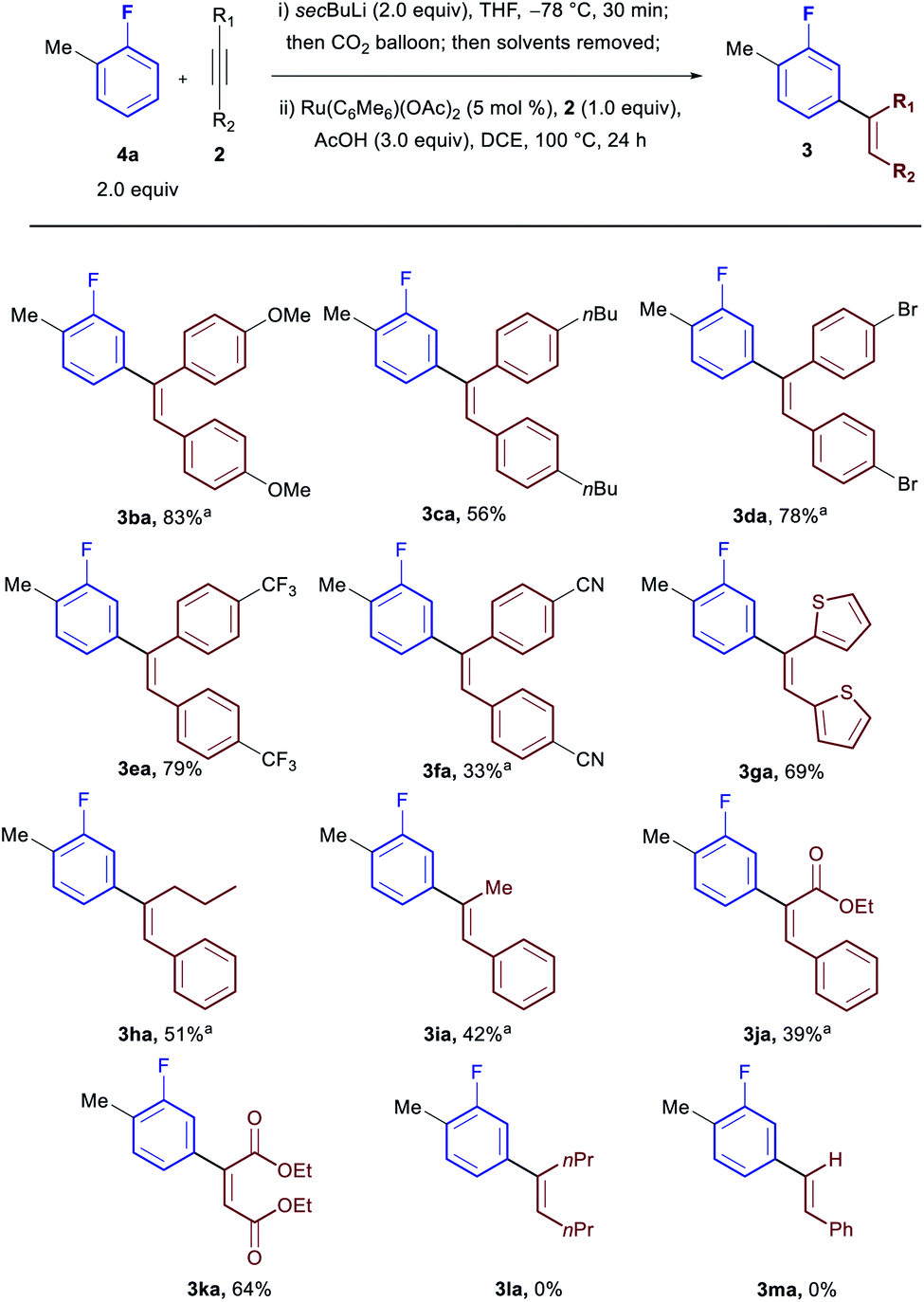 | ||
| Scheme 3 Scope in acetylene. a10 mol% catalyst used. | ||
This new meta-olefination methodology can be easily scaled up with, for example, 3aa being formed in 70% yield (1.10 g) without any changes to the protocol.
