
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic aspects of saccharide dehydration to furan derivatives for reaction media design
Thibaut Istasse
 * and
Aurore Richel
* and
Aurore Richel
Laboratory of Biomass and Green Technologies, University of Liege – Gembloux Agro-Bio Tech, Passage des Déportés 2, B-5030 Gembloux, Belgium. E-mail: thibaut.istasse@uliege.be
First published on 22nd June 2020
Abstract
The conversion of abundant hexoses (e.g. glucose, mannose and galactose) and pentoses (e.g. xylose and arabinose) to 5-hydroxymethylfurfural (5-HMF) and 2-furfural (2-F) is subject to intensive research in the hope of achieving competitive production of diverse materials from renewable resources. However, the abundance of literature on this topic as well as the limited number of studies systematically comparing numerous monosaccharides hinder progress tracking. Herein, we compare and rationalize reactivities of different ketoses and aldoses. Dehydration mechanisms of both monosaccharide types are reviewed regarding the existing experimental evidence. Ketose transformation to furan derivatives likely proceeds through cyclic intermediates and is hindered by side-reactions such as isomerization, retro-aldol reactions and polymerization. Different strategies can improve furan derivative synthesis from ketoses: limiting the presence of water, improving the dehydration rate, protecting 5-HMF and 2-F reactive moieties with derivatization or solvent interactions and extracting 5-HMF and 2-F from the reaction medium. In contrast to ketoses, aldose conversion to furan derivatives is not favored compared to polymerization reactions because it involves their isomerization or a ring contraction. Enhancing aldose isomerization is possible with metal catalysts (e.g. CrCl3) promoting a hydride shift mechanism or with boric/boronic acids promoting an enediol mechanism. This catalysis is however far more challenging than ketose dehydration because catalyst activity depends on numerous factors: Brønsted acidity of the medium, catalyst ligands, catalyst affinity for monosaccharides and their accessibility to several chemical species simultaneously. Those aspects are methodically addressed to support the design of new monosaccharide dehydration systems.
 Thibaut Istasse | Thibaut Istasse obtained a bioengineering degree in chemistry and bioindustries in 2015. He is currently working in the Laboratory of Biomass and Green Technologies at the University of Liège (Belgium). His research is conducted as part of the Low Carbon Footprint Materials (BIOMAT) project supported by the Walloon Region and the European Regional Development Fund. The project aims to develop new plastic materials from plant feedstock. For this purpose, Thibaut Istasse's research is mainly focused on the synthesis of furan building blocks from poly- and monosaccharides at moderate temperature in eutectic solvents based on cheap and safe components. |
 Aurore Richel | Aurore Richel has a PhD in chemistry. She is currently a full Professor and head of the Laboratory of Biomass and Green Technologies at the University of Liege (Belgium). The laboratory is engaged in research and education in the field of chemistry of renewable resources and associated technologies. A. Richel is involved in more than 15 national and international projects related to the production of bioenergy and bioproducts, including the design of innovative pretreatment protocols for the cracking of lignocellulosic materials, the formulation of novel “biomass-energy” solutions (including biofuels and energy vectors) as well as the upgrade of lignin for high-added value applications. She set up in 2018 an international platform dedicated to biomass and related technologies in collaboration with Kobe University (Japan), and Texas A&M (US). She is the author of more than 120 publications in international journals, several book chapters, patents, and a book dedicated to “Lignin and Hemicelluloses In Biorefinery” (CRC press). She is also active in the popularization of chemistry, notably via her personal website (http://www.chem4us.be). |
1. Introduction
Nowadays, most industrial chemicals are obtained from fossil resources. However, depletion and price fluctuations of these resources motivated the search for alternative and renewable sources of building blocks for the chemical industry.In 2004, the report “Top Value-Added Chemicals From Biomass” of the US department of energy led to a renewed interest in biobased building blocks.1 Among these platform molecules, 5-hydroxymethylfurfural (5-HMF) and 2-furfural (2-F) have been the subject of numerous studies to understand their synthesis and assess their potential for plastic and fuel production.
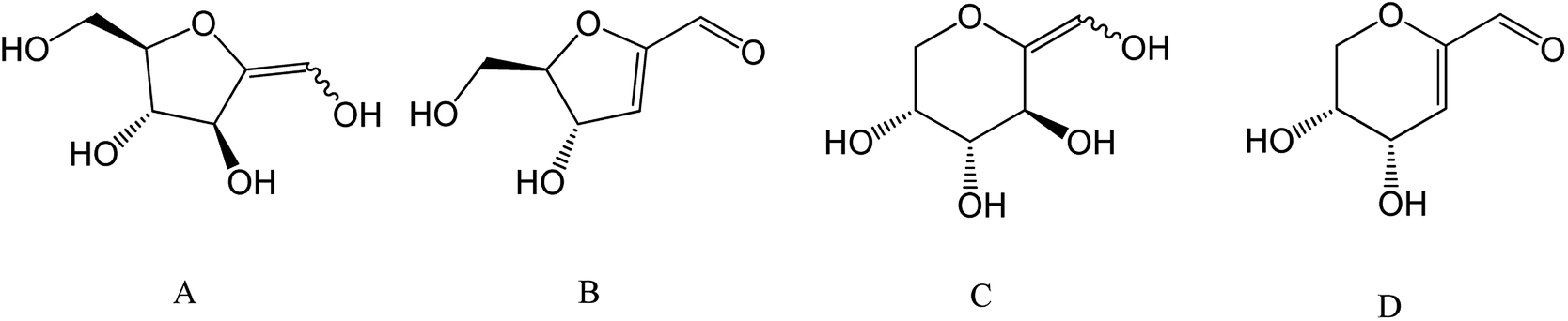
5-HMF and 2-F are both dehydration products of monosaccharides obtained through the loss of three water molecules during acid-catalyzed reactions. 5-HMF is specifically generated from hexoses such as fructose, glucose, mannose and galactose while 2-F is produced from pentoses such as xylulose, xylose or arabinose. Several of those monosaccharides are commonly found in plants cells wall as components of cellulose (glucose) and hemicellulose (glucose, mannose, xylose, arabinose, …) as depicted in Fig. 1.
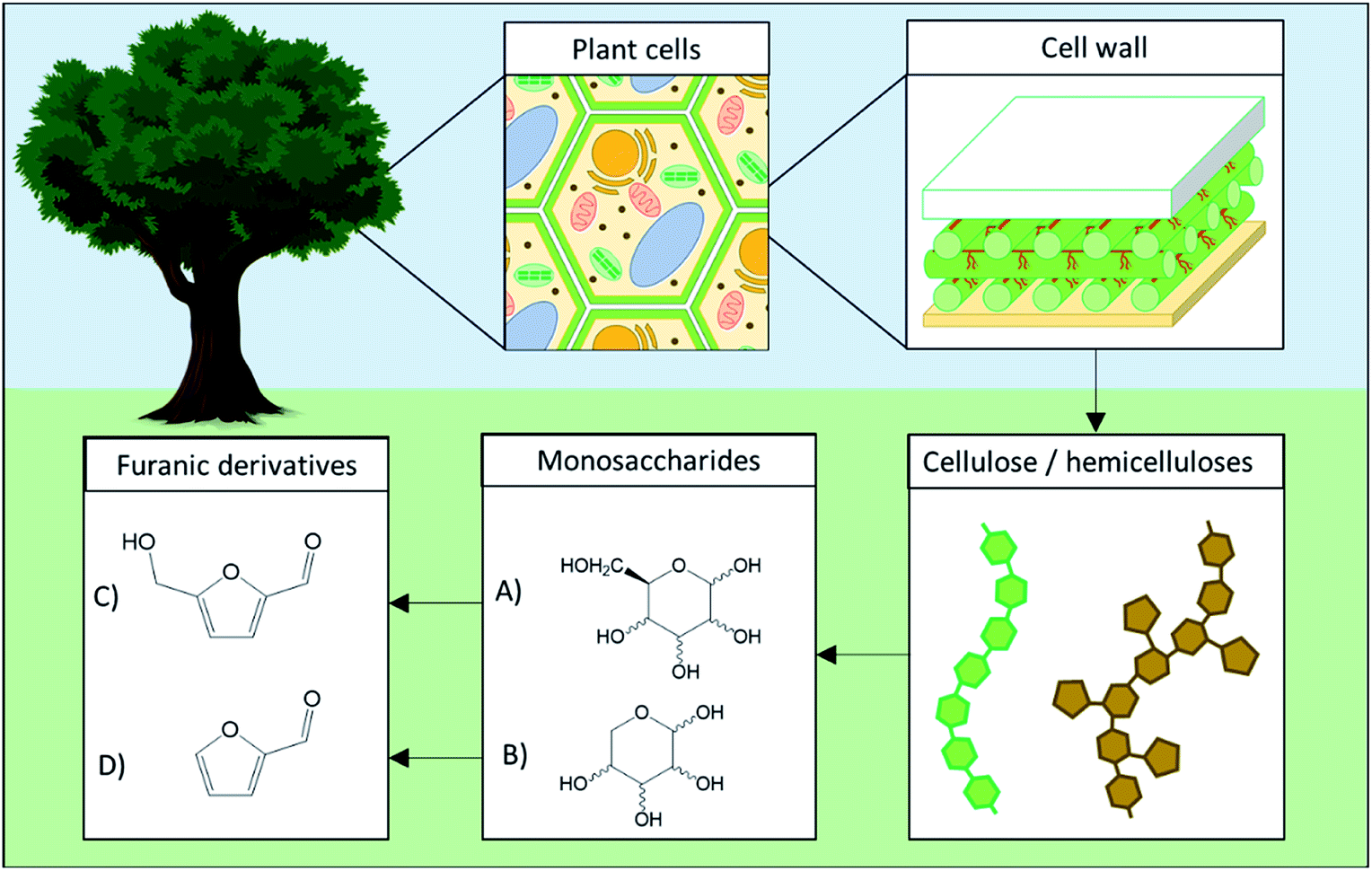 | ||
| Fig. 1 Overview of plant cell wall polysaccharides transformation to monosaccharides and their corresponding furan derivatives. (A) Hexose, (B) pentose, (C) 5-hydroxymethylfurfural, (D) 2-furfural. | ||
5-HMF can be converted into molecules of interest via diverse reaction pathways. Selective oxidation of the formyl, hydroxyl or both groups respectively leads to 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), 2,5-diformylfuran (DFF) or 2,5-furandicarboxylic acid (FDCA) (Rosatella 2011).3 While HMFCA is only an intermediate, DFF can find applications as fungicides, pharmaceutics, organic conductors and macrocyclic ligands. Regarding FDCA, the compound is cited in the TOP 12 biobased opportunities of the US department of energy and could serve as monomer in the synthesis of new plastics.2
The selective reduction of 5-HMF formyl group generates 2,5-bis(hydroxymethyl)furan which has potential for polymers and polyurethane foams production. Reduction of both formyl and hydroxyl groups leads to the formation of 2,5-dimethylfuran with potential application as biofuel.3
If 5-HMF has been presented as a promising renewable building block, its industrial production is still limited by major hurdles. Its synthesis from glucose, the cheapest and most abundant hexose, suffers from a lack of selectivity. Technologies allowing to achieve high selectivity exist but are generally complex or too expensive. 5-HMF synthesis consequently remains at pilot scale and is based on fructose dehydration. Avantium (the Netherlands) and Ava-Biochem (Switzerland) are examples of companies active in this field. Their annual production is in the order of several dozens of tons, targeting notably FDCA for poly(ethylene furanoate) manufacture.4,5 The current pilot plant of Avantium reaches a FDCA production capacity of 20–40 ton per year but the company is considering the creation of a flagship facility with a FDCA production capacity of 5000 ton per year.6,7 Avantium process for the dehydration of fructose is performed in methanol to generate a more stable derivative of 5-HMF, methoxymethylfurfural (MMF).8 Ava-Biochem rather developed a water-based process.5
Regarding the furan counterpart obtained from pentoses (e.g. xylose, arabinose), 2-F, its market is already well established with a global production of several hundred thousand ton per year.9,10 2-F is produced by acid hydrolysis of pentosan in agricultural residues (oat hulls, cornstalks, wheat straw, sugar cane bagasse, …). 2-F is mainly transformed to furfuryl alcohol for the production of thermosetting resins but can also be used in the manufacture of fuels, pharmaceuticals, plastics, fungicides and nematocides.11 China is the main producer of 2-F (e.g. Hebei Xingtai Chunlei Furfuryl Alcohol Co.).9,10 The central Romana Corporation in the Dominican Republic is also an important actor, producing 2-F (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ton per year) from sugar-cane bagasse.12 2-F is then transformed into furfuryl alcohol by Transfurans Chemicals in Belgium, again mainly to produce thermoset resins for the foundry industry.13 Although the market exists, production processes are still constrained by 2-F yields around 50%, long reaction time and large consumption of vapor (Quaker Oats technology).14,15
000 ton per year) from sugar-cane bagasse.12 2-F is then transformed into furfuryl alcohol by Transfurans Chemicals in Belgium, again mainly to produce thermoset resins for the foundry industry.13 Although the market exists, production processes are still constrained by 2-F yields around 50%, long reaction time and large consumption of vapor (Quaker Oats technology).14,15
While tremendous research efforts have been made to improve furan derivatives synthesis, the abundance of literature about the subject makes progress-tracking as well as mechanistic understanding difficult. The mechanisms explaining hexoses and pentoses dehydration to 5-HMF and 2-F are still not clear. Several chemical pathways have been proposed, sometimes with experimental evidences, but no consensus has been achieved. It is quite surprising that different dehydration mechanisms have been proposed for pentoses and hexoses whereas those substrates and their corresponding furan derivatives share a lot of similarities. Elucidation of dehydration mechanisms is challenging since a tremendous number of solvents and catalysts, often arbitrarily selected, have been tested and compared.
A first purpose of this review is to harmonize monosaccharides dehydration mechanisms regarding the last experimental investigations. Dehydration of ketoses (e.g. D-fructose, D-tagatose, D-xylulose, D-psicose, D-sorbose) to furan derivatives is much faster than dehydration of aldoses (e.g. D-glucose, D-mannose, D-galactose, D-xylose, D-arabinose). The first part of this work explains why ketoses are readily dehydrated and how selectivity for furan products can be enhanced simply through solvent choice.
However, ketoses are generally not the main components of most plant polysaccharides. Polysaccharides of vegetal biomass rather contains aldohexoses such as glucose, mannose, galactose and aldopentoses like xylose and arabinose. Understanding aldoses dehydration is consequently crucial because they represent a more abundant resource than ketoses. However, lower 5-HMF and 2-F yields are achieved from aldoses (typically less than 5% for D-glucose at 200 °C during 5 min without catalyst) than from ketoses (around 40% for D-fructose treated in similar conditions).16 Aldoses are also notably less reactive than ketose, requiring higher temperature to reach similar dehydration rate. The second part of this work details why aldoses are difficult to convert to furan products compared to ketoses and how catalyst/solvent combinations come into play to favor their conversion to 5-HMF or 2-F.
Both those parts highlight the required elements to constitute an efficient reaction medium for monosaccharide dehydration. A close attention is paid to the latest developments about ionic liquids and deep eutectic solvents in this context as well as potential synergy between catalysts.