A plausible mechanism for this transformation is shown in Scheme 4, based on the mechanistic studies performed by Hartwig and Zhao.20ortho-Lithiation and carboxylation of fluoroarene 4 affords lithium benzoate 5. ortho-C–H activation of lithium benzoate 5 with ruthenium complex 6a affords cyclometallated complex 6b. Insertion of alkyne 2a into the Ru–C of 6b forms complex 6c, which can in turn decarboxylate to from the 5-membered metallocycle in complex 6d. Protonation of this complex with 2 equiv. of AcOH liberates the final product 3 and reforms complex 6a, thus closing the catalytic cycle.
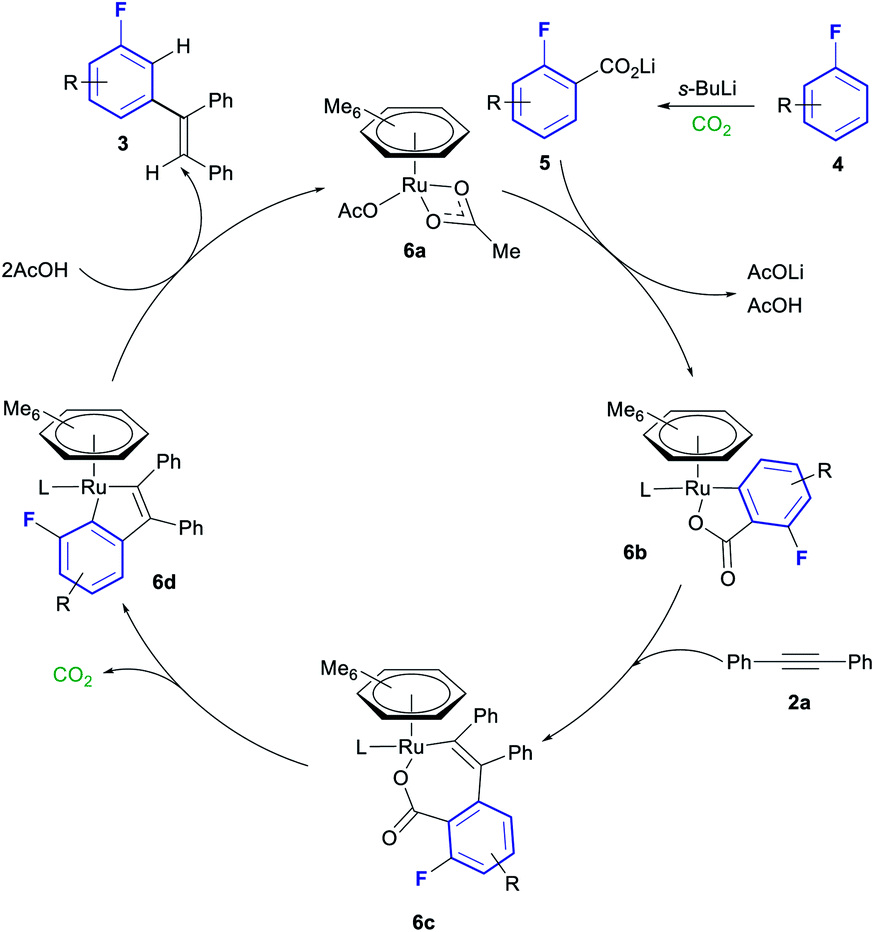 | ||
| Scheme 4 Plausible mechanism for the Ru catalysed meta olefination of fluoroarenes. | ||
Conclusions
In conclusion, we have developed the first example of a methodology for the meta-selective olefination of fluoroarenes. The natural ortho, para-reactivity of this class of substrates has been overcome by employing CO2 as a traceless directing group, that can be installed, used to control reactivity and then seamlessly removed in a one-pot process. Good to excellent yields can be obtained with a variety of functional groups and substitution patterns in both fluoroarene and alkyne, and in all cases complete meta-regioselectivity is observed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Engineering and Physical Sciences Research Council (EPSRC, EP/L014017/2) and the Marie Skłodowska Curie actions (IF-656841 to MF) for funding.Notes and references
- (a) R. H. Crabtree, Chem. Rev., 1985, 85, 245–269 CrossRef CAS; (b) A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed; (c) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS; (d) R. G. Bergman, Nature, 2007, 446, 391–393 CrossRef CAS; (e) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed; (f) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (g) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (h) C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS; (i) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC; (j) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS; (k) M. Tobisu and N. Chatani, Science, 2014, 343, 850–851 CrossRef CAS PubMed; (l) M. Simonetti, D. M. Cannas and I. Larrosa, Adv. Organomet. Chem., 2017, 67, 299–399 CrossRef CAS.