2. Dehydration of ketoses
Fructose, psicose, sorbose and tagatose are ketohexoses of which dehydration leads to 5-HMF. Xylulose and ribulose are ketopentoses and their dehydration consequently results in 2-F formation (Fig. 2). While those ketoses are scarce in vegetal biomass, understanding their fast conversion to furan derivatives brings light on dehydration mechanism of monosaccharides in general.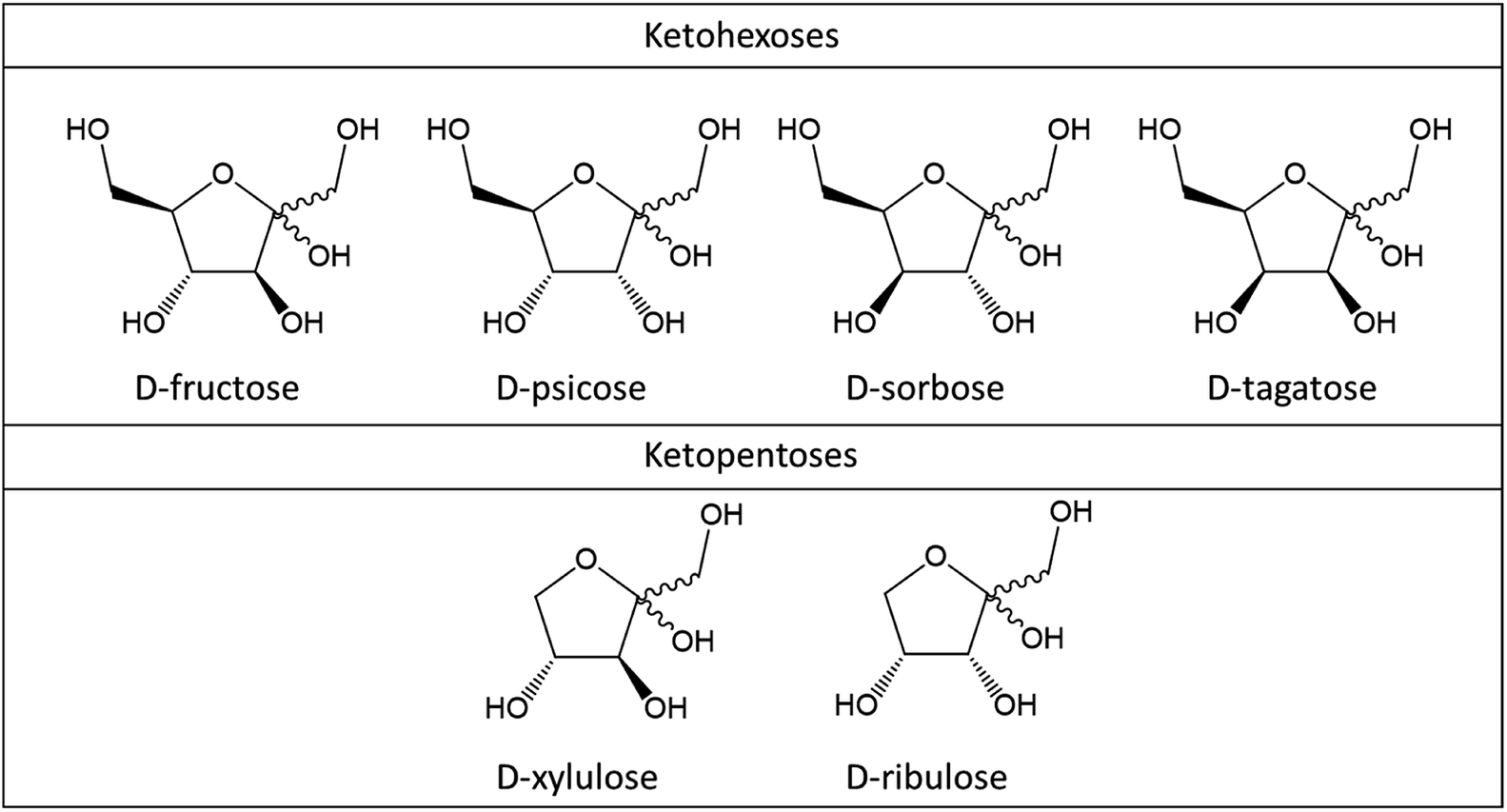 | ||
| Fig. 2 Chemical structures (Haworth's projections) of ketohexoses and ketopentoses. | ||
2.1. Dehydration mechanism
Although numerous mechanisms were proposed to explain dehydration of ketoses to 5-HMF and 2-F, all reaction paths involve three protonation steps and the loss of three water molecules.17 Mechanistic studies about ketopentoses dehydration being scarce, the following discussion is mainly based on ketohexoses dehydration completed by kinetic studies on ketopentoses.The first attempts to describe D-fructose dehydration mechanisms were initially based on reaction paths involving acyclic intermediates like 1,2-enediol and 3-deoxyhexosulose as depicted in Fig. 3(A).18–20 Since 1,2-enediol is also an intermediate of the isomerization of D-glucose into D-fructose, D-glucose and D-fructose acyclic dehydration pathways toward 5-HMF were considered similar. Because acyclic D-fructose is relatively more abundant in aqueous solution than acyclic D-glucose, an acyclic dehydration mechanism was thought consistent regarding the higher 5-HMF yields obtained with D-fructose.21
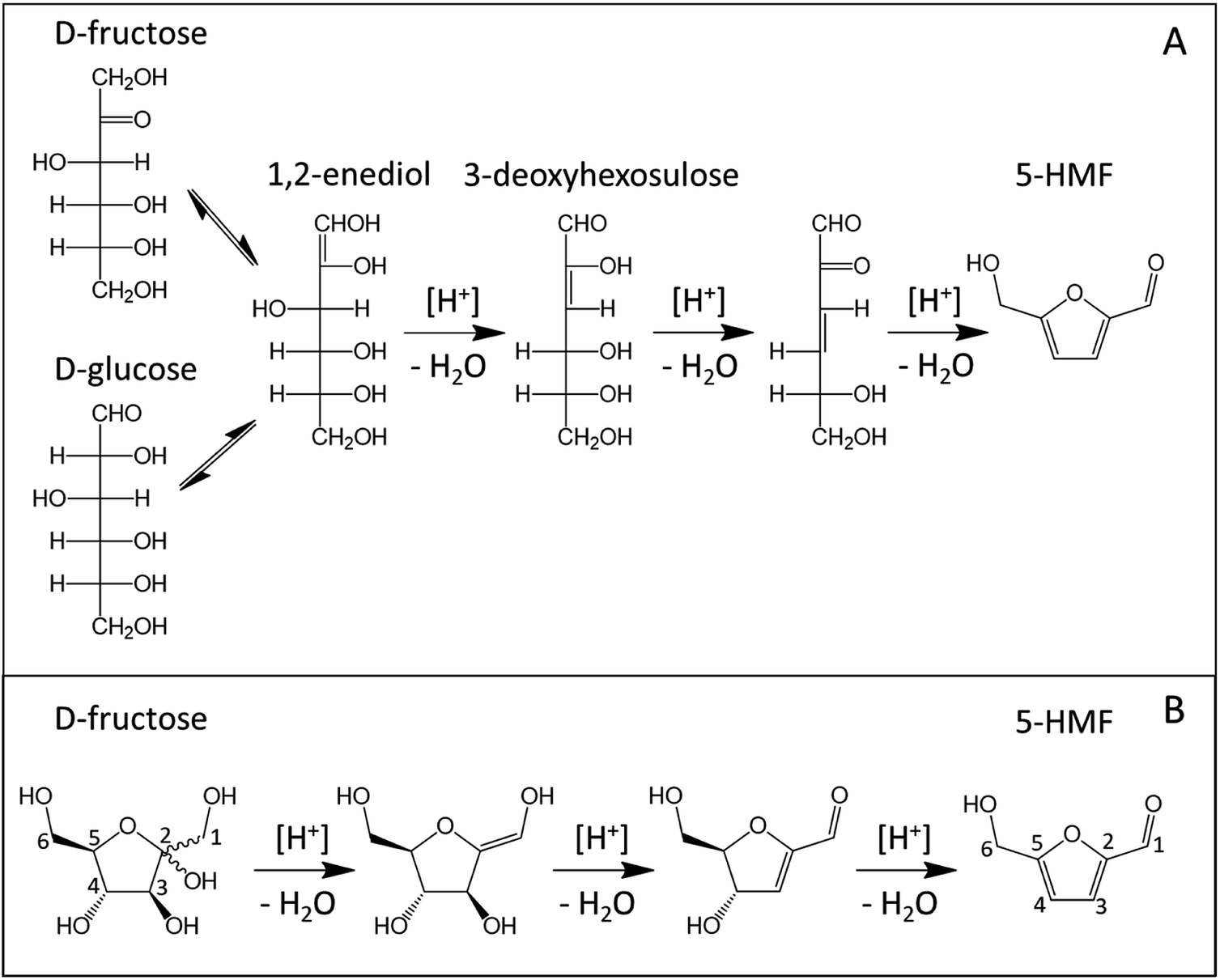 | ||
| Fig. 3 Acyclic (A) vs. cyclic (B) D-fructose dehydration pathway toward 5-HMF (carbon atoms labelling displayed in the cyclic path). | ||
More recent works now support reaction paths with cyclic intermediates to explain the dehydration of D-fructose into 5-HMF (Fig. 3B).18,19,22–24 Cyclic paths toward 5-HMF are initiated by protonation of the C2 hydroxyl group on fructose which is the most favored protonation site because of the high stability of the formed fructofuranosyl carbocation (carbon numbers are given in Fig. 3). The cationic species seems stabilized by its resonant structure (Fig. 4). The protonations of other oxygens lead to non-resonant structures and less stable carbocations.23
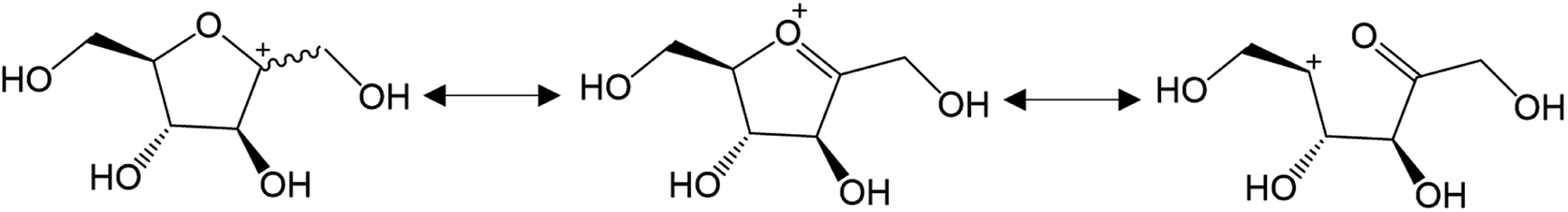 | ||
| Fig. 4 Carbocation resonance hybrids after first protonation and dehydration of D-fructose at O2. | ||
Cyclic mechanisms for 5-HMF formation are supported by several experimental observations. Firstly, dehydration of D-fructose was performed in deuterated water and no deuterium incorporation to 5-HMF was observed while acyclic pathways imply this incorporation because of the equilibrium between 3-deoxyhexosulose and its enolic tautomer. Secondly, 5-HMF yield calculated with the fructofuranosyl unit of sucrose (53%) is higher than the 5-HMF yield obtained with fructose (42%) at 250 °C after 32 seconds with sulfuric acid (1 mM). This observation suggests that the fructofuranosyl cation released from sucrose is an important intermediate during 5-HMF synthesis.18
In the gas phase, a mass spectrometry investigation highlighted the formation of a diagnostic ion at m/z 85 typical of ring structures (produced by a cross-ring bond cleavage), further supporting a dehydration pathway through cyclic intermediate.25 Dehydration product ions were observed at m/z 163, 145 and 127 formed through the loss of one, two and three water molecules respectively.
Cyclic intermediates mechanisms are also supported by the difference in reactivity between ketoses. Fructose, sorbose, psicose and tagatose differ from each other by the position of C3 and C4 hydroxyl groups. In aqueous sulfuric acid (100–160 °C, 33–300 mM H2SO4), tagatose and psicose are more reactive (higher conversion rate) than fructose and sorbose. However, 5-HMF formation is more selective from psicose and fructose.26,27 For acyclic pathways involving 1,2-enediol and 3-deoxyhexosulose as intermediates, positions of C3 and C4 hydroxyl groups on ketoses are not relevant to explain the observed differences in reactivity and selectivity, which is why cyclic mechanisms are favored.26
The higher reactivity of tagatose and psicose could be explained by cis orientation of C3–OH and C4–OH. This might cause a higher torsional strain as well as steric hindrance, leaving C2 more vulnerable to reaction on the opposite side of the furanose ring.28
Interestingly, several cyclic intermediates of the dehydration pathway in Fig. 3(B) have been identified by NMR monitoring of normal and 13C-labelled D-fructose dehydration in DMSO as well as ESI-MS (compounds A and B of Fig. 5).29–32 Both compounds are described as intermediates of the reaction since they progressively appear at the beginning of the reaction then disappear at the end of the treatment.29,30 Compounds C and D of Fig. 5 are intermediates obtained from fructopyranose and hypothetically lead to polymerized side-products (humins). The only major products observed in DMSO during D-fructose dehydration were 5-HMF, 2,6-anhydro-beta-D-fructofuranose and fructose dianhydrides.
 | ||
| Fig. 5 Intermediate species observed by NMR during dehydration of D-fructose in DMSO at 150 °C. | ||
NMR dehydration experiments on 13C labelled D-fructose confirmed that the ketose C1 becomes the carbon of the aldehyde moiety in the furan structure.32
As well as experimental evidences, reactions simulations support cyclic pathways from D-fructose to 5-HMF since ketoses dehydration acyclic mechanisms involve very high activation barriers.33
To our knowledge, no mechanistic insight has been provided yet regarding ketopentoses (xylulose and ribulose) dehydration. However, the higher reactivity of xylulose and ribulose compared to xylose and ribose has been confirmed. After 45 min at 145 °C in an aqueous solution of HCl (pH 1), 66% of xylulose is converted to 2-F. In the same conditions, xylose only leads to a 29% 2-F yield.34 The same trend was observed by Li et al. (2013) in close conditions (68% 2-F from xylulose after 25 min at 130 °C in 0.1 M HCl).35 Energy barrier of xylulose dehydration is lower (23 kcal mol−1) than energy barrier for xylose dehydration (30–32 kcal mol−1).34 In deuterated water at pH 1.5 at 96 °C, xylulose and ribulose are converted to 40–60% after 10 min while xylose and ribose are converted to only 0–10% in the same conditions.36
In brief, ketohexoses and ketopentoses are readily converted to furan derivatives because their cyclisation involves the C2 carbon (bearing the ketone moiety). Consequently, a carbocation stabilized by resonance can be formed after protonation of C2–OH and a first dehydration step, leaving the C1–OH available to constitute the future aldehyde moiety of 5-HMF or 2-F after further dehydrations.
2.2. Side-products of ketoses dehydration
Furan derivatives are major products of ketoses degradation in acidic water but many other side products are also generated limiting the selectivity. Ketoses reactions consist in tautomerization/isomerization, dehydration, retro-aldol reactions and polymerization.18,19,23,26,37–43 Those reactions are illustrated with D-fructose in Fig. 6.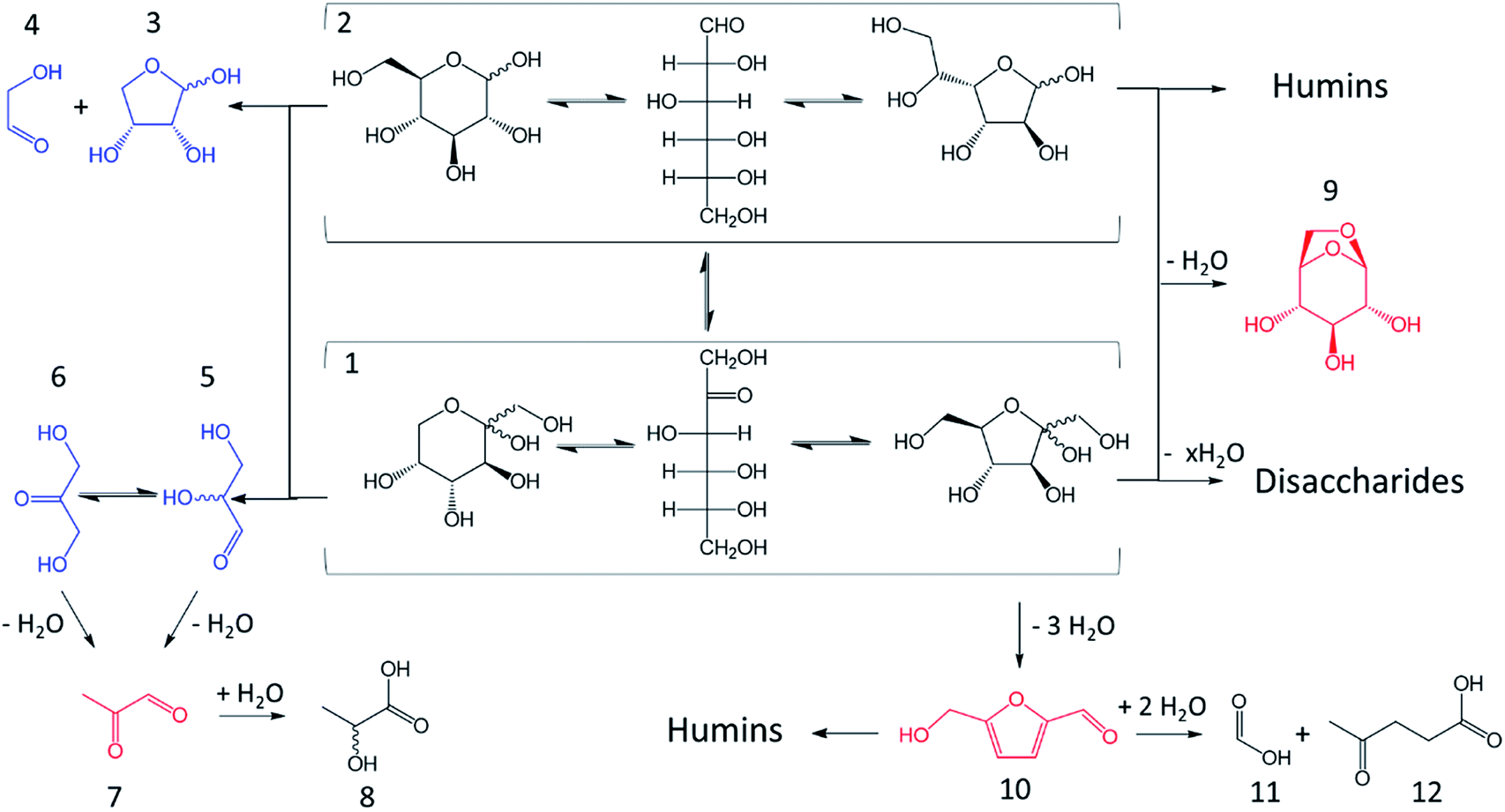 | ||
| Fig. 6 Typical compounds observed during subcritical treatment of a D-fructose aqueous solution with Brønsted acid. 1: fructopyranose, acyclic fructose, fructofuranose, 2: glucopyranose, acyclic glucose, glucofuranose, 3: erythrose, 4: glycolaldehyde, 5: glyceraldehyde, 6: dihydroxyacetone, 7: pyruvaldehyde, 8: lactic acid, 9: 1,6-anhydroglucose (1,6-anhydrofructose also observed in subcritical treatment), 10: 5-hydroxymethylfurfural, 11: formic acid, 12: levulinic acid. Blue = retro aldol reaction products, red = dehydration reaction products. | ||
Isomerization of ketose to aldose is a first possible side-reaction. Its rate is however limited compared to other reactions under Brønsted acidic conditions. In that respect, the molar yield of D-glucose from D-fructose is generally lower than 5% (32 s, 250 °C H2SO4 0–5 mM).18 Tautomerization rate between pyranose and furanose forms of ketoses is significantly larger than the isomerization rate to aldose or the dehydration rate to furan compounds according to in situ 13C NMR kinetic study.38
Dehydration of ketoses mainly leads to 5-HMF or 2-F. 5-HMF molar yield is typically between 20 and 50% for D-fructose dehydration in water at 200–250 °C after 0.5 to 5 minutes with Brønsted acids like sulfuric, hydrochloric, phosphoric or formic acids.16,23,44,45 5-HMF is nevertheless susceptible to rehydration leading to formic and levulinic acids as depicted in Fig. 7 (above). Ketoses as well as aldoses can also be dehydrated to anhydro-monosaccharides after the loss of only one water molecule (e.g. 1,6-anhydrofructose, 1,6-anhydroglucose, 1,5-anhydroxylose).
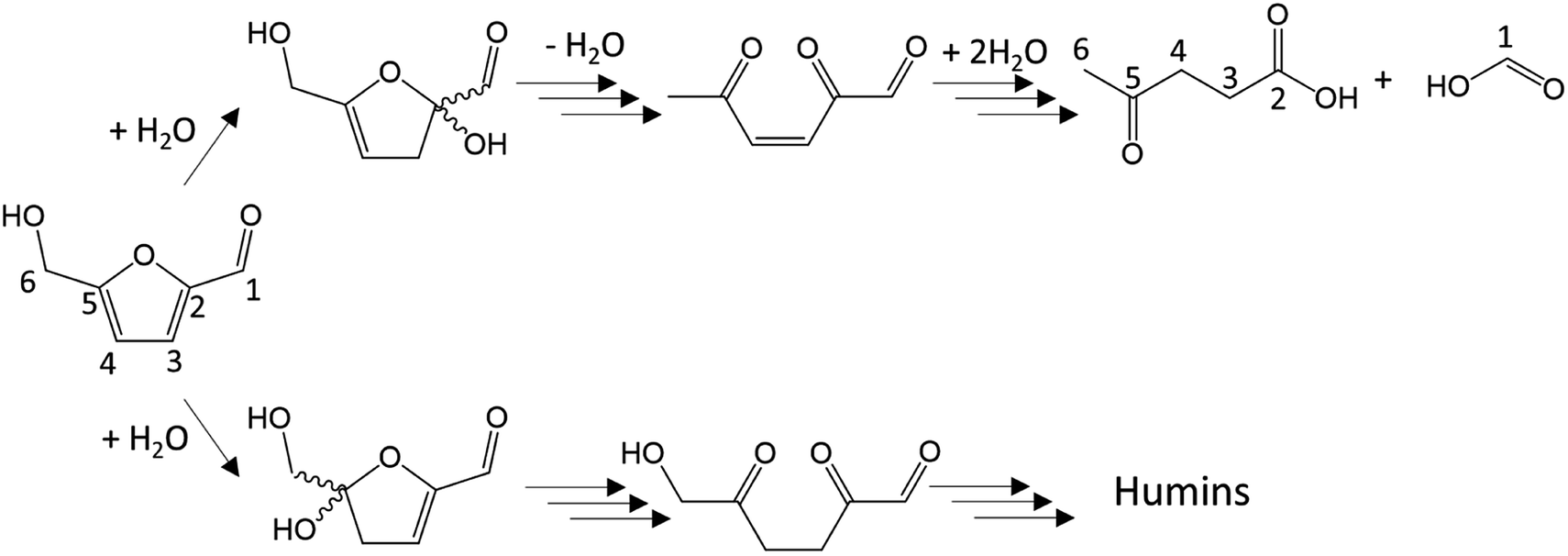 | ||
| Fig. 7 Conversion of 5-HMF to levulinic acid and formic acid (above)32,41,46/conversion of 5-HMF to humins through 2,5-dioxo-6-hydroxy-hexanal.43 | ||
Retro-aldol reactions give rise to many aldehyde or ketone products, fragmenting the initial monosaccharide in molecules of different sizes. Glyceraldehyde is a three-carbons product in isomerization equilibrium with dihydroxyacetone. Both compounds can be dehydrated to pyruvaldehyde which can itself undergoes benzylic acid rearrangement to produce lactic acid.37 Through retro-aldol reactions, hexoses can also be converted to erythrose (four carbon atoms) and glycolaldehyde (two carbon atoms). While not confirmed yet, the formation of pentoses (five carbon atoms) and formaldehyde (one carbon atom) has been suggested as well to explain the appearance of furfural during hexoses treatments. In a similar manner, pentoses can be converted to glyceraldehyde and glycolaldehyde.18,39,40
Regarding polymerization, disaccharides formation is frequently observed, especially at high monosaccharide concentration. In ketoses case, dimerization can be advantageous. Two molecules of D-fructose can reversibly form di-D-fructose dianhydrides. Under this form, the reducing group of D-fructose sensitive to cross-polymerization reactions is blocked, partially limiting humins growth.19 This is not the case for aldose as discussed further. A particularly problematic reactions chain is the polymerization to humins. Humins are dark polymers originating from polymerization and/or cross-polymerization of monosaccharides and furan derivatives. Their chemical structure, likely variable, is not fully identified but those polymers are generated through aldol addition/condensation reactions potentially threatening every molecule with a carbonyl moiety.19,23 Humins formation pathway involving the transformation of 5-HMF into 2,5-dioxo-6-hydroxy-hexanal has been suggested (Fig. 7, below) following a parallel road to levulinic and formic acid production. 2,5-Dioxo-6-hydroxy-hexanal is suspected to react with 5-HMF by aldol addition then condensation to initiate the polymerization.41–43 This mechanism involves the aldehyde group of 5-HMF, what will be of importance when the effects of organic solvents on 5-HMF generation will be discussed thereafter. Since humins are cross-polymerization products, their abundance will increase with monosaccharides and furan derivatives concentrations.47 Kuster et al. (1990) mentioned a humins yield of 20 wt% from a 0.25 M solution of fructose. At higher initial fructose concentration (1 M), the humins yield reaches 35 wt%.19
2.3. Limitation of side-products formation through solvent choice
Given the different possible degradation paths of ketoses, the selectivity for 5-HMF and 2-F is generally limited in aqueous media. Without catalyst, 5-HMF molar yields range between 0 and 22% for temperatures comprised between 150 to 250 °C and time of 30 seconds to 2 hours.16,18,45,48In the same temperature conditions, the addition of Brønsted acid like sulfuric, hydrochloric, phosphoric or formic acid allows to reach higher 5-HMF molar yields comprised between 20–60% since protonations are critical for the dehydration reaction.16,18,44,45 2-F yields between 60–70% are reached from xylulose dehydration at 110–150 °C with aqueous HCl (pH 1) but undesirable insoluble brown solids are generated during the treatment.35
With the development of dehydration systems with increased complexity, high ketoses conversion and high selectivity towards 5-HMF and 2-F have been achieved. These improved performances mainly result from the use of non-aqueous media. Organic solvents with high or medium polarity are generally preferred since they allow to work at higher D-fructose concentrations. The use of numerous protic solvents (e.g. alcohols and organic acids) as well as aprotic solvents (e.g. acetone, dimethyl sulfoxide, N,N-dimethyl acetamide or 1-methyl-pyrrolidin-2-one) has been reported.39,48–54 Organic solvents suppresses side-reactions in several ways:
- Reduced 5-HMF/2-F conversion to humins and organic acids due to the limited presence of water
- Improvement of ketoses dehydration rate to 5-HMF/2-F
- Protection of furan derivatives aldehyde moiety through reversible derivatization or solvent interactions
- Simultaneous extraction of furan product from the reaction medium
The first benefit of organic solvent use is to prevent side-reactions of Fig. 7 enabled by the presence of water. Shi et al. (2019) compared humins formation from 5-HMF and 2-F in water and ethyl acetate after 5 h at 220 °C. Respectively 65 and 23% of solid humins (carbon yield) were obtained from 5-HMF and 2-F in water while no solid formation was observed after treatment in ethyl acetate. If organic solvents seem to suppress furan derivatives degradation, they still enable humins formation from monosaccharides. However, they limit the extend of this side reaction compared to water. Humins production from D-fructose and D-sorbose reaches 50–55% (carbon yield) in water but is reduced to 30–35% in ethyl acetate (5 h, 220 °C).55
A second improvement of ketoses dehydration is based on reaction rate enhancement. Dehydration rate of D-fructose to 5-HMF is improved in various organic solvents. In a polar protic solvent like ethanol (78 °C) with sulfuric acid, D-fructose dehydration rate constant reaches 1.7 × 10−3 s−1. When ethanol is partially replaced with water in the same conditions (76:24 mass ratio), the reaction rate constant dramatically decreases to 5 × 10−5 s−1.56 Mellmer et al., (2019) highlighted the same trend for polar aprotic solvents. They compared D-fructose dehydration in water, γ-valerolactone/water mixture, tetrahydrofuran/water mixture and dioxane/water mixture in the presence of hydrochloric acid (0.5 M in water, 5 mM in solvents mixtures) at 373 K. The corresponding rate constants are 0.14 ± 0.01, 62 ± 4, 28 ± 5 and 95 ± 6 M−1 ks−1 respectively. The use of organic solvents consequently increases dehydration rate of several order of magnitude. Mellmer et al. (2019) also determined 5-HMF yield at 90% D-fructose conversion and obtained a 40% yield in water compared with more than 70% in polar aprotic solvents/water mixtures, which support a positive effect of the dehydration rate on the reaction yield.57 van Putten et al. (2017) observed a higher reactivity in methanol than in water for fructose, sorbose, tagatose and psicose. For instance, tagatose conversion is 48% after 75 min in 33 mM H2SO4. In methanol, such conversion is reached after only 15 min in milder conditions (100 °C, H2SO4 17 mM).27
The increased dehydration rate of ketohexoses in the presence of organic solvents rather than water could be partially related to the tautomeric distribution. Formation of furan derivatives from ketoses has been recognized as a pseudo-first order reaction, meaning that the reaction rate depends on ketoses concentration. Recent researches support ketofuranose forms as the reaction substrate for 5-HMF formation. Consequently, solvents promoting ketofuranose rather than ketopyranose tautomers are likely to improve the reaction rate. Bicker and coworkers (2005) showed that around 40% of the monosaccharide was in furanoid forms at 25 °C in pure methanol while in water, there is only 25% of D-fructose in these forms.58 The promotion of furanoid forms of D-fructose was further confirm by Shi et al. (2018) and extended to ethanol, n-propanol, n-butanol, 2-propanol, isobutanol, 2-butanol and tert-butanol.59 This effect could be further improved since the furanoid forms are promoted at higher temperatures.60
Compared to water, acetone also shifts the tautomerization equilibrium towards the furanoid forms of D-fructose. Bicker and coworkers observed around 50% of fructofuranose in a mixture of acetone and water (90:10 v/v) at 25 °C. The addition of acetone in water improves 5-HMF molar yield until 60–70% for temperatures between 150 and 180 °C in less than 15 minutes.39,49 In these conditions, no insoluble side-products were detected which implies a limited generation of humins. Although acetone seems to limit humins formation and 5-HMF rehydration, the solubility of monosaccharides decreases with its concentration. The solubility of D-fructose in pure acetone is only of 0.5 g L−1 at 25 °C. The addition of around 10 to 30 wt% of water increases the solubility of D-fructose to several dozens of grams per liter but allows the appearance of levulinic and formic acids (molar yields until 20% and 2% respectively). Despite this compromise, the low boiling point of acetone (56 °C at 1 atm) is an important advantage for solvent separation.39,49 However, reaction of acetone with 2-F has also been reported, leading to the formation trans-furfurylideneacetone followed by appearance of soluble then insoluble polymers. A similar phenomenon was observed with hydroxyacetone and cyclopentanone. This observation suggests that solvents with carbonyl moieties are susceptible to reaction with the furan derivatives in accordance with humins formation by aldol reactions.61
Among all the solvents evaluated for D-fructose dehydration, dimethyl sulfoxide (DMSO) has been described as an excellent medium for the generation of 5-HMF.50,52,62 Without catalyst, 72% of D-fructose (molar yield) are converted into 5-HMF after 4 hours at 130 °C in pure dimethyl sulfoxide. Furanose forms of D-fructose are greatly favored (72%) compared to the pyranose forms (28%) in dimethyl sulfoxide at 23 °C while the pyranose forms are predominant in water. As mentioned earlier, this shift towards furanose forms increases with temperature. At 150 °C, 79% of D-fructose is in the furanose forms. As other organic solvent, DMSO improves the rate of D-fructose dehydration to 5-HMF. Replacing water by a mixture of water and DMSO (80:20 v/v) at 160 °C in the presence of aluminium chloride and maleic acid multiplies the reaction rate by three.63 Psicose, tagatose and sorbose were also successfully converted to 5-HMF in DMSO in the presence of H2SO4 and after 120 min, 5-HMF yields of 82%, 61% and 60% were achieved respectively.64 For comparison, dehydration of fructose, tagatose and sorbose in water (H2SO4 33–300 mM, 100–140 °C) did not allow yields higher than 50%.26
If tautomeric distribution possibly affects ketohexose dehydration rate, it should not influence ketopentoses (xylulose, ribulose) dehydration rate because they are only present as furanose forms.
A third way to favor 5-HMF/2-F rather than side-products is to prevent their aldehyde moiety from reacting. Alcohols and organic acids are known to form ethers and esters with 5-HMF and this phenomenon is thought to limit humins formation, which seem consistent regarding humins formation path proceeding through aldol addition/condensation as proposed by Patil and Lund (2011).43
Fig. 8 reports experimentally observed reaction products of D-fructose in alcohols in the presence of a strong acid catalyst (HCl or H2SO4).48,54,56,58,65,66 Those products originate from ethers formation and acetalization reactions.
 | ||
| Fig. 8 Major products generated from D-fructose in alcohols in the presence of strong acid.56,65 This figure has been adapted from ref. 65 with permission from John Wiley and Sons, copyright 2020. | ||
Interestingly, the nature of the alcohol heavily impacts products selectivity. After a 2 h treatment of D-fructose in methanol at 120 °C in the presence of HCl (5 mol%), a mixture of 5-HMF with compounds C–E (Fig. 8) is obtained. In similar conditions in ethanol, propan-1-ol and butan-1-ol, only 5-HMF and compound C were identified, and no acetal seem to form. In isopropyl alcohol and tert-butyl alcohol, 5-HMF is the major product and no ether or acetal was observed in the tested conditions. Small amounts of isoproxymethylfurfural (3%) can appear after 12 h at 120 °C in isopropyl alcohol with ammonium chloride but 5-HMF remains by far the dominant furan derivative (68%).54 These observations suggest that the bulkiness of the alcohol is a key factor in reaction selectivity. The best 5-HMF yield (83%) was obtained in isopropyl alcohol after 2 h at 120 °C (HCl 5 mol%). The reaction still proceeds at 80 °C and a 67% 5-HMF yield is achieved after 8 hours.65 This surprising performance of bulky alcohol in the absence of 5-HMF etherification or acetalization could imply a protection of the molecule through specific solvation structure as discussed further with dimethyl sulfoxide.
Like 5-HMF, alkoxymethylfurfural can be used in bioplastics or biofuel synthesis. If alcohols limit humins formation (less than 1.9 wt% after 5 h at 78 °C in ethanol with sulfuric acid), the hydration of alkoxymethylfurfural may still take place since molar yields of ethyl levulinate (Fig. 8 compound F) as high as 15% were reported, probably because of in situ generated water.56
Mixtures of water and concentrated organic acids (at least 20 wt%) are another option to improve D-fructose dehydration and 5-HMF molar yields as high as 43–64% have been achieved at 150 °C in 2 hours.48 The use of pure organic acids gives rise to the corresponding hydroxymethylfurfural-ester. In subcritical acetic acid, Bicker and coworkers (2005) produced 5-acetoxymethylfurfural with selectivity and conversion of 38 and 98% respectively after 120 s at 180 °C with 10 mM sulfuric acid. The solubility of D-fructose in this medium is however limited to only 21.5 g L−1 at 25 °C.58
Interactions between 5-HMF aldehyde moiety and solvent molecules are also suspected to suppress side-reactions. During solvation, DMSO binds to 5-HMF more strongly than water, what reduces its susceptibility to nucleophilic attack and limits hydration as well as humins formation.67 In the solvation structure of 5-HMF in mixture of water and DMSO, DMSO coordinates best with the carbon of the carbonyl moiety of 5-HMF protecting the group from reactions.68 A similar phenomenon was suggested for alcohols such as 1-propanol, iso-propanol, 1-butanol and 2-butanol which could prevent 2-F degradation through solvation and steric hindrance compared to reaction in water.61
The protective effect of DMSO was also demonstrated on 2-F. In aqueous HCl (pH 1) at 130 °C, 21% of 2-F is lost after 2 h and insoluble brown solids are observed. When 2-F is treated in a mixture of aqueous HCl and DMSO (50:50) in the same conditions, only 3% of 2-F is lost.35 Several authors suggested that DMSO could also act as a catalyst.30,31 Acidic species originating from DMSO decomposition in the presence of oxygen interact with the carbocation obtained after the first dehydration step of D-fructose and likely catalyze the formation of intermediate A (Fig. 5). A possible mechanism is depicted in Fig. 9.
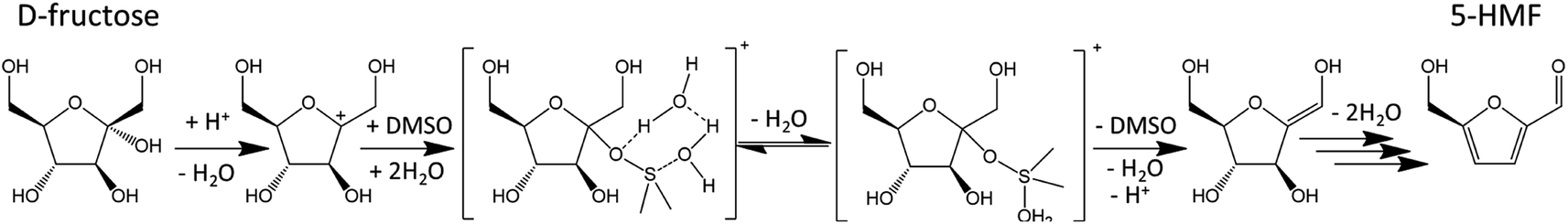 | ||
| Fig. 9 Dehydration of D-fructose catalyzed by DMSO as proposed by Zhang et al. (2016).30 This figure has been adapted from ref. 30 with permission from Elsevier, copyright 2020. | ||
If several organic media are very effective for the dehydration of D-fructose into 5-HMF, their use at industrial scale can be compromised for different reasons. At first, solubility of monosaccharides in polar organic solvents remains lower than their solubility in water (e.g. 0.5 g of D-fructose per liter of acetone).39 Larger amounts of solvent are consequently required for the dehydration step. Regarding polar aprotic solvents such as DMSO, DMAc and 1-methylpyrrolidin-2-one, their boiling point is high (around 190 °C, 165 °C and 203 °C respectively), which makes the recovery of 5-HMF difficult.49
To avoid these drawbacks, biphasic reaction media have been developed. Dehydration of D-fructose occurs in the aqueous phase and the produced 5-HMF is directly extracted in an apolar organic phase to prevent its decomposition into organic acids.14,20,44,69 Biphasic mixtures allow the use of higher D-fructose concentrations (0.56 to 1.67 M) compared to monophasic organic mixtures (0.06 to 0.56 M).14,20,39,44,48–54,62
Solvents commonly used as extractive phase are methyl isobutyl ketone, butanol, hexanol, toluene and tetrahydrofuran.14,20,44,69
The critical factor in biphasic “organic solvent/water” mixture is the partition coefficient (defined as the ratio between 5-HMF concentrations in organic and aqueous phases) of 5-HMF since it directs the solvent choice. The higher is the partition coefficient, the more 5-HMF is extracted in the organic phase.70
Partition coefficients of 5-HMF in previously mentioned apolar solvents are generally lower than two.14,69,71 This coefficient can be increased by a factor of 1.5 to 3 thanks to a salting out effect when salts like sodium or potassium chlorides are added to saturation.69–72 In solution, these salts alter intermolecular forces between both phases and enhance their immiscibility which increases 5-HMF proportion in the organic phase.72 In that respect, a partition coefficient as high as 7.3 is reached for an aqueous phase saturated with sodium chloride and tetrahydrofuran as the apolar organic phase. This solvents combination results in a 78% 5-HMF molar yields after 50 minutes at 160 °C.69 In more complex solvents mixtures comprising water, dimethyl sulfoxide, methyl isobutyl ketone and butan-2-ol (14/13/51/22), an even higher 5-HMF molar yield is achieved (85%) after only 4 minutes at 170 °C.14 Regarding xylulose dehydration, 2-F yield can be increased from 68 to 90% simply by simultaneously extracting it with MIBK (110 °C, HCl (0.1 M), water/MIBK = 1/3).35
This overview of solvents effects on ketoses dehydration demonstrates that molecular organic solvents can be wisely selected to promote 5-HMF and 2-F formation through dehydration rate enhancement and 5-HMF/2-F functional groups protection. In terms of selectivity, polar aprotic solvents and bulky alcohols (like isopropyl alcohol) are solvents of choice for ketoses dehydration. In a practical point of view, bulky alcohols could present advantages compared to polar aprotic solvents, like their lower boiling points which could facilitate extraction/purification processes.