- Selected examples of aryl olefins as intermediates for the synthesis of bioactive molecules (a) T. Nishimata, Y. Sato and M. Mori, J. Org. Chem., 2004, 69, 1837–1843 CrossRef CAS PubMed; (b) L. V. White, B. D. Schwartz, M. G. Banwell and A. C. Willis, J. Org. Chem., 2011, 76, 6250–6257 CrossRef CAS PubMed; (c) C. Singh, M. Hassam, V. P. Verma, A. S. Singh, N. K. Naikade, S. K. Puri, P. R. Maulik and R. Kant, J. Med. Chem., 2012, 55, 10662–10673 CrossRef CAS PubMed.
- Examples of aryl olefins in bioactive molecules (a) J. Joseph-Charles and M. Bertucat, Anal. Chim. Acta, 1993, 284, 45–52 CrossRef CAS; (b) D. B. Mendel, A. D. Laird, X. Xin, S. G. Louie, J. G. Christensen, G. Li, R. E. Schreck, T. J. Abrams, T. J. Ngai, L. B. Lee, L. J. Murray, J. Carver, E. Chan, K. G. Moss, J. Ö. Haznedar, J. Sukbuntherng, R. A. Blake, L. Sun, C. Tang, T. Miller, S. Shirazian, G. McMahon and J. M. Cherrington, Clin. Cancer Res., 2003, 9, 327–337 CAS; (c) S. Hirankarn, J. S. Barrett, N. Alamuddin, G. A. FitzGerald and C. Skarke, Clin. Pharmacol. Drug Dev., 2013, 2, 379–386 CrossRef CAS PubMed; (d) J. Qian, Y. Wang, J. Cao and J. Li, J. Pharm. Biomed. Anal., 2013, 80, 173–179 CrossRef CAS PubMed.
- Selected reviews on C–H olefination of arenes (a) C. Nevado and A. M. Echavarren, Synthesis, 2005, 2, 167–182 Search PubMed; (b) V. P. Boyarskiy, D. S. Ryabukhin, N. A. Bokach and A. V. Vasilyev, Chem. Rev., 2016, 10, 5894–5986 CrossRef PubMed; (c) W. Ma, P. Gandeepan, J. Li and L. Ackermann, Org. Chem. Front., 2017, 4, 1435–1467 RSC.
- Selected examples of ortho C–H olefination (a) G. Cai, Y. Fu, Y. Li, X. Wan and Z. Shi, J. Am. Chem. Soc., 2007, 129, 7666–7673 CrossRef CAS; (b) L. Wang, S. Liu, Z. Li and Y. Yu, Org. Lett., 2011, 13, 6137–6139 CrossRef CAS PubMed; (c) B. Liu, H.-Z. Jiang and B.-F. Shi, J. Org. Chem., 2014, 79, 1521–1526 CrossRef CAS PubMed; (d) A. Deb, S. Bag, R. Kancherla and D. Maiti, J. Am. Chem. Soc., 2014, 136, 13602–13605 CrossRef CAS PubMed; (e) G. Li, L. Wan, G. Zhang, D. Leow, J. Sprangler and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 4391–4397 CrossRef CAS PubMed.
- Selected reviews on meta selective C–H functionalisation (a) J. Yang, Org. Biomol. Chem., 2015, 13, 1930–1941 RSC; (b) J. Lie, S. De Sarkar and L. Ackermann, Top. Organomet. Chem., 2015, 55, 217–257 CrossRef; (c) A. Dey, S. Agasti and D. Maiti, Org. Biomol. Chem., 2016, 14, 5440–5453 RSC; (d) J. A. Leitch and C. G. Frost, Chem. Soc. Rev., 2017, 46, 7145–7153 RSC.
- D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed.
- (a) S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778–18781 CrossRef CAS PubMed; (b) S. Fang, X. Wang, F. Yin, P. Cai, H. Yang and L. Kong, Org. Lett., 2019, 21, 1841–1844 CrossRef CAS PubMed.
- R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215–220 CrossRef CAS PubMed.
- (a) M. Bera, A. Modak, T. Patra, A. Maji and D. Maiti, Org. Lett., 2014, 16, 5760–5763 CrossRef CAS PubMed; (b) Y. Deng and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 888–891 CrossRef CAS PubMed.