2.4. Anions catalysis of ketoses dehydration
The effects of salts on ketoses dehydration have been investigated in several media including water and polar aprotic solvents. The effect of alkali metals cations (K+, Li+, Na+) is generally insignificant.57 Transition and post-transition metal cations strongly affect ketose dehydration but their impact is addressed further with aldoses dehydration mechanisms. In water, potassium chloride, bromide, iodide and nitrate accelerate D-fructose conversion and slightly improve selectivity for 5-HMF.73 Halides are known to promote D-fructose dehydration in numerous dehydration media. In water, effect of halides on ketose dehydration is limited because of their solvation which limits their interactions with the monosaccharides. A great amount of halide salt (>100 mM) is consequently required. This high amount of halide salts required in protic solvents can be exploited to facilitated 5-HMF extraction by salting-out effect.In polar aprotic solvents, the interaction of halides with monosaccharides is easier and concentration as small as 5 mM can drastically increase the rate constant of the dehydration (over 10 time).57 In γ-valerolactone, chloride alone could not catalyze D-fructose dehydration, but greatly enhanced the reaction combined to an acid catalyst. From computational simulations, it was suggested that the highly localized charge on chloride anions allows them to stabilize carbocations which are dehydration intermediate as well as their deprotonation transition state. Rate constants of D-fructose dehydration in γ-valerolactone (373 K) with triflic acid (5 mM) were determined in the presence of different halide salts and the following trend was obtained: KCl (64 ± 5 M−1 ks−1) > KBr (22 ± 3 M−1 ks−1) > KI (4 ± 1 M−1 ks−1) > KF (2 ± 1 M−1 ks−1). A very similar trend was observed when halide anions were provided by acids (HCl > HBr > HI).57
Halide effects were also observed in N,N-dimethyl acetamide with lithium chloride (90/10 w/w%). This reaction medium can dehydrate more than 60% of D-fructose into 5-HMF at 120 °C in 2 hours. By addition of sulfuric acid, this result is achieved in one hour.51 In the N,N-dimethyl acetamide–lithium chloride mixture, lithium ions form macrocations with N,N-dimethyl acetamide. Chloride anions are consequently weakly paired with them. These weakly paired chloride anions are also observed in some ionic liquids known to be very effective to produce 5-HMF.74–77 A direct intervention of chloride anions in the dehydration reaction is suspected, as depicted in Fig. 10, which results in a limited appearance of side-products.51 When lithium chloride is replaced by a salt containing bromide (lithium, sodium or potassium bromide) or iodide (lithium, sodium or potassium iodide), 5-HMF molar yield can exceed 90% at 100 °C with sulfuric acid after 2 hours. The intermediates depicted in Fig. 5 were also observed during D-fructose dehydration in N,N-dimethyl acetamide with lithium chloride (90/10 w/w%) and products distribution was similar to that in DMSO suggesting analogous dehydration paths.29
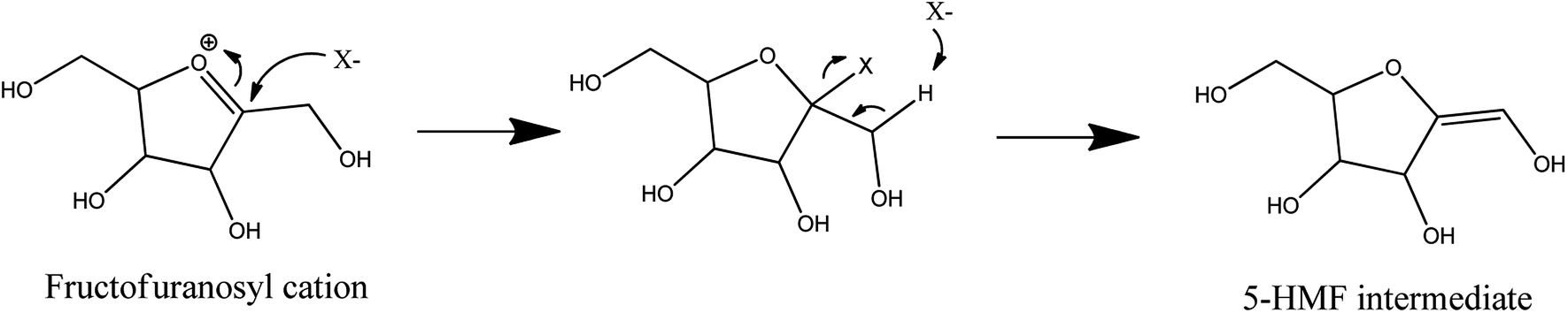 | ||
| Fig. 10 Dehydration of D-fructose to 5-HMF in the presences of halides (X−) as proposed by Binder et al. 2009.51 This figure has been adapted from ref. 51 (https://pubs.acs.org/doi/10.1021/ja808537j) with permission from ACS Publications, copyright 2020. Further permissions related to the material excerpted should be directed to the ACS. | ||
Halide anions roles were further investigated comparing fructose and tagatose dehydration in the presence of several sodium salts (100 g L−1, H2SO4 0.1 M, 105 °C). All salts (NaCl, NaBr, NaI, NaClO4, NaOMs, NaOTs, NaHSO4) accelerated the initial 5-HMF formation compared to salt-free samples excepted for nitrates which completely inhibited 5-HMF formation above a salt concentration of 0.8 M. The reaction order with respect to the anion concentration was between 0.12 and 0.68, which suggests a direct participation of the anions. NaHSO4 had the strongest effect on reaction rate followed by halide salts. Interestingly, tagatose dehydration rate was independent of the halide type on the contrary of fructose whose dehydration was most promoted by chloride, bromide then iodide. This observation suggests once again that C3 and C4 hydroxyl groups position is a key factor influencing the reactivity even in the presence of halide salts.28
From the measured order of reaction in the presence of different salts as well as LC-MS experiments studying the insertion of 18O from labelled water into fructose, nucleophile substitutions likely play an important role in halides dehydration catalysis. Anions could efficiently accelerate dehydration if they have a good leaving group quality, are good nucleophiles and have a small size.28
Nucleophilicity is of importance since this property is strongly affected by the solvent type especially in the case of ionic liquids as discussed in further sections. Solvent choice has consequently an impact on anions choice for dehydration catalysis. Nevertheless, chloride and bromide anions generally appear as good dehydration catalysts in numerous molecular solvents probably because of their appropriate balance between nucleophilicity, leaving group quality and size. F− anions should be carefully considered because they are the conjugated base of a weak acid (pKa = 3.2) on the contrary of the other mentioned halide anions. In an acidic medium (required condition for dehydration reaction), the hydrogen fluoride form will be predominant, which explains the observed inefficiency of F− to catalyze fructose dehydration.57 The use of iodide is also delicate since the halide is susceptible to oxidation to iodine which could limit its efficiency.
Halides catalysis possesses however a serious drawback for industrial process: pitting corrosion. In acidic medium, halides are known to rapidly lead to cracks formation in numerous alloys. The aggressiveness of halides considering pitting corrosion is fortunately not similar from one anion to another and follows this order: Cl− > Br− > F− > I−.78 Iodide, being the less corrosive anion, should therefore be further investigated in the absence of oxygen, especially in polar protic solvents where it has a higher nucleophilicity.
One way to reduce corrosion by halides is the development of treatments at moderate temperature. In this context, several low-transition-temperature mixtures including ionic liquids and deep eutectic solvents enable monosaccharides dehydration below 100 °C, which opens new possibilities to limit corrosion, even the use of non-metallic reactor.
2.5. D-Fructose dehydration in low-temperature-transition mixtures (LTTMs)
Ionic liquids (ILs) and deep eutectic solvents (DES) share common advantages as reaction media such as a low vapor pressure and tunable physical (melting point, solubility, viscosity, density) or chemical (hydrophobicity, catalytic activity) properties. Those mixtures are however formed through different kinds of interactions. ILs can be described as liquid compounds displaying ionic–covalent crystalline structures. They are commonly mentioned as liquid electrolytes composed entirely of ions and associated to low melting temperature (often <100 °C).79 Imidazolium based ILs have been largely investigated for monosaccharides dehydration.75–77,80–84Regarding eutectic mixtures, they can be defined as mixtures of two or more compounds which, at a well-defined composition, display a unique and minimum melting point in the phase diagram. When the melting point of the eutectic mixture is substantially lower than those of pure components, the “deep eutectic” term is applied. This melting point drop is associated to non-covalent affinities as hydrogen bonding or van der Waals' interactions.85 DES can be obtained by mixing a wide diversity of components, even relatively cheap and abundant substances like choline derivatives, organic acids and monosaccharides.
With the constant development of new LTTMs, the frontier between ILs and DES is sometimes unclear. Therefore, the LTTMs general designation was preferred to ILs and DES to describe advances in D-fructose dehydration in the next part of this document.
The dehydration of D-fructose in imidazolium-based LTTMs was investigated.86 Several cations in combination with chloride anions were compared at 120 °C (50 min). The longer was the alkyl chain on the imidazolium cation (1-butyl-3-methylimidazolium, 1-hexyl-3-methylimidazolium, 1-octyl-3-methylimidazolium), the lower was D-fructose conversion (93.4, 65.7, 42.5%) and 5-HMF yield (63.1, 7.3, 0%). In nearly similar conditions (120 °C, 60 min), the 5-HMF yield gradually decreased for longer alkyl chains from nearly 80% with 3-methylimidazolium chloride (n = 0) to less than 5% with 1-decyl-3-methylimidazolium chloride (n = 10).55 The performance of 3-methylimidazolium chloride (MIMCl) was further confirmed in another test reaching a 92% 5-HMF yield at only 90 °C after 45 min. Moreover, the LTTM was able to solubilize a significant amount of D-fructose (250 g of D-fructose per kg).87 MIMCl enabled D-fructose dehydration without addition of a Brønsted acid evidencing its potential as solvent and catalyst. It should be noted that the effect of imidazolium side-chain length was measured comparing similar masses of LTTMs. This implies that chloride anions concentration significantly changes from one test to another. For a same mass of LTTM, chloride concentration is twice higher in MIMCl than in 1-octyl-3-methylimidazolium chloride (OMIMCl), which could strongly affect the rate and selectivity of the reaction as discussed in point 1.4. Imidazolium cation structural effect should be assessed on mole basis.
Besides the alkyl chain, the C-2 hydrogen seem to play a critical role in D-fructose dehydration since a poor D-fructose conversion (44.3%) is achieved and no 5-HMF is obtained if the 1-butyl-3-methylimidazolium (BMIM+) cation is replaced by 1-butyl-2,3-dimethylimidazolium (BDMIM+). C-2 hydrogen of imidazolium is thought to play the role of an acid catalyst. When sulfuric acid is added to BMIMCl and BDMIMCl, their performances become similar (82.9 and 77.6% 5-HMF yields respectively) after 50 min at 100 °C. Moreover, the reaction can still be conducted at 40 °C. At this temperature, dehydration of D-fructose to 5-HMF in BMIMCl (fructose/IL weight ratio = 1/10) in the presence of sulfuric acid (24 mol%) leads to a 5-HMF yield as high as 83.3%.86 In both cases, humins were generated as the main side-product. Without the additional acid catalyst, BMIMCl do not enable the formation of 5-HMF at 100 °C showing that the alkyl chain on imidazolium also affects the minimum possible temperature for the reaction to proceed.86
The effects of anions in LTTMs are likely similar to the trends highlighted for the common molecular solvents of the previous section and halide anions have been proven to effectively support the dehydration reaction. Among HSO4−, Cl−, BF4−, PF6−, OTf− and SCN− combined to BMIM+, only HSO4− and Cl− allowed a significant production of 5-HMF from D-fructose with around 70% and 50% yields respectively.87
Choosing the appropriate halide for a specific ionic liquid is however much more complicated than for molecular solvents. In a previous section, nucleophilicity of halides was presented as one of the key factors explaining their efficiency to catalyze D-fructose dehydration. This property being solvent dependent, nucleophilicity of halide anions in ILs will depends on their interactions with the associated cations as demonstrated in several work provided by Lancaster et al. (2001, 2002, 2004).88–90
If acidic LTTMs containing halides anions are efficient media/catalyst to perform D-fructose dehydration, the minimum reaction temperature remains limited by their melting or freezing point as well as their viscosity. For example, 1-ethyl-3-methylimidazolium chloride (EMIMCl) melts around 89 °C and will remain liquid until 33 °C (supercooling).91 Several works overcame this problem by the addition of cosolvent to LTTMs.32,49,65,92 In pure DMSO with GeCl4 as a catalyst, a max 5-HMF yield of 40% was reached from D-fructose at 25 °C and did not improve after 12 h. Adding 0.5 g of BMIMCl to 2.5 g of DMSO increased reaction rate more than twice without enhancing the yield. However, increasing BMIMCl mass to 1.5 g (with 1.5 g of DMSO) enabled a 70% 5-HMF yield after 12 h.32 Similarly, the addition of small amounts (0.9 mmol) of acetone, DMSO, methanol, ethanol or ethyl acetate to BMIMCl (5.73 mmol) enabled efficient dehydration of D-fructose (around 80% 5-HMF yield) at 25 °C after 6 hours. The cosolvents decreased the reaction medium viscosity by around 6800 mPa s. Room temperature (23 °C)49 D-fructose dehydration was also achieved thanks to the addition of HCl and few drops of CHCl3 to BMIMCl. A 5-HMF yield of 72% was reached after 24 h.65 This effect of cosolvent was also observed in mixture of choline chloride and maleic acid. In previous work, we showed that addition of water to the choline chloride–maleic acid mixture strongly inhibited D-fructose conversion to 5-HMF at 60 °C. However, replacing water by an organic solvent (methanol, ethanol, isopropyl alcohol) still enabled the formation of a liquid reaction medium at this temperature as well as 5-HMF formation (62, 73 and 79% 5-HMF yields respectively after 5 h).92
In appropriate ratio, mixtures of organic solvents with LTTMs present promising catalytic performances associated to a reduced viscosity and a potentially reduced energy consumption.
A careful choice of LTTMs components can provide a selective process at moderate temperature with efficient furan compounds isolation. Simeonov et al. (2012) demonstrated that 5-HMF could be synthesized and isolated in very high yield (97%) and purity (99%) from a LTTM (Fig. 11). They produced 5-HMF in tetraethylammonium bromide (TEAB) at 100 °C in only 15 min in the presence of water (10–15%) and a heterogeneous Brønsted acid catalyst (Amberlyst-15, 10–15%). The LTTM was then dissolved in hot ethanol and precipitated by the addition of ethyl acetate at room temperature. LTTM could be re-used seven time before catalyst was added to maintain the performances.93
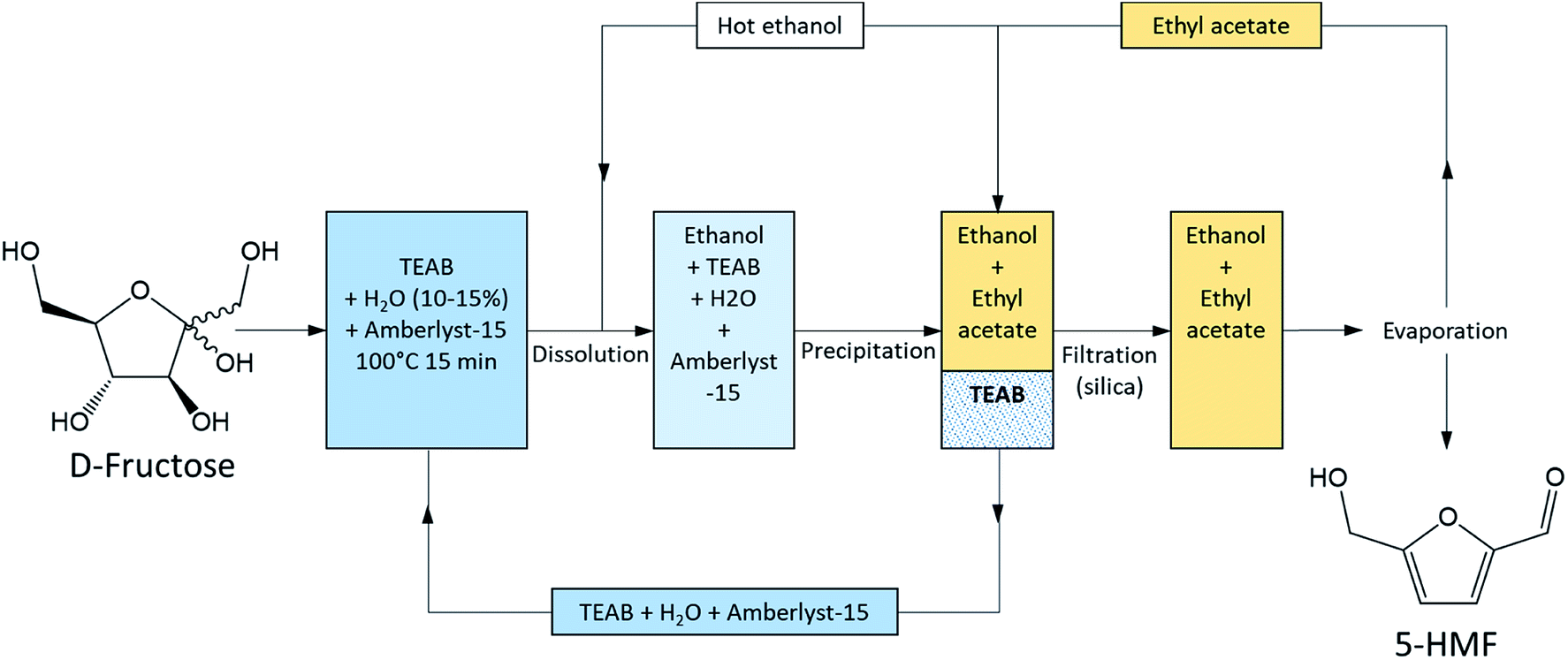 | ||
| Fig. 11 Process for D-fructose dehydration and 5-HMF isolation according to Simeonov et al. 2012.93 This figure has been adapted from ref. 93 with permission from John Wiley and Sons, copyright 2020. | ||
3. Dehydration of aldoses
Glucose, mannose and galactose (Fig. 12) are examples of aldohexoses of which dehydration leads to 5-HMF. Xylose, lyxose and arabinose (Fig. 12) are aldopentoses and their dehydration consequently results in 2-F formation. Glucose and xylose are major components of plant cell walls.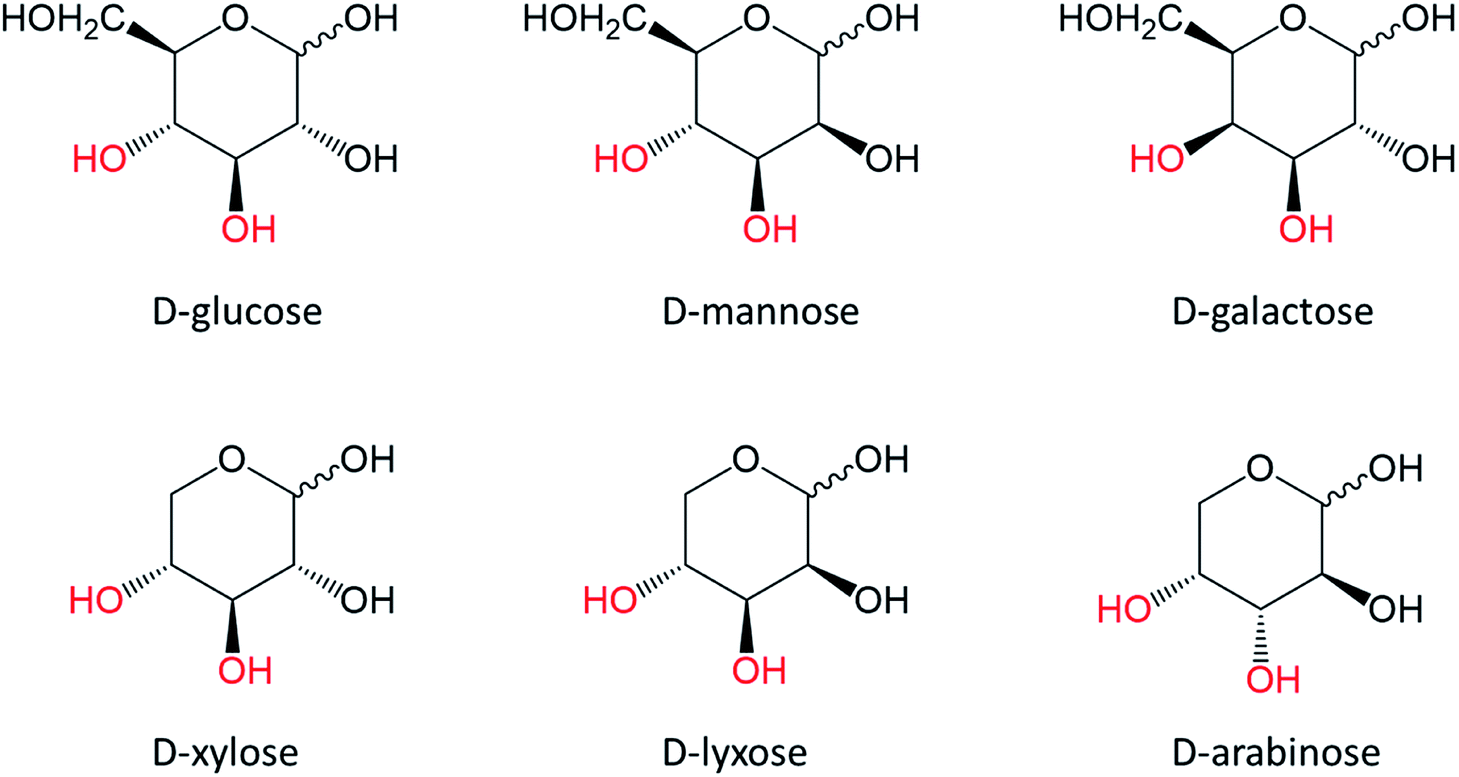 | ||
| Fig. 12 Aldoses chemical structures with highlighted C3 and C4 hydroxyl groups. | ||
However, they cannot be selectively converted to 5-HMF and 2-F using reaction media effective for ketose dehydration (e.g. Brønsted acid in organic solvents or LTTM). Understanding their dehydration is therefore a key to develop biorefining process targeting furan products.
3.1. Mechanism of aldoses dehydration and limitations
Because aldoses possess an aldehyde moiety rather than a ketone moiety, C1 carbon is necessarily a part of the pyranose or furanose ring on the contrary of ketose. Consequently, a resonance stabilized carbocation is formed after protonation of C1–OH rather than C2–OH. This carbocation further reacts and leads preferentially to reversion products and humin precursors rather than 5-HMF.23,46,94 This favored pathway is consistent with experimental observations of the major compounds generated from D-glucose in hot water or hot acidic aqueous solution. A 1 M solution of D-glucose containing sulfuric acid (0.01 M) can reach a yield in insoluble humins of 35 wt% after 6 hours at 180 °C.95 Considering reversion products, the condensation of D-glucose into disaccharides was observed in water at 100–170 °C with sulfuric acid (1.2 wt%).96 Up to 12 wt% of D-glucose was transformed into disaccharides at high sugar loading (300 g L−1). In contrast to di-D-fructose dianhydrides, D-glucose disaccharides are engaged in irreversible cross-polymerization reactions responsible for humins growth.19Attempts to find pathways resulting in 5-HMF and 2-F formation led to several possibilities. Aldoses dehydration mechanisms can be sorted in two categories: reaction paths with at least some acyclic intermediates (Fig. 13) and reaction paths with exclusively cyclic intermediates (Fig. 14). While valid for all the described mechanisms, isotopic labelling demonstrated that carbon atoms positions in aldoses were maintained in the furan derivatives. For instance, glucose C1 correspond to 5-HMF C1 bearing the aldehyde moiety.33,46,97
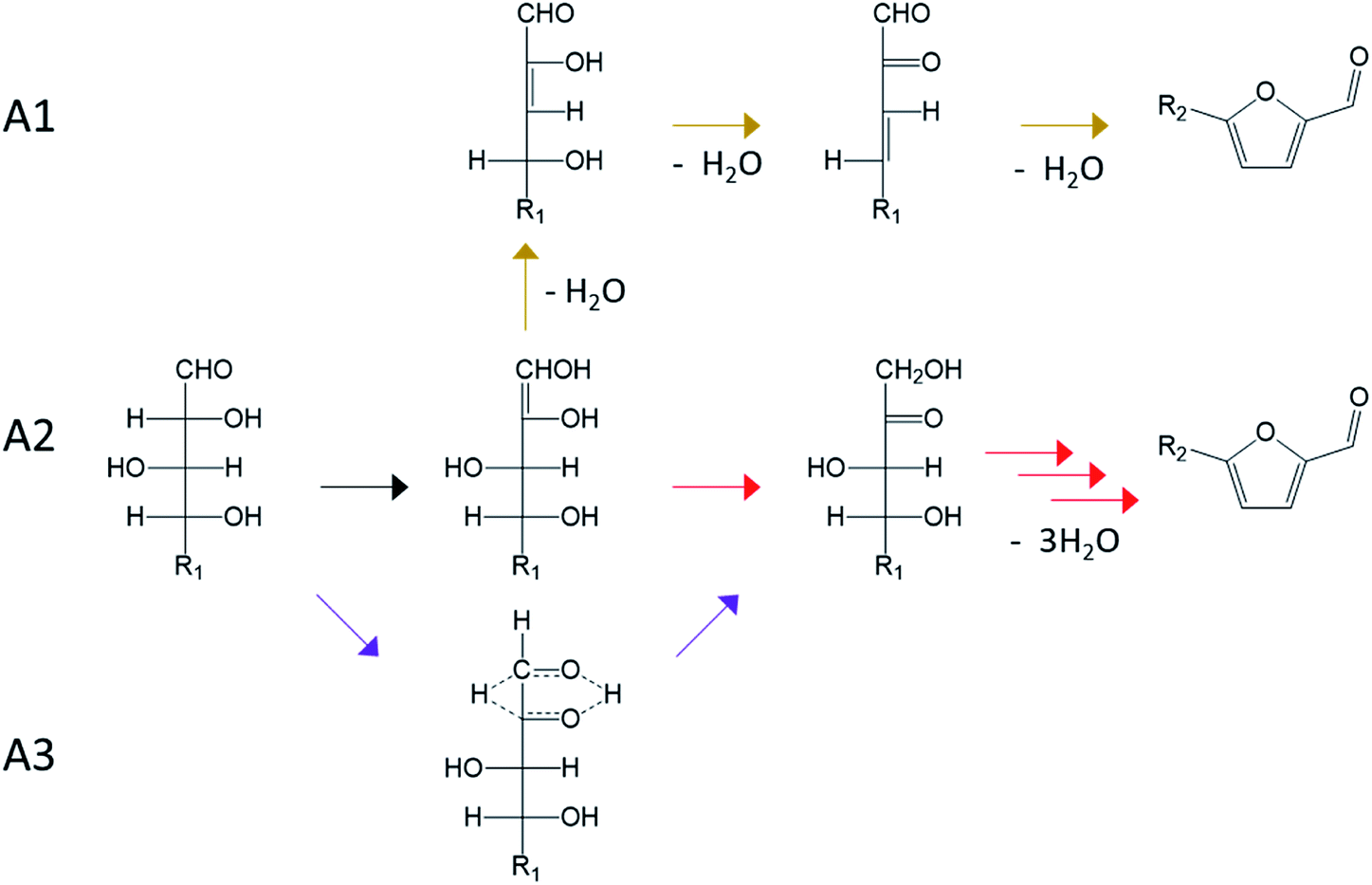 | ||
| Fig. 13 Acyclic pathways for aldoses conversion to furan derivatives. A1 (brown path): reaction path through enediol and other acyclic dehydration intermediate, A2 (red path): isomerization to ketose through an enediol intermediate, A3 (purple path): isomerization to ketose by hydride shift. | ||
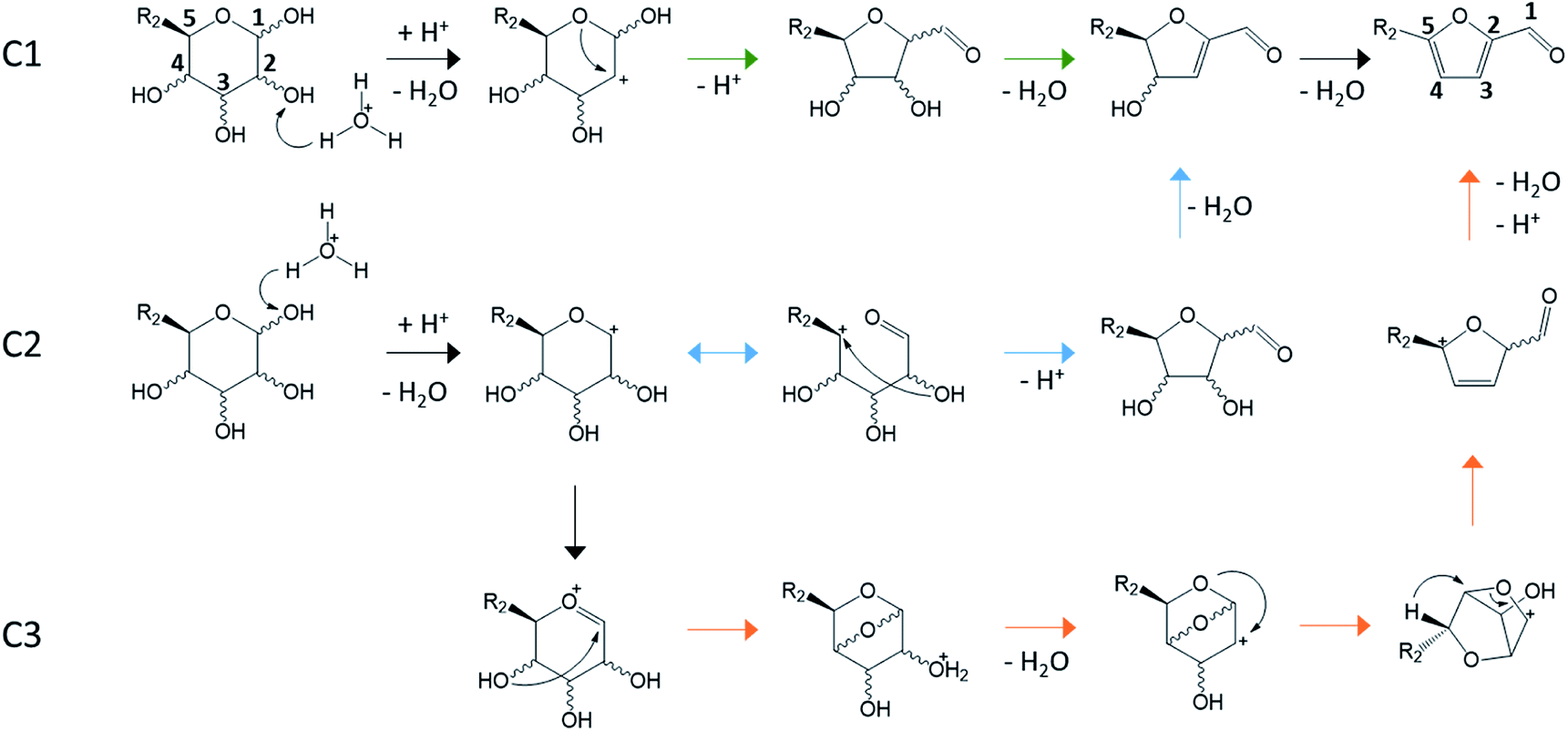 | ||
| Fig. 14 Cyclic pathways for aldoses conversion to furan derivatives. C1 (green path): protonation of C2 hydroxyl and ring contraction after ring O attack on C2,24,33,46,94,97,102 C2 (blue path): protonation of C1 hydroxyl and ring contraction after C2 O attack on C5,103 C3 (orange path): protonation of C1 hydroxyl and ring contraction assisted by formation of a bicyclic cation (gas phase study).104 | ||
Regarding dehydration mechanism involving acyclic intermediates, a fully acyclic path was firstly considered (Fig. 13, A1). This path proceeds through the formation of an enediol intermediate followed by two dehydration steps leading to acyclic intermediates. A2 mechanism consist in isomerization of the aldose to the corresponding ketose by formation of the enediol intermediate. A3 pathway also involves isomerization to ketose but through a hydride shift from C2 to C1.
Dehydration pathways of Fig. 14 involve only cyclic intermediates. All paths consist in a ring contraction of aldoses pyranose form enabling the formation of an aldehyde group on C1.
While no consensus has been achieved yet, some reactions pathways are supported by experimental evidences. The mechanism A1 is generally ruled out because its intermediates are subject to keto–enol tautomerism. Consequently, dehydration reactions performed in D2O should result in the incorporation of deuterium in the formed furan compounds. However, this incorporation of deuterium is low (<5%) or absent.36,64 Compared to the other acyclic pathways (A2 and A3), the fully acyclic path A1 shows a higher effective free energy barrier (34 kcal mol−1 compared to 26 and 24 kcal mol−1 in Brønsted acid catalyzed dehydration under ambient temperature).98
Mechanism A2 is associated to the Lobry de Bruyn–van Ekenstein transformation and leads to the ketose formation. Ketoses formation during aldoses treatment in acidic water has been reported, supporting the A2 path.38,99–101 However, this reaction path is mainly active under base catalysis and involves proton exchange with the solvent because of keto–enol tautomerism. The absence of such exchange in strong acidic conditions implies that A2 path is probably limited in the presence of Brønsted acid. A3 mechanism involving an intramolecular hydride shift is supported by tritium labelling of D-glucose and D-xylose at the C2 position. After the treatment of D-glucose-2-3H in sulfuric acid (2 N) under reflux during 16 h, D-fructose-1-3H is obtained, confirming the hydride shift.100 Similarly, the formation of D-xylulose-1-3H was observed from the treatment of D-xylose-2-3H in sulfuric acid (1 N) at 100 °C during 3 h.101 A3 pathway is compatible with the limited deuterium incorporation from the reaction medium as well as with ketose appearance.
Cyclic reaction paths (Fig. 14) are mainly supported by DFT calculations and gas phase studies with mass spectrometry.24,33,46,94,103–105 Decomposition of many aldoses (8 aldohexoses, 3 hexoketoses, 4 aldopentoses) has been studied by mass spectrometry.97,103–105 Typical observed fragments are m/z 181, 163, 145, 127 for hexoses and m/z 151, 133, 115, 97 for pentoses, which corresponds to the loss of three water molecules from the protonated monosaccharides. A diagnostic ion (m/z 85) obtained by cross ring cleavage is generated from m/z 181, 163, 145, 151, 133, 115 ions suggesting that all the intermediates of the degradation pathways are cyclic.103–105 18O–C1 labelling experiment supports that the dehydration is initiated by C1–OH protonation. However, the presence of solvent is likely to strongly influence the dehydration activation energy.33 Several DFT calculations support the C1 mechanism starting from C2–OH protonation.24,33,46,94 This less energetically favorable protonation site compared to C1–OH is consistent with the low selectivity to produce 5-HMF and 2-F from aldoses. However, cyclic mechanisms do not explain the observed hydride shift from C2 to C1 positions. Interestingly, when 18O-labelled D-xylose (ring oxygen) was dehydrated in aqueous HCl (140 °C, 50 mM), 69% of the obtained 2-F has 18O in the ring while 31% of 18O was observed at the aldehyde moiety. In the presence of NaCl (5 M), 48% of 18O was at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group.106 This oxygen transfer from D-xylose ring to 2-F aldehyde group could be explained by the C2 cyclic mechanism. This mechanism could possibly explain the higher reactivity of D-xylose compared to D-glucose in aqueous Brønsted acid solution.107 Considering the C2 mechanism, the ring contraction occurs through C2–O attack on C5. For hexose, C5 is more substituted than for pentose which could affect the reactivity according to this cyclic mechanism.
O group.106 This oxygen transfer from D-xylose ring to 2-F aldehyde group could be explained by the C2 cyclic mechanism. This mechanism could possibly explain the higher reactivity of D-xylose compared to D-glucose in aqueous Brønsted acid solution.107 Considering the C2 mechanism, the ring contraction occurs through C2–O attack on C5. For hexose, C5 is more substituted than for pentose which could affect the reactivity according to this cyclic mechanism.
Consequently, the most likely pathway to explain furan derivatives formation from aldose in Brønsted acid solution is currently the prior isomerization to ketose through hydride shift. Other mechanisms cannot be excluded at very high temperature or in neutral pH conditions, but more experimental evidences are still required to support them. Further 18O isotopic labelling experiments should be performed to assess the occurrence of C2 cyclic mechanism during the dehydration of different aldohexoses and aldopentoses in Brønsted acid solutions.
In conclusion for this second part, aldopentoses and aldohexoses are not readily dehydrated to 2-F and 5-HMF because their ring includes the C1 carbon as illustrated in Fig. 15. Consequently, the most stable carbocation is formed after protonation of C1–OH. The involvement of C1 in the carbocation ring eliminate the possibility of the furan derivative aldehyde function direct formation.