- (a) M. Bera, A. Maji, S. K. Sahoo and D. Maiti, Angew. Chem., Int. Ed., 2015, 54, 8515–8519 CrossRef CAS PubMed; (b) M. Brochetta, T. Borsari, S. Bag, S. Jana, S. Maiti, A. Porta, D. B. Werz, G. Zanoni and D. Maiti, Chem.–Eur. J., 2019, 25, 10323–10327 CrossRef CAS PubMed.
- S. Li, L. Cai, H. Ji, L. Yang and G. Li, Nat. Commun., 2016, 7, 10443 CrossRef PubMed.
- S. Xie, S. Li, W. Ma, X. Xu and Z. Jin, Chem. Commun., 2019, 55, 12408–12411 RSC.
- A. F. Williams, A. J. P. White, A. C. Spivey and C. J. Cordier, Chem. Sci., 2020, 11, 3301–3306 RSC.
- H.-J. Xu, Y.-S. Kang, H. Shi, P. Zhang, Y.-K. Chen, B. Zhang, Z.-Q. Liu, J. Zhao, W.-Y. Sun, J.-Q. Yu and Y. Lu, J. Am. Chem. Soc., 2019, 141, 76–79 CrossRef CAS PubMed.
- (a) P. Kirsch and M. Bremer, Angew. Chem., Int. Ed., 2000, 39, 4216–4235 CrossRef CAS; (b) W. R. Dolbier, J. Fluorine Chem., 2005, 126, 157–163 CrossRef CAS; (c) C. Isanbor and D. O'Hagan, J. Fluorine Chem., 2006, 127, 303–319 CrossRef CAS; (d) F. Babudri, G. M. Farinola, F. Naso and R. Ragni, Chem. Commun., 2007, 1003–1022 RSC; (e) M. Hird, Chem. Soc. Rev., 2007, 36, 2070–2095 RSC; (f) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (g) P. Jeschke, Pest Manage. Sci., 2010, 66, 10–27 CrossRef CAS PubMed; (h) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC; (i) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (j) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed; (k) T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS; (l) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- (a) K. S. Kanyiva, N. Kashihara, Y. Nakao, T. Hiyama, M. Ohashi and S. Ogoshi, Dalton Trans., 2010, 39, 10483–10494 RSC; (b) C.-H. Ying, S.-B. Yan and W.-L. Duan, Org. Lett., 2014, 16, 500–503 CrossRef CAS PubMed; (c) L. G. Y. Chung, N. A. B. Juwaini and J. Seayad, ChemCatChem, 2015, 7, 1270–1274 CrossRef CAS.
- (a) H. U. Vora, A. P. Silvestri, C. J. Engelin and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 2683–2686 CrossRef CAS PubMed; (b) P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K.-S. Yeung and J.-Q. Yu, Nature, 2017, 551, 489–493 CrossRef CAS PubMed.
- (a) J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109–4112 CrossRef CAS PubMed; (b) J. Luo, S. Preciado, S. O. Araromi and I. Larrosa, Chem.–Asian J., 2016, 11, 347–350 CrossRef CAS PubMed; (c) M. Font, A. R. A. Spencer and I. Larrosa, Chem. Sci., 2018, 9, 7133–7137 RSC.
- J. Zhang, R. Shrestha, J. F. Hartwig and P. Zhao, Nat. Chem., 2016, 8, 1144–1151 CrossRef CAS PubMed.
- N. Y. P. Kumar, A. Bechtoldt, K. Raghuvanshi and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 6929–6932 CrossRef CAS PubMed.
- L. Huang, A. Biafora, G. Zhang, V. Bragoni and L. J. Gooßen, Angew. Chem., Int. Ed., 2016, 55, 6933–6937 CrossRef CAS PubMed.
- (a) A. Maehara, H. Tsurugi, T. Satoh and M. Miura, Org. Lett., 2008, 10, 1159–1162 CrossRef CAS PubMed; (b) S. Mochida, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 12, 5776–5779 CrossRef CAS PubMed; (c) S. Mochida, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2011, 76, 3024–3033 CrossRef CAS PubMed.
- M. Simonetti, G. J. P. Perry, X. C. Cambeiro, F. Juliá-Hernández, J. N. Arokianathar and I. Larrosa, J. Am. Chem. Soc., 2016, 138, 3596–3606 CrossRef CAS PubMed.
- A. Biafora, B. A. Khan, J. Bahri, J. M. Hewer and L. J. Gooβen, Org. Lett., 2017, 19, 1232–1235 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01138j |
| This journal is © The Royal Society of Chemistry 2020 |