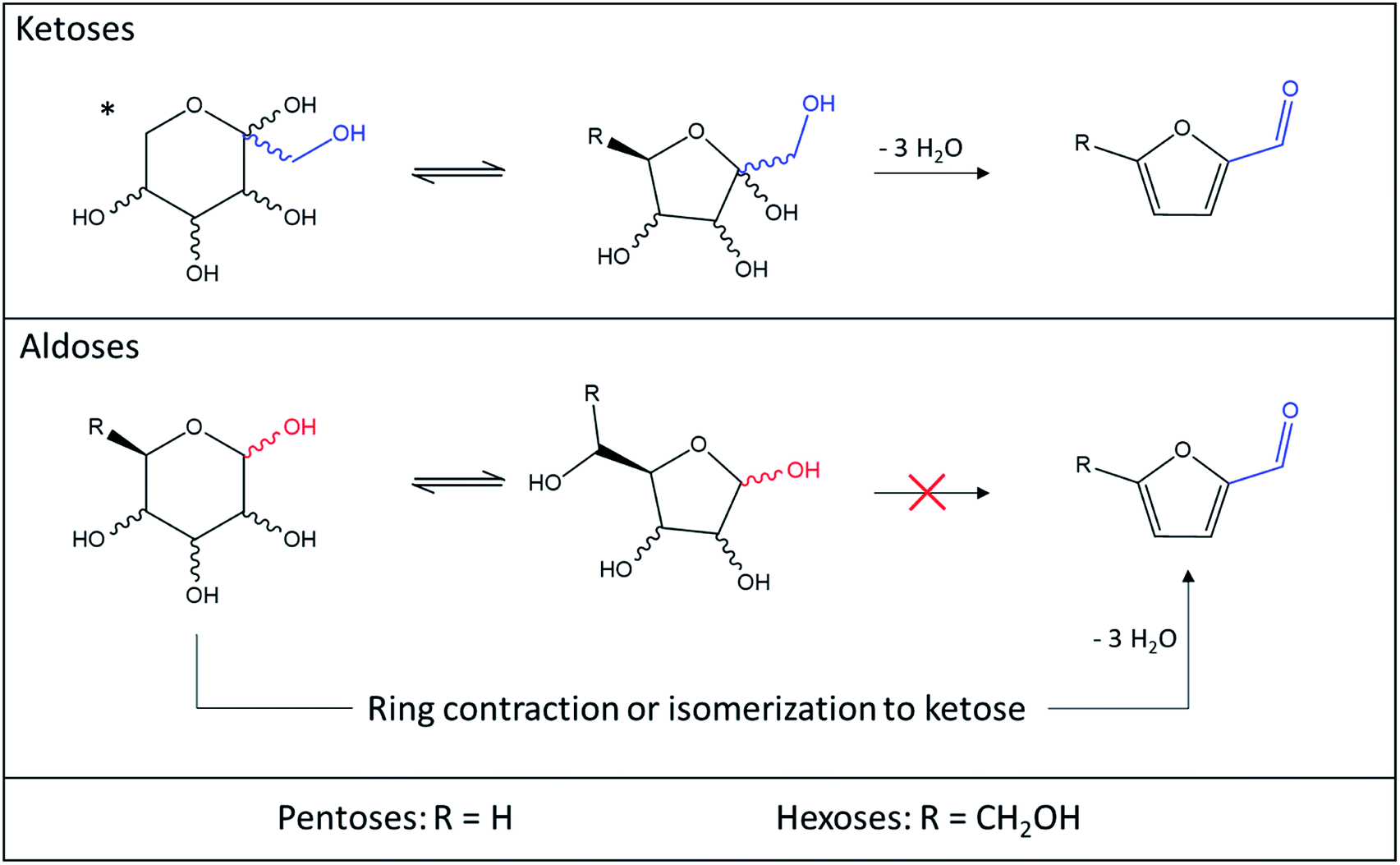 | ||
| Fig. 15 Comparison of ketoses and aldoses dehydration to 5-HMF and 2-F. *This form is only valid for hexoses since ketopentoses only exist in furanose form. | ||
Isomerization or cycle contraction being required to make the formation of the carbonyl moiety possible, other products more energetically favored such as humins or oligosaccharides are generated. The reactivity of hexoses and pentoses towards the dehydration reaction to furan products is therefore a direct consequence of the position of their carbonyl moiety. Whether the conversion of aldoses to 5-HMF and 2-F is direct or proceeds through isomerization to ketoses, it is not favored in most solvents compared to the generation of polymers. It is however certain that promoting aldoses isomerization to ketoses enhances their conversion to furan derivatives.
Understanding both aldoses and ketoses dehydration is therefore crucial. It is illustrated by a less selective formation of 5-HMF from galactose compared to glucose and mannose which are generally converted to 5-HMF in similar yields in the presence of isomerization catalysts.64,108–110 This phenomenon could arise from the intermediate ketose. Glucose and mannose are both isomerized to fructose whereas galactose is isomerized to tagatose. Tagatose, being more reactive and less selective than fructose, could therefore result in a limited 5-HMF formation. A similar trend is observed for arabinose conversion to 2-F compared to xylose and lyxose.34,108,111 Xylose and lyxose share a similar selectivity for 2-F likely because they isomerize to the same ketose: xylulose. Arabinose is rather converted to ribulose. This trend should be further investigated because it means that all aldoses do not share similar selectivity for furan derivatives in the presence of isomerization catalysts.
3.2. Improving aldoses isomerization through catalyst/solvent combination
There are two main pathways to promote aldoses isomerization to ketoses: the Lobry Debruin–van Ekenstein (LDVE) path, mainly promoted in the Brønsted base route, and the Lewis acid path. LDVE pathway requiring an alkaline reaction medium, the simultaneous acid-catalyzed dehydration of ketoses to furan derivatives is not possible. Both reaction steps must be performed in two different media. This problem becomes even greater when more complex feedstock such as polysaccharides are considered to produce 5-HMF and 2-F because their depolymerization generally requires acidic conditions. This alternation of acidity conditions being undesirable in the perspective of an industrial process, Lewis acid catalyzed isomerization has gained a lot of attention for the past few years.A large number of Lewis acids have been investigated including compounds based on transition metals (Cr, Mo, W, Fe, Ru, Cu, Mn, Pd, Pt, V), post-transition metals (Al, Zn, Ga, In, Sn), lanthanides (La, Dy, Yb) and metalloids (B, Ge) elements.74,81,112–114 Given the tremendous number of explored catalysts in different reaction conditions (temperature, solvents), reactivity trends are not easily identified. It is however possible to illustrate important aspects of this catalysis with transition metal, post-transition metal and lanthanides chlorides.
Aldoses isomerization is assisted by coordination of their C1O and C2–OH moieties to the metallic center of the metal chloride, what facilitates the hydride transfer from C2 to the C1. This hydride transfer mechanism has been confirmed for many Lewis acids (CrCl3·6H2O, AlCl3, InCl3, GaCl3, LaCl3, DyCl3, YbCl3) thanks to conversion of D-glucose-2-2H to D-fructose-1-2H and 5-HMF-1-2H as well as the absence of hydrogen/deuterium exchange with the solvent.64,115,116 The same mechanism has been demonstrated for Sn-β zeolites catalyzed isomerization.115 This catalysis is firstly detailed considering an aqueous reaction medium.
Some Lewis acids (e.g. CrCl3·6H2O, AlCl3, GaCl3, InCl3) are known to be water sensitive and will rapidly dissociate in water.112 They are then solvated to form complex ions which releases H3O+, decreasing therefore the pH of the solution. In this sense, Lewis acids also produce Brønsted acidity, useful for dehydration catalysis of the formed ketoses. Dissociation of chromium and aluminum trichlorides in water is depicted in Fig. 16 as well as the observed dominant species.115 The dissociation equilibrium is shifted to the right when temperature increases meaning that the Brønsted acidity can be modulated with temperature. Attention should be paid to anhydrous CrCl3. This chromium species is known as kinetically inert and will not dissolve in solvents. However, CrCl3·6H2O and CrCl2 can be readily dissolved and are generally used as catalyst. The in situ generated Brønsted acidity in aqueous media explains why some Lewis acids (e.g. CuCl2, FeCl3), while inefficient for aldoses conversion to 5-HMF, have a strong ability to catalyze ketose dehydration.114
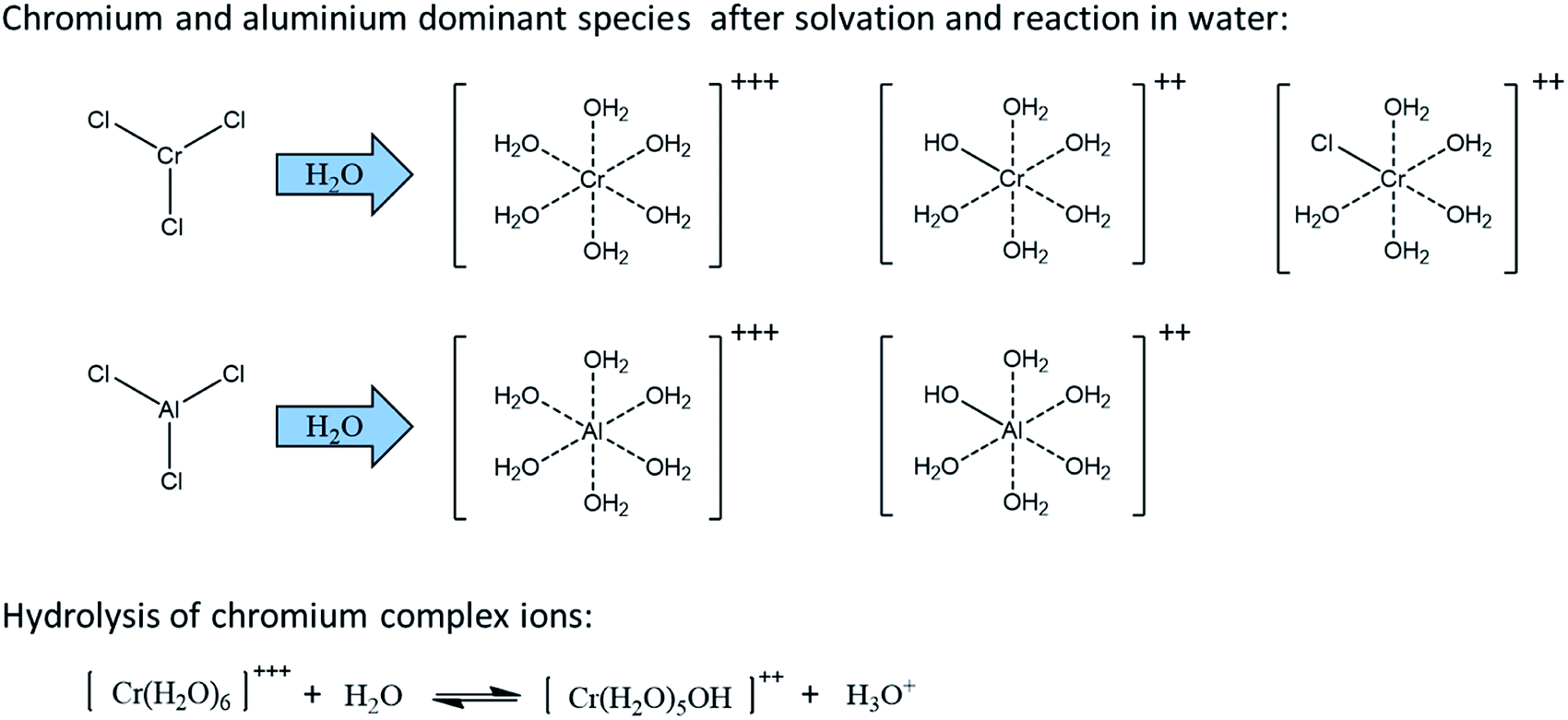 | ||
| Fig. 16 Dominant species of some Lewis acid in water. | ||
Some Lewis acid (LaCl3, DyCl3, YbCl3) are said “water compatible” and are less susceptible to dissociation.112 The Lewis acid–solvent combination is consequently crucial for isomerization catalysis. Following the hydrolysis equation depicted in Fig. 16, the pH will likely influence the catalysis in aqueous media, especially if the different metal species possess different isomerization abilities. The addition of Brønsted acids to a CrCl3 aqueous solution decreased D-glucose conversion rate and 5-HMF yield which confirms this hypothesis.117,118 The phenomenon was explained by an increased fructose dehydration rate thanks to HCl and a decreased glucose isomerization rate related to a shift of Fig. 16 equilibrium to the left.117 However, the opposite trend was observed with AlCl3, InCl3 and GaCl3. The conversion rate of D-glucose to 5-HMF in the presence of AlCl3 was far superior when a Brønsted acid was added to lower the initial pH. To demonstrate that conversion rate increase was not due to the higher initial Brønsted acid concentration, the reaction was conducted in the presence of YbCl3 in different pH conditions. With this water compatible Lewis acid, no significant change of the conversion rate was observed.112 The effect of Brønsted acid addition on Lewis acid reactivity was also investigated for xylose dehydration. HCl addition to a CrCl3·6H2O solution decreased xylose conversion rate but enhanced 2-F yield compared to HCl and CrCl3·6H2O separately. In a biphasic system (water, toluene at 140 °C, 120 min), a 76.3% 2-F yield is achieved with the combination of HCl (0.1 M) and CrCl3·6H2O (6 mM) while 2-F yield was limited to 26.7 and 34.7% with HCl (0.1 M) or CrCl3·6H2O (6 mM) alone respectively.34
Given the different effects of Brønsted acid addition for different metal chlorides, it is likely that Brønsted and Lewis acidities have to be specifically optimized for each catalyst in order to balance isomerization and dehydration rates and limit side products formation.
Isomerization catalysis is illustrated in Fig. 17. The depicted metal (M) species could for instance correspond to [Cr(H2O)5OH]++, thought to be one of the possible active species generated by reaction of CrCl3·6H2O with water. The metal center is coordinated to oxygen atom and has a covalently bonded hydroxyl group.115 The isomerization reaction comprises several steps. Firstly, a proton transfer from C2–OH is enabled by the covalently bonded hydroxyl of Cr(III), acting as a Brønsted base. Then, the hydride shift from C2 to C1 can occur followed by another proton transfer from the metal species to C1–O. The metal species acts therefore as a bifunctional catalyst possessing a Lewis acid site (the metal center) and a Brønsted base site (the ligand).115 Both these aspects should be considered to enable efficient isomerization to ketoses.
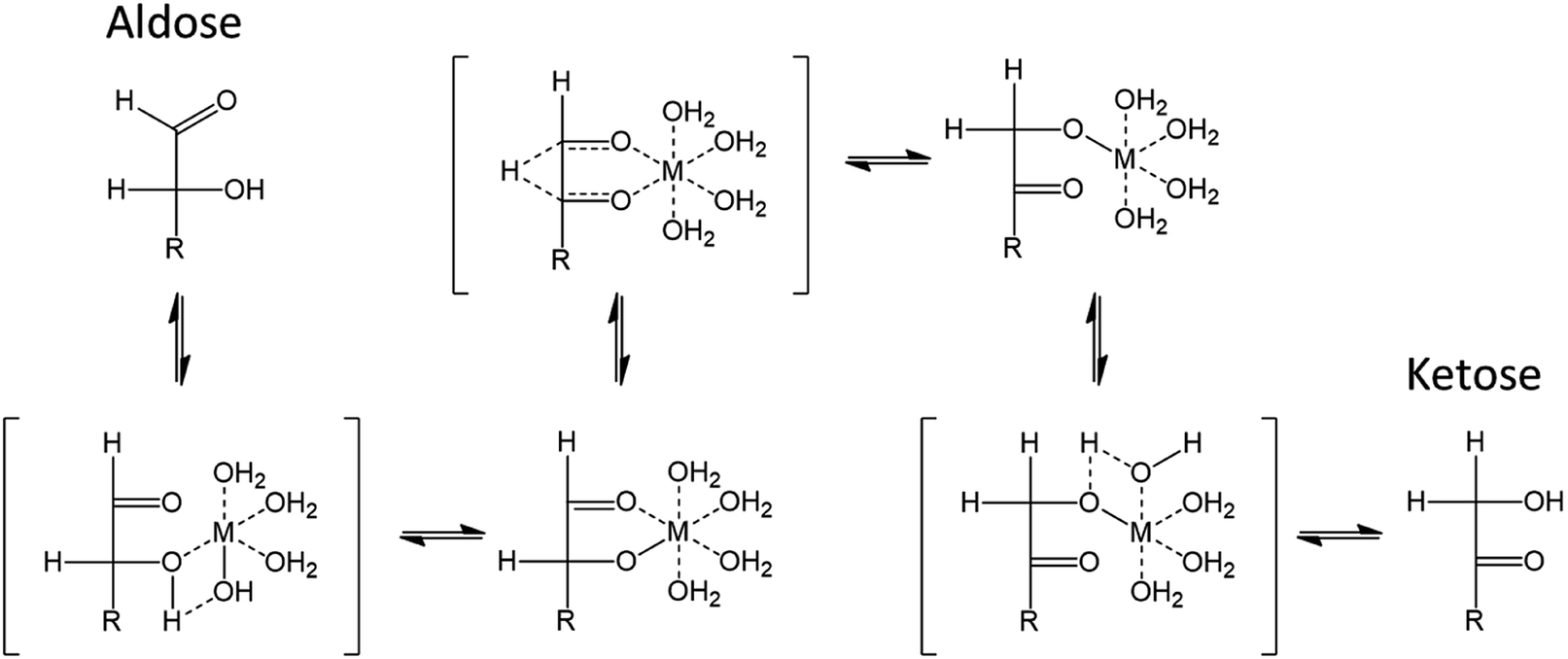 | ||
| Fig. 17 Isomerization of aldoses to ketoses assisted by a metallic Lewis acid in water115 This figure has been adapted from ref. 115 with permission from John Wiley and Sons, copyright 2020. | ||
Considering first ligand properties, coordination strength and basicity are critical for the isomerization. During the reaction, –OH and O groups of aldoses must replace ligand molecules in the coordination sphere. Strongly coordinating ligands will limit this exchange and lead to poor catalytic performances.119 This has been illustrated in imidazolium based LTTMs. In EMIMCl, CrCl3·6H2O will form a CrCl63− octahedrally coordinated complex. This complex is surrounded by EMIM+ cations which compensate the negative charge. Replacement of Cl− ligands by –OH groups is energetically favored, which explain how isomerization takes place in ionic liquids. Cl− can assist proton transfers like –OH bonded to the metal center for the catalysis in aqueous phase.119 However, strongly coordinating ligands drastically limit the catalysis. In EMIMCl, several chromium complexes were compared. All catalysts were activated in the LTTM during 20 min at 150 °C prior to use. This step could be specific to chromium(III) which is again known for its substitution inertness meaning that high temperatures are required for continuous introduction of other species into the coordination sphere.120 Compared to CrCl3·6H2O which resulted in a 48% 5-HMF yield (3 h, 100 °C), chromium species with acetate (Cr2(OAc)4), ethylenediamine (Cr(en)3Cl3) and acetylacetonate (Cr(acac)3) led to 5-HMF yields of 15, 7 and 4% respectively. In contrast, the introduction of weakly coordinating ligand such as THF (CrCl3(THF)3) and n-butanol (CrCl3(n-BuOH)3) improved 5-HMF yield to 71 and 69% respectively.120 The insignificant 5-HMF yield obtained from glucose with GeCl4 in 1-butyl-3-methylimidazolium acetate (BMIMOAc) could further support the inhibition by strongly coordinating ligand.121
Ligand basicity has also been suggested as an important parameter and weakly coordinating non basic PF6− and BF4− anions are expected to be inefficient, which could be supported by the poor conversion to 5-HMF in BMIMBF4 (around 5% yield after 75 min at 100 °C with GeCl4).119,121
Beside ligand properties, the nature of the metallic center obviously affects aldoses isomerization catalysis. Metal affinity for carbohydrates generally increases in the order of univalent, divalent and trivalent metals.116 This is a first element explaining why alkali and alkaline earth metals cannot efficiently promote isomerization of aldose. The bond strength between metal center and –OH, O groups of aldoses is a key factor.122 Glucose conversion to 5-HMF was studied in BMIMCl at 100 °C during one hour in the presence of different metal chlorides: CrCl3, VCl3, FeCl3 and PtCl2. 5-HMF approximative yields of 65%, 30%, <5% and 5% were achieved. To understand those results, far infrared spectroscopy was used in experiments with cyclohexanone, n-butanol and glycolaldehyde as model compounds representative of aldose functional groups. Adding cyclohexanone or butanol during the treatment with CrCl3 did not affect the reaction but the introduction of glycolaldehyde strongly inhibited the formation of 5-HMF (around 15% yield), which confirmed that the metal interacted with aldose through their carbonyl and hydroxyl moieties at C1 and C2 position. In VCl3 case, n-butanol did not change the reaction result, but cyclohexanone suppressed 5-HMF formation (5% yield). Compared to chromium, vanadium binds carbonyl moieties more strongly. Moreover, vanadium is more likely to coordinate with glycolaldehyde and an additional carbonyl moiety than chromium, favoring polymerization reactions leading to humins formation. For PtCl2, the replacement of Pt–Cl bond by Pt–O bond is less favored. The metal thus possesses a low catalytic activity for aldose conversion. Regarding FeCl3, the metal strongly binds to the different oxygen sources (alcohol, carbonyl) which do not enable a selective reaction. In addition, it also binds strongly with water oxygen, leading to complete reaction inhibition in the presence of water. These experiments highlight the efficiency of chromium compared to other metals and show that isomerization performances are directly linked to a fine tuning of coordination strength.122 Similarly to PtCl2, CuCl2 do not likely exchange Cu–Cl bonds to Cu–O bonds. Cu(II) is also susceptible to reduction to Cu(I) by glucose.123
Besides chromium, aluminum and tin chlorides are also generally reported as particularly effective catalysts for aldoses conversion to furan derivatives in water, polar aprotic solvents and LTTMs.81,113,114,124–127 Their coordination to the previous model compounds should be further investigated. In this sense, galactose dehydration to 5-HMF by AlCl3·6H2O (130 °C in DMSO/H2O 9/1 v/v) was completely inhibited by addition of glyceraldehyde confirming the coordination to C1O and C2–OH for the aluminum catalyst. Without glyceraldehyde, galactose transformation to 5-HMF was again less selective than glucose transformation achieving a 34.6% 5-HMF yield after 60 min vs. 54.3% for glucose. This limited selectivity was rationalized by dehydration of tagatose, in similar conditions. Only a 30–35% 5-HMF yield was obtained from the ketose at a nearly complete conversion explaining the poor dehydration selectivity of galactose.110
Among Lewis acids, boric and boronic acid derivatives that possesses a non-metallic center stand apart from a mechanistic point of view. They are known to form borate esters with 1,2- and 1,3-diols (Fig. 18), including monosaccharides as well as with several organic acids (e.g. salicylic, oxalic, glycolic, tartaric acids).128–130 The formation of borate esters in aqueous solution is associated with a pH decrease.131
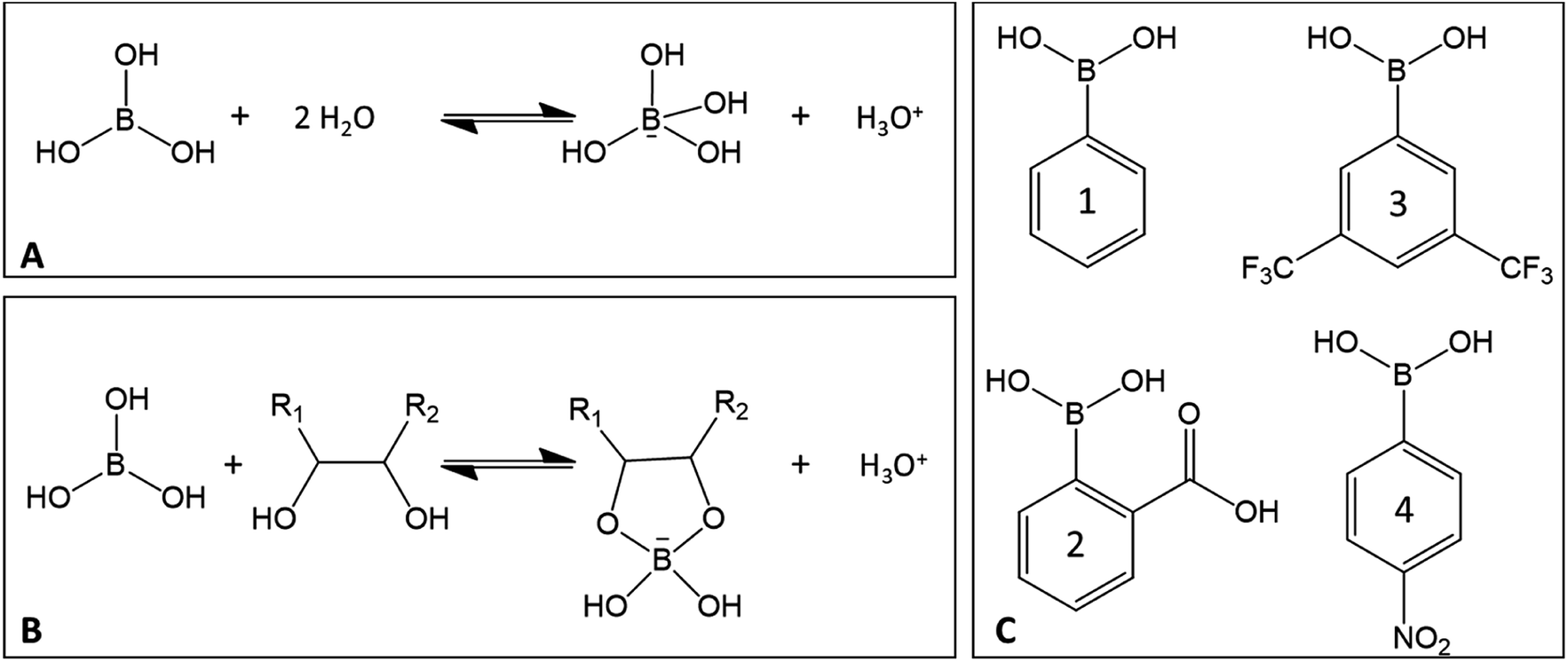 | ||
| Fig. 18 (A) Reaction of boric acid with water, (B) reaction of boric acid with a cis-diol, (C) examples of boronic acids: phenylboronic acid (1), 2-carboxyphenylboronic acid (2), 3,5 bis(trifluoromethyl)phenylboronic acid (3), (4-nitrophenyl)boronic acid (4). | ||
Using boric acid as a catalyst, a 42% 5-HMF yield is achieved from D-glucose after 3 h at 120 °C in EMIMCl.77 DFT studies suggest that chelation of boric acid with D-glucose (coordination to C3–OH and C4–OH) results in a stabilization of the open chain glucose. The relative energy of acyclic glucose compared to β-glucopyranose was 20 kJ mol−1. β-Glucopyranose coordinated to boric acid at C3, C4 positions has a relative energy of −50 kJ mol−1 and the acyclic coordinated form – 90 kJ mol−1. Protonation of O1 becomes more favorable than in the absence of boric acid (probably because of the negative charge of boric acid). This protonation step is then followed by proton transfer resulting in the formation of an enediol intermediate still coordinated to boron. An additional proton transfer leads to the formation 3,4-borofructose. Fructose can then be dehydrated to 5-HMF.77
An enediol mechanism (A2, Fig. 13) was confirmed by the reaction of D-glucose-2-2H. Less than 5% deuterium remained in the produced 5-HMF contrarily to the expected 50% for a 1,2-hydride shift mechanism (considering 100% incorporation of deuterium at D-fructose C1 position, theoretically 50% of deuterium should be lost in the solvent through the formation of the dehydration intermediate A of Fig. 5).77 This study implies that boric acid catalyzed isomerization proceeds through a mechanism different from other metallic Lewis acid for which the hydride shift is validated.
The enediol mechanism is also supported for boronic acid derivatives. Dehydration of D-glucose-2-2H to 5-HMF was performed in dimethyl acetamide with MgCl2·6H2O and 2-carboxyphenylboronic acid (molecule 2, Fig. 18C) as the catalyst. No deuterium was retained in the produced 5-HMF. Moreover, substantial amounts of deuterium are incorporated in 5-HMF at C1 position when D2O is present during the treatment.132
Boric and boronic acids have been used in different media (imidazolium based LTTMs, water, dimethylacetamide, dimethyl sulfoxide) resulting in furan derivatives yields comprised between 5 and 60% from various aldoses (glucose, mannose, galactose, allose, xylose, arabinose).77,132–134
Boric and boronic acids catalyzed aldoses dehydration being based on complex formation, the stability of those complexes strongly affects the reaction rate and selectivity. This stability depends on several factor including: the amount of boron catalyst, its associated organic chemical structure and aldose nature.
An excessive amount of boric or boronic acids (e.g. a 2/1 molar ratio with the aldose) will produce complexes including two boron entities limiting the aldose reactivity.77,134
Additionally, the organic structure of phenylboronic acid (molecule 1, Fig. 18C), especially the position and nature of substituents on the aromatic ring, modulates the catalytic activity. Phenylboronic acids with an ortho carboxylic acid or ester enhanced 5-HMF yield from glucose in dimethylacetamide with MgCl2·6H2O compared to other substitution patterns. A 54% 5-HMF yield was obtained from D-glucose after 4 h at 120 °C in DMA, MgCl2·6H2O thanks to the use of 2-carboxyphenylboronic acid (10 wt% glucose, 2 eq. MgCl2·6H2O, 5 eq. of added water, 1 eq. of boronic acid). The presence of MgCl2·6H2O greatly enhanced the reaction (54% 5-HMF with MgCl2·6H2O and only 2% 5-HMF without the salts) as expected from a chloride anions source. The oxygen of the ortho carboxyl group is suspected to supply electron density to the empty p orbital of boron decreasing the strength of the complex.132 A similar trend was also observed in BMIMCl (105 °C, 2 h). 24%, 11% and 7% 5-HMF yield were achieved from glucose with ortho-carboxyphenyl, meta-carboxyphenyl and phenylboronic acids respectively. Ortho-carboxyphenylboronic acid enabled a 58% 5-HMF yield when used in DMF containing BMIMCl (0.19 M) at only 95 °C for one hour.135
The substituents effects on phenylboronic acid catalyzed dehydration of aldoses was further studied in EMIMCl (120 °C, 3 h). Without catalyst, no 5-HMF was obtained from glucose. The introduction of phenylboronic acid to the system enabled a limited formation of the product (17%). Many substituted derivatives were then compared, and a 44% 5-HMF yield was reached using 3,5-bis(trifluoromethyl)phenylboronic acid (molecule 3, Fig. 18C). The presence of non-resonance-donating electron withdrawing groups could strongly activate arylboronic acids.134
Boric and boronic acids dehydration catalysis is also impacted by hydroxyl groups positions on aldoses ring. The dehydration of glucose, mannose, galactose and allose with 3,5-bis(trifluoromethyl)phenylboronic acid leads to the following 5-HMF yields: 50, 37, 19 and 5%. When the same procedure was repeated with CrCl2 rather than the boronic acid, 5-HMF yields of 66, 61, 13 and 44% were reached respectively. As discussed earlier, galactose resulted in a poor yield probably because of the tagatose intermediate. However, glucose and mannose conversion to 5-HMF are similar with CrCl2 and different with the boronic acid. Strikingly, the transformation of allose is strongly limited in the presence of the boronic acid.134 This phenomenon is hypothetically due to the favored complexation of cis vicinal diol compared to trans vicinal diol.130,134 Considering hydroxyl positions on C2, C3 and C4 of aldoses, glucose possesses two trans vicinal diol sites while mannose and galactose only possess one. Allose only possesses cis-vicinal diol sites. The complexation of a boronic acid with a trans vicinal diol would induce a distortion of the pyranose ring and could in this way favor ring opening, explaining the different dehydration yield achieved from the different aldoses.134
Boric and boronic acids interacting with diols, those catalyst are inefficient in solvents with a diol moiety (e.g. ethylene glycol, glycerol).134
Boron catalysts promoting the enediol pathway, a possible synergy with other Lewis acids acting on hydride shift could be expected. While limited, the synergy has been experimented in several works. Hu et al. (2012) compared the isomerization/dehydration of D-glucose in the presence of boric acid or hydrated chromium trichloride in BMIMCl. They demonstrated that chromium trichloride had a far superior ability to catalyze glucose isomerization compared to boric acid alone (60.3% of HMF against 1.4%). However, when both Lewis acids were combined, the 5-HMF yield was enhanced to 69.1% suggesting a synergy between both catalysts.136 52% of glucose was converted to 5-HMF with a combination of boric acid with tungstophosphoric acid in BMIMCl (140 °C, 40 min). Only 0.8 and 23.5% 5-HMF were obtained with the isolated catalysts.137 The use of boric acid also enhanced dehydration of D-glucose by AlCl3·6H2O in water with NaCl at 170 °C.138
Despite improvement of aldose isomerization to ketose, Lewis-acid catalysis is still confronted to the major problem of humins formation. This phenomenon is widely reported for various catalysts (even CrCl3) in many reaction media: water, BMIMCl, DMSO, NMP, DMA, DMF.112,117,122,124,125,139 In water, as much as 40% of glucose is transformed to humins (carbon yield) at 75% glucose conversion (130–150 °C) in the presence of CrCl3.117 Humins formation has also been reported during catalysis by Hβ-zeolith (Si/Al = 25) progressively covering the heterogeneous catalyst.139 This phenomenon may be surprising considering that solvents like DMSO protect ketoses from degradation during treatment but the polymerization to humins is likely inherent to the coordination to the metal center. Through coordination, metals can bring reactive species with carbonyl or alcohol moieties closer to aldoses. Blocking only a part of the coordination sites with strong ligands could reduce humins formation by limiting the access to the metal center. This could be suggested in the work of Yong et al. (2008) which highlighted that the conversion of D-glucose to 5-HMF was enhanced using chromium complexed with bulky N-heterocyclic carbene (81% 5-HMF yield after 6 h at 100 °C in BMIMCl).74 This strategy would probably affect the reaction rate but should be further investigated in attempt to suppress humins formation.
4. Conclusions
Through a cyclic dehydration mechanism initiated by protonation of C2–O, ketoses can be selectively converted to 5-HMF and 2-F with the help of halide anions in different media, preferably alcohols for their protective effect and low boiling point or LTTMs which enable treatment temperature below 100 °C. Ketopentoses dehydration path should be further investigated with isotopic labelling to complete the current knowledges. A deeper understanding of C3–OH and C4–OH orientation effect on ketoses reactivity is still required.Globally, ketoses dehydration has been largely improved and is now possible at moderate temperature (<100 °C) with high selectivity (>80%). The current understanding of dehydration catalysis and solvents effects should enable the development of selective low cost 5-HMF and 2-F synthesis processes which is why D-fructose dehydration to 5-HMF or derivatives is on the verge of reaching commercial scale.
Understanding and improving aldoses conversion to 5-HMF and 2-F remains challenging, but progresses importance is undeniable. Currently, the most supported mechanism explaining aldoses dehydration by Brønsted acids proceeds through isomerization to ketoses via a hydride shift. However, 18O labelling experiments suggests that ring contraction remains a possibility. The extent of this mechanism compared to the isomerization path should be further explored in a systematic study on several hexoaldoses and hexopentoses. The main way to improve 5-HMF and 2-F formation from aldoses consists in enhancing their isomerization to ketoses. In this purpose, two categories of catalysts have been explored in literature: metallic Lewis acids promoting the isomerization through hydride shift and boric/boronic acids promoting the isomerization through an enediol intermediate. This catalysis is however far more complex than ketoses dehydration because catalysts activity depends on numerous factors: the Brønsted acidity of the medium, catalysts affinity for monosaccharides, catalysts accessibility to several chemical species and catalysts ligands. Consequently for further development of aldoses dehydration systems, a particular attention should be paid to the following elements:
- The acidity of the medium can strongly impact the reaction and should be estimated or compared for different catalysts/solvent systems, especially in LTTMs. Some efficient catalysts may not be identified if not used under optimized acidic conditions.
- Testing isomerization catalysts in the presence of model compounds representative of monosaccharides reactive moieties (simple alcohols, aldehydes/ketones, diols, α-hydroxycarbonyl compounds) can rapidly provide useful information regarding catalyst affinity or the risk of side reactions.
- The activity of metallic Lewis acids is affected by ligands type. Special care should consequently be taken regarding catalysts preparation. The different possibilities of ligands exchange in the reaction medium should also be assessed to prevent catalyst inhibition. Similarly, the activity of boronic acid-based catalysts can be modulated via their associated organic structure. Dehydration systems involving boric and boronic acids are also susceptible to interferences of the reaction medium (e.g. competition with diols or diketones).
Humins formation during Lewis acid catalysis remains an important issue for furan products synthesis. The polymers are probably formed because of the acidity conditions or the coordination of multiple chemical species to the catalyst. Additional researches should focus on humins formation mechanisms. Humins precursors likely derive from furan compounds in aqueous media but other origins are possible as suggested by aldoses degradation in organic solvents.
From the accumulated knowledges concerning monosaccharides dehydration, selective furan derivatives synthesis from aldoses appears feasible provided that all potential interactions of catalysts with monosaccharides and the reaction medium are considered. Further investigations on humins formation will likely provide the tools to inhibit their appearance, enabling the development of efficient aldoses dehydration media.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the European Regional Development Fund and the Walloon Region for their financial support (LCFM – Low Carbon Footprint Materials F_436 – project BIOMAT_3).References
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Volume I – Results of Screening for Potential Candidates from Sugars and Synthesis Gas, United States, 2004, DOI:10.2172/15008859.
- D. X. Martínez-Vargas, J. Rivera De La Rosa, L. Sandoval-Rangel, J. L. Guzmán-Mar, M. A. Garza-Navarro, C. J. Lucio-Ortiz and D. A. De Haro-Del Río, Appl. Catal., A, 2017, 547(2), 132–145, DOI:10.1016/j.apcata.2017.08.035.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- https://www.avantium.com/technologies/yxy/, 03/06/2020.
- https://ava-biochem.com/, 03/06/2020.
- https://www.chemicals-technology.com/projects/avantiumyxy/, 03/06/2020.
- https://www.avantium.com/2020/avantium-to-build-fdca-flagship-plant-at-chemie-park-delfzijl-netherlands/, 03/06/2020.
- S. S. Shannon and L. A. Paul, Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives, John Wiley & Sons, New Jersey, 2016, p. 314 Search PubMed.
- S. Saravanamurugan, L. Hu, R. Anders and P. Ashok, Biomass, Biofuels, Biochemicals: Recent Advances in Development of Platform Chemicals, Elsevier, Amsterdam, 2019, p. 286 Search PubMed.
- B. Thallada, P. Ashok, M. S. Venkata, L. Duu-Jong and K. Samir Kumar, Waste Biorefinery: Potential and Perspectives, Elsevier, Amsterdam, 2018, p. 287 Search PubMed.
- A. E. Eseyin and P. H. Steele, Int. J. Adv. Chem., 2015, 3, 42 CrossRef.
- http://centralromana.com.do/estructura-corporativa/manufacturing/?lang=en, 03/06/2020.
- https://www.polyfurfurylalcohol.com/, 03/06/2020.
- J. N. Chheda, Y. Román-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- A. Mittal, S. K. Black, T. B. Vinzant, M. O'Brien, M. P. Tucker and D. K. Johnson, ACS Sustainable Chem. Eng., 2017, 5, 5694–5701 CrossRef CAS.
- M. Watanabe, Y. Aizawa, T. Iida, T. M. Aida, C. Levy, K. Sue and H. Inomata, Carbohydr. Res., 2005, 340, 1925–1930 CrossRef CAS PubMed.
- I. Agirrezabal-Telleria, Y. Guo, F. Hemmann, P. L. Arias and E. Kemnitz, Catal. Sci. Technol., 2014, 4, 1357–1368 RSC.
- M. J. Antal, W. S. L. Mok and G. N. Richards, Carbohydr. Res., 1990, 199, 91–109 CrossRef CAS PubMed.
- B. F. M. Kuster, Starch/Staerke, 1990, 42, 314–321 CrossRef CAS.
- C. Moreau, R. Durand, S. Razigade, J. Duhamet, P. Faugeras, P. Rivalier, P. Ros and G. Avignon, Appl. Catal., A, 1996, 145, 211–224 CrossRef CAS.
- H. E. van Dam, A. P. G. Kieboom and H. van Bekkum, Starch/Staerke, 1986, 38, 95–101 CrossRef CAS.
- S. Caratzoulas and D. G. Vlachos, Carbohydr. Res., 2011, 346, 664–672 CrossRef CAS PubMed.
- G. Yang, E. A. Pidko and E. J. M. Hensen, J. Catal., 2012, 295, 122–132 CrossRef CAS.
- R. S. Assary, T. Kim, J. J. Low, J. Greeley and L. A. Curtiss, Phys. Chem. Chem. Phys., 2012, 14, 16603–16611 RSC.
- F. Pepi, A. Ricci, S. Garzoli, A. Troiani, C. Salvitti, B. Di Rienzo and P. Giacomello, Carbohydr. Res., 2015, 413, 145–150 CrossRef CAS PubMed.
- R. J. Van Putten, J. N. M. Soetedjo, E. A. Pidko, J. C. Van Der Waal, E. J. M. Hensen, E. De Jong and H. J. Heeres, ChemSusChem, 2013, 6, 1681–1687 CrossRef CAS PubMed.
- R. J. van Putten, J. C. van der Waal, E. de Jong and H. J. Heeres, Carbohydr. Res., 2017, 446–447, 1–6 CrossRef CAS PubMed.
- P. Körner, S. Beil and A. Kruse, React. Chem. Eng., 2019, 4, 747–762 RSC.
- G. R. Akien, L. Qi and I. T. Horváth, Chem. Commun., 2012, 48, 5850–5852 RSC.
- J. Zhang, A. Das, R. S. Assary, L. A. Curtiss and E. Weitz, Appl. Catal., B, 2016, 181, 874–887 CrossRef CAS.
- A. S. Amarasekara, L. T. D. Williams and C. C. Ebede, Carbohydr. Res., 2008, 343, 3021–3024 CrossRef CAS PubMed.
- J. Zhang and E. Weitz, ACS Catal., 2012, 2, 1211–1218 CrossRef CAS.
- M. R. Nimlos, X. Qian, M. Davis, M. E. Himmel and D. K. Johnson, J. Phys. Chem. A, 2006, 110, 11824–11838 CrossRef CAS PubMed.
- V. Choudhary, S. I. Sandler and D. G. Vlachos, ACS Catal., 2012, 2, 2022–2028 CrossRef CAS.
- B. Li, S. Varanasi and P. Relue, Green Chem., 2013, 15, 2149–2157 RSC.
- T. Ahmad, L. Kenne, K. Olsson and O. Theander, Carbohydr. Res., 1995, 276, 309–320 CrossRef CAS.
- T. M. Aida, Y. Sato, M. Watanabe, K. Tajima, T. Nonaka, H. Hattori and K. Arai, J. Supercrit. Fluids, 2007, 40, 381–388 CrossRef CAS.
- H. Kimura, M. Nakahara and N. Matubayasi, J. Phys. Chem. A, 2011, 115, 14013–14021 CrossRef CAS PubMed.
- M. Bicker, J. Hirth and H. Vogel, Green Chem., 2003, 5, 280–284 RSC.
- B. M. Kabyemela, T. Adschiri, R. M. Malaluan and K. Arai, Ind. Eng. Chem. Res., 1999, 38, 2888–2895 CrossRef CAS.
- J. Horvat, B. Klaid, B. Metelko and V. Sunjid’, Tetrahedron Lett., 1985, 26(17), 2111–2114 CrossRef CAS.
- J. Horvat, B. Kuuc, B. Metelko and V. Sunjic, Croat. Chem. Acta, 1986, 59, 429–438 CAS.
- S. K. R. Patil and C. R. F. Lund, Energy Fuels, 2011, 25, 4745–4755 CrossRef CAS.
- Y. Román-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef PubMed.
- P. Daorattanachai, S. Namuangruk, N. Viriya-empikul, N. Laosiripojana and K. Faungnawakij, J. Ind. Eng. Chem., 2012, 18, 1893–1901 CrossRef CAS.
- L. Yang, G. Tsilomelekis, S. Caratzoulas and D. G. Vlachos, ChemSusChem, 2015, 8, 1334–1341 CrossRef CAS PubMed.
- I. Van Zandvoort, Y. Wang, C. B. Rasrendra, E. R. H. Van Eck, P. C. A. Bruijnincx, H. J. Heeres and B. M. Weckhuysen, ChemSusChem, 2013, 6, 1745–1758 CrossRef CAS PubMed.
- R. L. de Souza, H. Yu, F. Rataboul and N. Essayem, Challenges, 2012, 3, 212–232 CrossRef.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Green Chem., 2008, 10, 799–805 RSC.
- Z. Huang, W. Pan, H. Zhou, F. Qin, H. Xu and W. Shen, ChemSusChem, 2013, 6, 1063–1069 CrossRef CAS PubMed.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS PubMed.
- H. Yan, Y. Yang, D. Tong, X. Xiang and C. Hu, Catal. Commun., 2009, 10, 1558–1563 CrossRef CAS.
- X. Tong, M. Li, N. Yan, Y. Ma, P. J. Dyson and Y. Li, in Catalysis Today, Elsevier B.V., 2011, vol. 175, pp. 524–527 Search PubMed.
- J. Liu, Y. Tang, K. Wu, C. Bi and Q. Cui, Carbohydr. Res., 2012, 350, 20–24 CrossRef CAS PubMed.
- N. Shi, Q. Liu, H. Cen, R. Ju, X. He and L. Ma, Biomass Convers. Biorefin., 2020, 10, 277–287 CrossRef CAS.
- T. Flannelly, S. Dooley and J. J. Leahy, Energy Fuels, 2015, 29, 7554–7565 CrossRef CAS.
- M. A. Mellmer, C. Sanpitakseree, B. Demir, K. Ma, W. A. Elliott, P. Bai, R. L. Johnson, T. W. Walker, B. H. Shanks, R. M. Rioux, M. Neurock and J. A. Dumesic, Nat. Commun., 2019, 10, 1132 CrossRef PubMed.
- M. Bicker, D. Kaiser, L. Ott and H. Vogel, J. Supercrit. Fluids, 2005, 36, 118–126 CrossRef CAS.
- K. Shi, C. M. Pedersen, Z. Guo, Y. Li, H. Zheng, Y. Qiao, T. Hu and Y. Wang, J. Mol. Liq., 2018, 271, 926–932 CrossRef CAS.
- B. Schneider, F. W. Lichtenthaler, G. Steinle and H. Schiweck, Liebigs Ann. Chem., 1985, 2443–2453 CrossRef CAS.
- X. Hu, R. J. M. Westerhof, D. Dong, L. Wu and C. Z. Li, ACS Sustainable Chem. Eng., 2014, 2, 2562–2575 CrossRef CAS.
- H. Wang, T. Deng, Y. Wang, Y. Qi, X. Hou and Y. Zhu, Bioresour. Technol., 2013, 136, 394–400 CrossRef CAS PubMed.
- J. C. Overton, X. Zhu and N. S. Mosier, ACS Sustainable Chem. Eng., 2019, 7, 12997–13003 CrossRef CAS.
- J. B. Binder, A. V. Cefali, J. J. Blank and R. T. Raines, Energy Environ. Sci., 2010, 3, 765–771 RSC.
- L. Lai and Y. Zhang, ChemSusChem, 2011, 4, 1745–1748 CrossRef CAS PubMed.
- X. Yu, X. Gao, R. Tao and L. Peng, Catalysts, 2017, 7(182), 1–10 Search PubMed.
- G. Tsilomelekis, T. R. Josephson, V. Nikolakis and S. Caratzoulas, ChemSusChem, 2014, 7, 117–126 CrossRef CAS PubMed.
- S. H. Mushrif, S. Caratzoulas and D. G. Vlachos, Phys. Chem. Chem. Phys., 2012, 14, 2637–2644 RSC.
- Y. Román-Leshkov and J. A. Dumesic, Top. Catal., 2009, 52, 297–303 CrossRef.
- F. Liu, S. Sivoththaman and Z. Tan, Sustainable Environ. Res., 2014, 24, 149–157 Search PubMed.
- S. Mohammad, C. Held, E. Altuntepe, T. Köse and G. Sadowski, J. Phys. Chem. B, 2016, 120, 3797–3808 CrossRef CAS PubMed.
- F. N. D. C. Gomes, L. R. Pereira, N. F. P. Ribeiro and M. M. V. M. Souza, Braz. J. Chem. Eng., 2015, 32, 119–126 CrossRef CAS.
- X. Wu, J. Fu and X. Lu, Bioresour. Technol., 2012, 119, 48–54 CrossRef CAS PubMed.
- G. Yong, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2008, 47, 9345–9348 CrossRef CAS PubMed.
- C. Moreau, A. Finiels and L. Vanoye, J. Mol. Catal. A: Chem., 2006, 253, 165–169 CrossRef CAS.
- S. Lima, P. Neves, M. M. Antunes, M. Pillinger, N. Ignatyev and A. A. Valente, Appl. Catal., A, 2009, 363, 93–99 CrossRef CAS.
- T. Ståhlberg, S. Rodriguez-Rodriguez, P. Fristrup and A. Riisager, Chem.–Eur. J., 2011, 17, 1456–1464 CrossRef PubMed.
- S. M. Salih, I. K. Shakir and A. M. A. Al-Sammarraie, Mater. Sci. Appl., 2017, 08, 889–898 CAS.
- R. L. Vekariya, J. Mol. Liq., 2017, 227, 44 CrossRef CAS.
- F. C. De Melo, R. F. De Souza, P. L. A. Coutinhob and M. O. De Souza, J. Braz. Chem. Soc., 2014, 25, 2378–2384 CAS.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS PubMed.
- J. Zhang, X. Yu, F. Zou, Y. Zhong, N. Du and X. Huang, ACS Sustainable Chem. Eng., 2015, 3, 3338–3345 CrossRef CAS.
- H. Ma, B. Zhou, Y. Li and D. S. Argyropoulos, BioResources, 2012, 7, 533–544 CAS.
- C. Li, Z. K. Zhao, A. Wang, M. Zheng and T. Zhang, Carbohydr. Res., 2010, 345, 1846–1850 CrossRef CAS PubMed.
- E. Durand, J. Lecomte and P. Villeneuve, Biochimie, 2016, 120, 119–123 CrossRef CAS PubMed.
- Q. Cao, X. Guo, S. Yao, J. Guan, X. Wang, X. Mu and D. Zhang, Carbohydr. Res., 2011, 346, 956–959 CrossRef CAS PubMed.
- C. Shi, Y. Zhao, J. Xin, J. Wang, X. Lu, X. Zhang and S. Zhang, Chem. Commun., 2012, 48, 4103–4105 RSC.
- N. L. Lancaster, T. Welton and G. B. Young, J. Chem. Soc., Perkin Trans. 2, 2001, 2267–2270 RSC.
- N. Llewellyn Lancaster, P. A. Salter, T. Welton and G. Brent Young, J. Org. Chem., 2002, 67, 8855–8861 CrossRef PubMed.
- N. L. Lancaster and T. Welton, J. Org. Chem., 2004, 69, 5986–5992 CrossRef CAS PubMed.
- H. L. Ngo, K. Lecompte, L. Hargens and A. B. Mcewen, Thermochim. Acta, 2000, 357–358, 97–102 CrossRef CAS.
- T. Istasse, L. Bockstal and A. Richel, ChemPlusChem, 2018, 83, 1135 CrossRef CAS PubMed.
- S. P. Simeonov, J. A. S. Coelho and C. A. M. Afonso, ChemSusChem, 2012, 5, 1388–1391 CrossRef CAS PubMed.
- X. Qian, M. R. Nimlos, M. Davis, D. K. Johnson and M. E. Himmel, Carbohydr. Res., 2005, 340, 2319–2327 CrossRef CAS PubMed.
- T. M. C. Hoang, E. R. H. Van Eck, W. P. Bula, J. G. E. Gardeniers, L. Lefferts and K. Seshan, Green Chem., 2015, 17, 959–972 RSC.
- H. M. Pilath, M. R. Nimlos, A. Mittal, M. E. Himmel and D. K. Johnson, J. Agric. Food Chem., 2010, 58, 6131–6140 CrossRef CAS PubMed.
- N. R. Vinueza, E. S. Kim, V. A. Gallardo, N. S. Mosier, M. M. Abu-Omar, N. C. Carpita and H. I. Kenttämaa, Biomass Bioenergy, 2015, 74, 1–5 CrossRef CAS.
- S. Kunnikuruvan and N. N. Nair, ACS Catal., 2019, 9, 7250–7263 CrossRef CAS.
- O. Ershova, J. Kanervo, S. Hellsten and H. Sixta, RSC Adv., 2015, 5, 66727–66737 RSC.
- D. W. Harris and M. S. Feather, J. Am. Chem. Soc., 1975, 97(1), 178–181 CrossRef CAS.
- S. Ramchander and M. S. Feather, Arch. Biochem. Biophys., 1977, 178, 576–580 CrossRef CAS PubMed.
- B. Danon, G. Marcotullio and W. De Jong, Green Chem., 2014, 16, 39–54 RSC.
- A. Ricci, B. Di Rienzo, F. Pepi, A. Troiani, S. Garzoli and P. Giacomello, J. Mass Spectrom., 2015, 50, 228–234 CrossRef CAS PubMed.
- L. Antonini, S. Garzoli, A. Ricci, A. Troiani, C. Salvitti, P. Giacomello, R. Ragno, A. Patsilinakos, B. Di Rienzo and F. Pepi, Carbohydr. Res., 2018, 458–459, 19–28 CrossRef CAS PubMed.
- K. P. Madhusudanan, J. Mass Spectrom., 2006, 41, 1096–1104 CrossRef CAS PubMed.
- K. R. Enslow and A. T. Bell, ChemCatChem, 2015, 7, 479–489 CrossRef CAS.
- B. Cinlar, T. Wang and B. H. Shanks, Appl. Catal., A, 2013, 450, 237–242 CrossRef CAS.
- S. Peleteiro, G. Garrote, V. Santos and J. C. Parajo, Afinidad LXXI, 2014, 567, 202–206 Search PubMed.
- J. Tuteja, S. Nishimura and K. Ebitani, Bull. Chem. Soc. Jpn., 2012, 85, 275–281 CrossRef CAS.
- S. Jia, X. He, J. Ma, K. Wang, Z. Xu and Z. C. Zhang, Catal. Sci. Technol., 2018, 8, 5526–5534 RSC.
- S. Wang, Y. Zhao, H. Lin, J. Chen, L. Zhu and Z. Luo, Green Chem., 2017, 19, 3869–3879 RSC.
- T. Wang, J. A. Glasper and B. H. Shanks, Appl. Catal., A, 2015, 498, 214–221 CrossRef CAS.
- S. Xiao, B. Liu, Y. Wang, Z. Fang and Z. Zhang, Bioresour. Technol., 2014, 151, 361–366 CrossRef CAS PubMed.
- I. K. M. Yu, D. C. W. Tsang, A. C. K. Yip, S. S. Chen, Y. S. Ok and C. S. Poon, Bioresour. Technol., 2016, 219, 338–347 CrossRef CAS PubMed.
- V. Choudhary, A. B. Pinar, R. F. Lobo, D. G. Vlachos and S. I. Sandler, ChemSusChem, 2013, 6, 2369–2376 CrossRef CAS PubMed.
- H. Nguyen, V. Nikolakis and D. G. Vlachos, ACS Catal., 2016, 6, 1497–1504 CrossRef CAS.
- T. Dallas Swift, H. Nguyen, A. Anderko, V. Nikolakis and D. G. Vlachos, Green Chem., 2015, 17, 4725–4735 RSC.
- P. Wrigstedt, J. Keskiväli, M. Leskelä and T. Repo, ChemCatChem, 2015, 7, 501–507 CrossRef CAS.
- Y. Zhang, E. A. Pidko and E. J. M. Hensen, Chem.–Eur. J., 2011, 17, 5281–5288 CrossRef CAS PubMed.
- S. Bali, M. A. Tofanelli, R. D. Ernst and E. M. Eyring, Biomass Bioenergy, 2012, 42, 224–227 CrossRef CAS.
- Z. Zhang, Q. Wang, H. Xie, W. Liu and Z. Zhao, ChemSusChem, 2011, 4, 131–138 CrossRef CAS PubMed.
- H. Li, W. Xu, T. Huang, S. Jia, Z. Xu, P. Yan, X. Liu and Z. C. Zhang, ACS Catal., 2014, 4, 4446–4454 CrossRef CAS.
- E. A. Pidko, V. Degirmenci, R. A. Van Santen and E. J. M. Hensen, Inorg. Chem., 2010, 49, 10081–10091 CrossRef CAS PubMed.
- C. B. Rasrendra, I. G. B. N. Makertihartha, S. Adisasmito and H. J. Heeres, in Topics in Catalysis, 2010, vol. 53, pp. 1241–1247 Search PubMed.
- C. B. Rasrendra, J. N. M. Soetedjo, I. G. B. N. Makertihartha, S. Adisasmito and H. J. Heeres, in Topics in Catalysis, 2012, vol. 55, pp. 543–549 Search PubMed.
- L. X. Zhang, H. Yu, H. B. Yu, Z. Chen and L. Yang, Chin. Chem. Lett., 2014, 25, 1132–1136 CrossRef CAS.
- Y. Nie, Q. Hou, W. Li, C. Bai, X. Bai and M. Ju, Molecules, 2019, 24(594), 1–18 Search PubMed.
- A. Queen, Can. J. Chem., 1977, 55, 3035–3039 CrossRef CAS.
- M. Rebstockova and M. Bartusek, Collect. Czech. Chem. Commun., 1977, 42, 627–636 CrossRef CAS.
- J. A. Peters, Coord. Chem. Rev., 2014, 268, 1–22 CrossRef CAS.
- T. S. Hansen, J. Mielby and A. Riisager, Green Chem., 2011, 13, 109–114 RSC.
- B. R. Caes, M. J. Palte and R. T. Raines, Chem. Sci., 2013, 4, 196–199 RSC.
- Q. Girka, B. Estrine, N. Hoffmann, J. Le Bras, S. Marinković and J. Muzart, React. Chem. Eng., 2016, 1, 176–182 RSC.
- D. H. Lukamto, P. Wang and T. P. Loh, Asian J. Org. Chem., 2013, 2, 947–951 CrossRef CAS.
- B. J. Graham and R. T. Raines, Biomass Convers. Biorefin., 2019, 9, 471–477 CrossRef CAS.
- L. Hu, Y. Sun, L. Lin and S. Liu, J. Taiwan Inst. Chem. Eng., 2012, 43, 718–723 CrossRef CAS.
- L. Hu, Y. Sun, L. Lin and S. Liu, Biomass Bioenergy, 2012, 47, 289–294 CrossRef CAS.
- Z. L. Xu, X. Y. Wang, M. Y. Shen and C. H. Du, Chem. Pap., 2016, 70, 1649–1657 CAS.
- X. Hu, R. J. M. Westerhof, D. Dong, L. Wu and C. Z. Li, ACS Sustainable Chem. Eng., 2014, 2(11), 2562–2575 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |