
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDFT screening of metallic single-replacements for lead-free perovskites with intrinsic photovoltaic functionalities†
Clark Zhang and
Xuan Luo*
and
Xuan Luo*
National Graphene Research and Development Center, Heming Avenue, Springfield, Virginia, USA
First published on 23rd June 2020
Abstract
Methylammonium lead triiodide perovskites, CH3NH3PbI3 (MAPbI3), are solution-processable materials with photovoltaic properties capable of surpassing those of silicon solar cells. However, concerns over lead toxicity and lack of exploration into transition metal perovskites drove this ab initio Density Functional Theory screening for environmentally friendly perovskite materials by incorporating transition and post-transition metals at the B-site of MAPbI3. This revealed fourteen replacements to be suitable: their band structures are highly dispersive while band gaps of such materials fall within ideal ranges for single-junction and tandem cells. Transition metal monoreplacements are shown to be viable perovskites after reducing the size of the halide, corroborating that tunability of the band gap is observed in halide replacement at the X-site. Strong peaks in the imaginary output of the dielectric function below 3.5 eV indicate high sunlight absorption efficiency for select materials. Excellent carrier mobility is expected of studied materials as their effective mass is low. This work helps gain further insight into the viability of transition metals for lower toxicity and higher absorption divalent perovskites.
1 Introduction
In an effort to reduce the impact of global warming, renewable energy reliance has increased. Recent reductions in manufacturing costs of solar cells are steps towards the future use of large-scale photovoltaics cells (PV). However, cost and efficiency are still challenges faced by the PV industry. A new family of hybrid organic–inorganic lead halide perovskites offer high prospects on both energy conversion efficiency and production cost, especially methylammonium lead triiodide (CH3NH3PbI3 or MAPbI3).MAPbI3 has an extremely high absorption coefficient, long charge diffusion length, high defect tolerance, and high ambipolar transport. It has an experimental band gap of 1.55 eV,1,2 close to that of the ideal band gap (which yields the highest theoretical conversion efficiency), 1.35 eV.3 The outstanding performance of these materials proves useful for other optoelectronic applications.4
However, there are two main issues: the stability and the toxicity of lead (Pb). The presence of Pb in high performing perovskites poses serious environmental and health concerns. Solar cells are placed in direct contact with nature, so exposure to rainfall and solar heating is inevitable. Degradation into volatile PbI2 is highly likely due to the low thermal stability of MAPbI3, causing severe chronic health problems.5 Lead pollution further impacts natural resources, diffusing into the atmosphere, contaminating water sources, and emitting greenhouse gases.6 Elimination of the Pb toxicity from perovskite cells requires complete replacement of Pb with other non-toxic elements.
The present study is a systematic search for stable methylammonium-metallic halide alternatives to Pb-based perovskites. Each suitable metal candidate was trialed as a single replacement of Pb at the B-site. Replacements of iodine at the X site with bromine were conducted for every B-site replacement, and with X-site replacement with chlorine for select perovskites. The paper details methods used to perform first-principles calculations, presents, discusses and compares results to other theoretical and experimental researches. The results show that proposed lead-free perovskite materials display heightened optoelectrical properties, greater stability, and much potential for use in single-junction and tandem solar cells.
2 Computational details
First-principles calculations were performed in the frame of Density Functional Theory (DFT) with the GGA-PBE exchange–correlation functional implemented in ABINIT7 code. PAW pseudopotentials,8 generated with the ATOMPAW code, were used to calculate the electronic structure of each perovskite, shown in Fig. 1. Originally, only 48 perovskites were to be considered, but MAZnCl3 was later included after analyzing how the halogen tunability of the band gap affected the methylammonium zinc tribromide perovskite (see Section 3.1).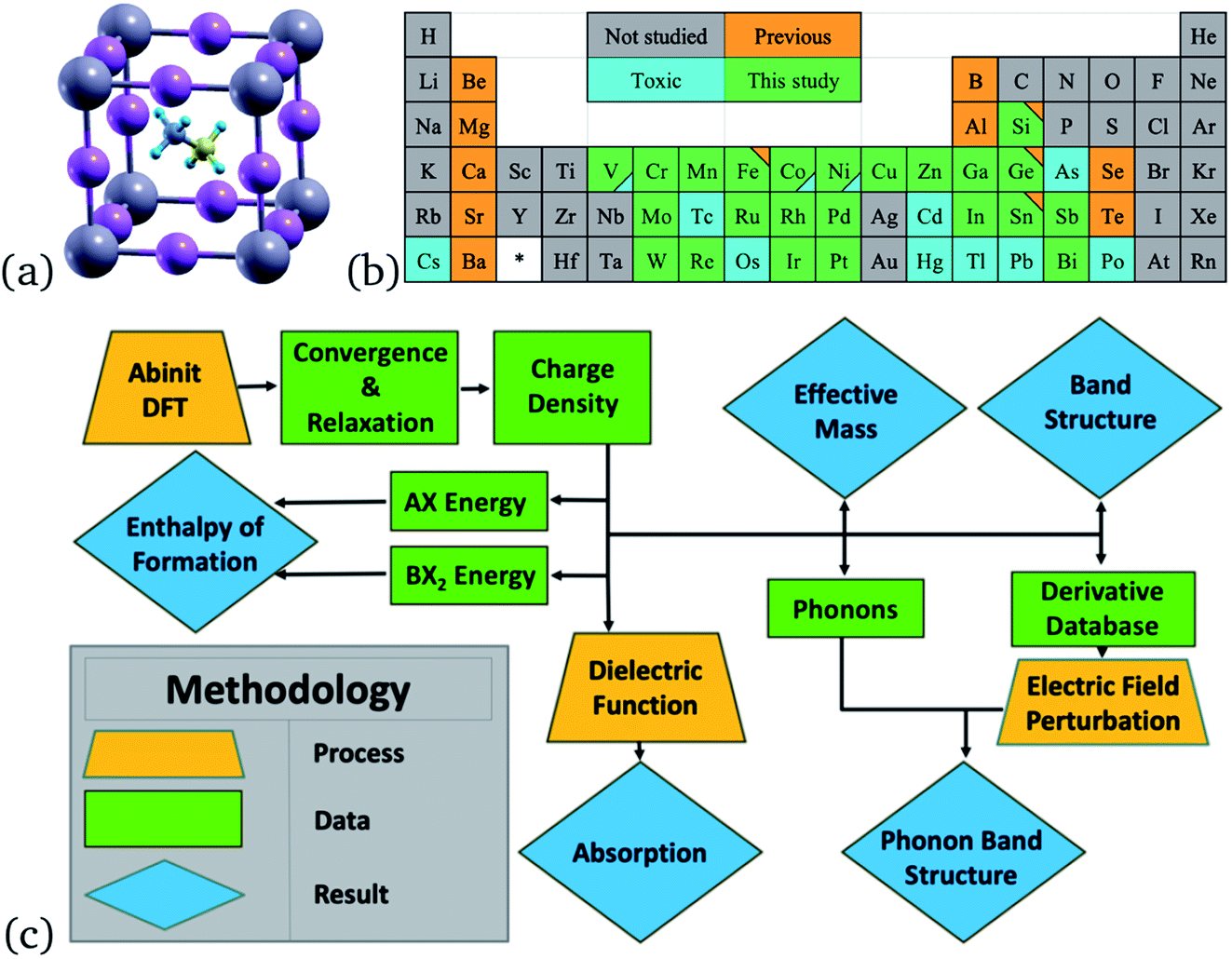 | ||
| Fig. 1 (a) Unit cell of MAPbI3 (ball and stick) from an isometric view, Pb represented in grey, I in pink, C in yellow, N in greyish blue, H in bright blue. (b) Elements used to replace lead at the B-site are colored green. Elements with Q3D Class 1 and 2A of toxicity16 are colored blue. Previously studied elements for single-replacement of Pb are colored orange. Triangles in the corner represent two colorings. (c) A diagram of the research process and order of calculations used in this project. Orange trapezoids represent major calculations and processes. Green rectangles represent obtained data. Blue rhombuses represent final results. | ||
Convergence studies (see Fig. S1†) in the energy cutoff and the density of the k-mesh revealed 25 hartree and a density of 6 × 6 × 6 to be sufficiently converged. A homogenous shift in the grid of k-points of 0.5, 0.5, 0.5 (reduced coordinates in the coordinate system defining the k-point lattice) was implemented to increasing the calculation efficiency. Perovskites were relaxed to a pseudo-cubic crystal structure with the tolerance on the maximal force of 5 × 105 hartree per bohr and with a tolerance on the differences in forces per self-consistent cycle of 5 × 106 hartree per bohr. Dilation of primitive vectors was conducted with the Broyden–Fletcher–Goldfarb–Shanno minimization.9–12
For the band structure, k-points S, X, U, R, Γ, Z, T, and Y were selected as the circuit to sample the irreducible first Brillouin zone, taking into account spin–orbit coupling.
Effective mass was calculated using a linear response function that eliminates the need for spin–orbit coupling (SOC) and temperature smearing.13 Degenerate bands were treated by using transport equivalent effective mass to avoid double perturbation.
For absorption, the Kohn–Sham band structure is further corrected using a one-shot G0W0 quasiparticle calculation14 for solids with an energy cutoff of the wavefunction, sigma exchange, and a dielectric energy cutoff of 25 hartree (an exact treatment of the exchange part can be achieved with these parameters set to the value of the energy cutoff15). The lowest occupied band considered was band number six, with a total of 44 bands for the Bethe–Salpeter calculation. This screening was formatted with an unsymmetrical k-mesh (shifted along the primitive axis by 0.11, 0.21, 0.31) and a 6 × 6 × 6 q-point mesh, and solved with Bethe–Salpeter equations. The imaginary output was plotted against light intensities from 0 to 4 eV, with intervals of 0.02 eV. The research process is depicted in Fig. 1c.
3 Results and discussion
3.1 Band structure
Table 1 shows the band gap values for MAPbI3 and all mono-replacements in comparison to previous calculated results (with pseudo-cubic structures and without quasi-particle corrections) and experimental results. Several calculated band gaps in this study were reported with previously calculated and/or experimental results. All calculated band structures are displayed in the ESI,† with each band structure centered at the Fermi energy.| Material | Fund. (eV) | Opt. (eV) | PBE prev. (eV) | Exp. (eV) |
|---|---|---|---|---|
| MAPbI3 | 1.59 | 1.59 | 1.63,1 1.59![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 17 |
1.5518 |
| MASiI3 | 1.70 | 1.70 | 0.141 |
— |
| MAVI3 | 0.00 | 0.00 | 0 | — |
| MACrI3 | 0.00 | 0.00 | — | — |
| MAMnI3 | 0.00 | 0.00 | — | — |
| MAFeI3 | 0.00 | 0.00 | — | — |
| MACoI3 | 0.00 | 0.00 | — | — |
| MANiI3 | 0.00 | 0.00 | — | — |
| MACuI3 | 0.85 | 1.25 | — | — |
| MAZnI3 | 1.89 | 1.89 | 2.721 |
— |
| MAGaI3 | 1.45 | 1.45 | 0.721 |
— |
| MAGeI3 | 1.38 | 1.38 | — | 1.901 |
| MAMoI3 | 0.00 | 0.00 | — | — |
| MARhI3 | 0.00 | 0.00 | — | — |
| MARuI3 | 0.00 | 0.00 | — | — |
| MAPdI3 | 0.00 | 0.00 | — | — |
| MAInI3 | 0.61 | 0.61 | — | — |
| MASnI3 | 1.52 | 1.52 | 0.501 |
1.101 |
| MASbI3 | 0.00 | 0.00 | — | — |
| MAWI3 | 0.00 | 0.00 | — | — |
| MAReI3 | 0.00 | 0.00 | — | — |
| MAIrI3 | 0.00 | 0.00 | — | — |
| MAPtI3 | 0.00 | 0.00 | — | — |
| MABiI3 | 0.00 | 0.00 | — | — |
In general, the VB and CB dispersion is similar for most transition metals (see Tables S1 and S2†), especially the conduction band edge. In general, the VBM is dominated by the 5px,y,z orbitals of iodine while the CBM is dominated by the P orbitals in the B cation, see Fig. S2.† For certain monoreplacements, their metallic nature dwarfed the effects of methylammonium and iodine, resulting in a loss of the semiconductor properties of those materials.
Most transition metal monoreplacements didn't form any band gaps. However, MACuI3 formed an indirect band gap with the VBM at point R and a CBM and point Z. MAZnI3 formed a direct band gap of 1.89 eV at point R, see Fig. S5(4a).† Partial experimental replacement of Pb with Zn (MAI[PbI2]0.97[ZnCl2]0.03) shows heightened conversion efficiency of 1.8% compared to MAPbI3.19 This indicates ZnCl is a viable PV material, and its other properties are explored throughout this study.
The post-transition metal monoreplacement band gaps mostly fell between 1.3 and 1.7 eV. From group XIII elements, MAGaI3 was studied by Ali et al.1 and they found it to have a band gap of 0.72 eV, while this study finds it to be 1.45 eV. This discrepancy is attributed to the denser k-point mesh used in this study. Both the gallium and indium replacements had a single very high energy valance band (near the Fermi level) compared to the other valance bands (see Fig. S5(4b) and S6(5c),† respectively), therefore, their conversion efficiencies would be lower due to the energy expended during the phonon-assisted recombination.
Group XIV consists of Si, Ge, Sn, and Pb. MASiI3 has the largest band gap of the group, at 1.70 eV. MASnI3 has a band gap of 1.52 eV, however, differs from the previously reported results. MAGeI3 is also a prospective alternative to MAPbI3. It has an experimental band gap of 1.90 eV, larger than the calculated band gap of 1.38 eV. A binary mixture of Sn/Ge or with other suitable replacements could result in very strong absorption efficiencies.
Group XV consists of metalloids As and Sb, and poor metal Bi. Arsenic was not studied due to its toxicity. Both Sb and Bi monoreplacements have an intermediate band. This is interesting for intermediate band solar cells, but not for the current study, so group XV materials are not suitable for making a direct band gap material (Table 2).
| Material | Fund. (eV) | Opt. (eV) | PBE prev. (eV) | Exp. (eV) |
|---|---|---|---|---|
| MAPbBr3 | 2.23 | 2.23 | 2.11 |
2.320 |
| MASiBr3 | 2.57 | 2.57 | — | — |
| MAVBr3 | 0.00 | 0.00 | — | — |
| MACrBr3 | 0.00 | 0.00 | — | — |
| MAMnBr3 | 0.14 | 0.11 | — | — |
| MAFeBr3 | 0.12 | 0.08 | — | — |
| MACoBr3 | 0.00 | 0.00 | — | — |
| MANiBr3 | 0.00 | 0.00 | — | — |
| MACuBr3 | 3.74 | 3.74 | — | — |
| MAZnBr3 | 1.49 | 1.49 | — | — |
| MAGaBr3 | 1.85 | 1.85 | — | — |
| MAGeBr3 | 2.45 | 2.45 | — | — |
| MAMoBr3 | 0.00 | 0.00 | — | — |
| MARhBr3 | 0.00 | 0.00 | — | — |
| MARuBr3 | 0.19 | 0.18 | — | — |
| MAPdBr3 | 0.00 | 0.00 | — | — |
| MAInBr3 | 1.57 | 1.57 | — | — |
| MASnBr3 | 1.97 | 1.97 | — | — |
| MASbBr3 | 0.00 | 0.00 | — | — |
| MAWBr3 | 0.00 | 0.00 | — | — |
| MAReBr3 | 0.00 | 0.00 | — | — |
| MAIrBr3 | 0.00 | 0.00 | — | — |
| MAPtBr3 | 0.00 | 0.00 | — | — |
| MABiBr3 | 0.75 | 0.25 | — | — |
Further replacement of the X triple anion was studied with bromine (X-site = Br).
Replacing iodine with lighter halogens increased the bandgap; methylammonium triiodide perovskites with direct band gaps had an increase in their band gap, much higher than the ideal band gap for single junction cells (1.35 eV), when the triiodide was replaced with tribromide. However, replacement of triiodide with tribromide opened up an optical band gap of some transition metal monoreplacements, indicating they have semimetal properties. This can be noted in MAMnBr3, MAFeBr3, and MARuBr3. The VBM and CBM is shown in more detail in Fig. S3,† represented in yellow fill. This is crucial for further research into the incorporation of transition metals in PV materials.
After noting how MAZnBr3 had a band gap of 1.49 eV, we decided to study MAZnCl3 and MAZnF3 as well because replacement at the X-site with a lighter halogen could result in another perovskite with a close-to-ideal band gap. As predicted, MAZnCl3 had a band gap of 1.69 eV, shown in Fig. 2, but MAZnF3 had a band gap of 3.55 eV. In all later sections, MAZnCl3 is another perovskite that we studied and found the properties of. Its prospects as a new foundation for experimental PV investigations will be made clear as its properties are discussed in later sections.
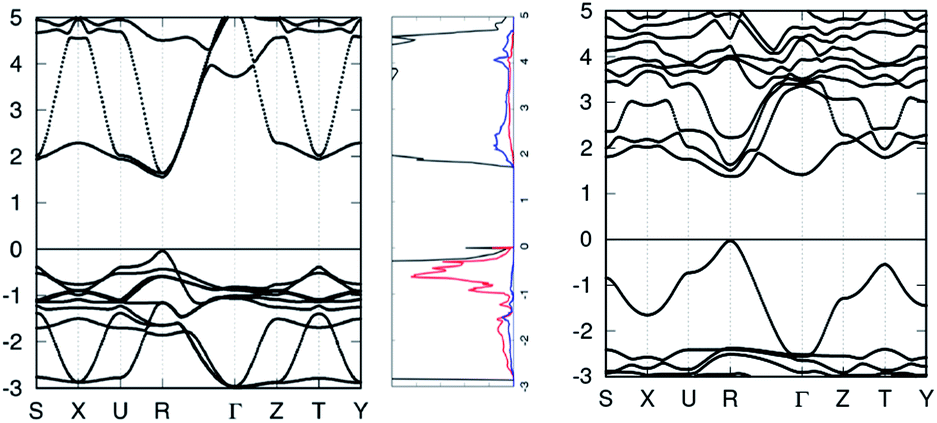 | ||
| Fig. 2 Band structure with its TDOS and PDOS (iodine in red, lead in blue) of MAPbI3 on the left and band structure of MAZnCl3 on the right. The Fermi energies are shifted to be at 0 eV in the band structures. | ||
No other trichloride materials were studied since the tunability nature of the band gap would increase the band gap of all trichloride monoreplacements beyond that of the ideal band gap. Therefore, studying them would be counterintuitive.
For monoreplacements, it can be concluded that Ge, Sn, Si, and Zn perovskites are the best replacements of Pb in MAPbI3 for high performing solar cells. However, perovskites with band gaps inconsistent with the single-junction ideal can still be used in creation of tandem solar cells.21,22
3.2 Structural and thermal stability
A main issue of MAPbI3 is its poor structural and thermal stability. According to Yang et al.12 and Li et al.,23 a material with Goldschmidt tolerance factor24 between 0.81 and 1.11 exists in a cubic to pseudocubic (lattice parameters relatively equal; dihedral angles relatively close to 90°) perovskite structure. The octahedral factor (μ) further indicates severity of octahedral tilting, the primary factor for structural distortions; the suitable range for the octahedral factor is 0.4 ≤ μ ≤ 0.90.
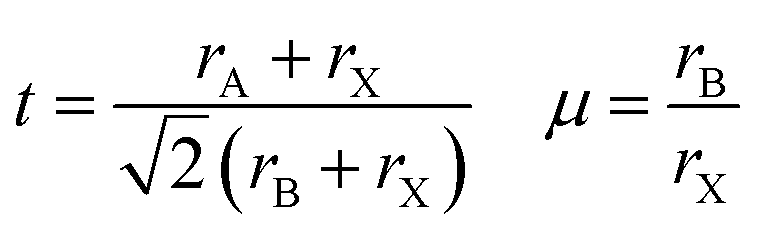 | (1) |
The thermal stability of each perovskite is determined via its energy of formation. MAPbI3 is thermally unstable, decomposing into the compounds MAI + PbI2 above 333 K.26 Similarly, this study considers the decomposition of all mono-replacements in the form ABX3 → AX + BX2. The enthalpy of formation can be computed through the following equation:
| ΔH = E(ABX3) − [E(AX) + E(BX2)] | (2) |
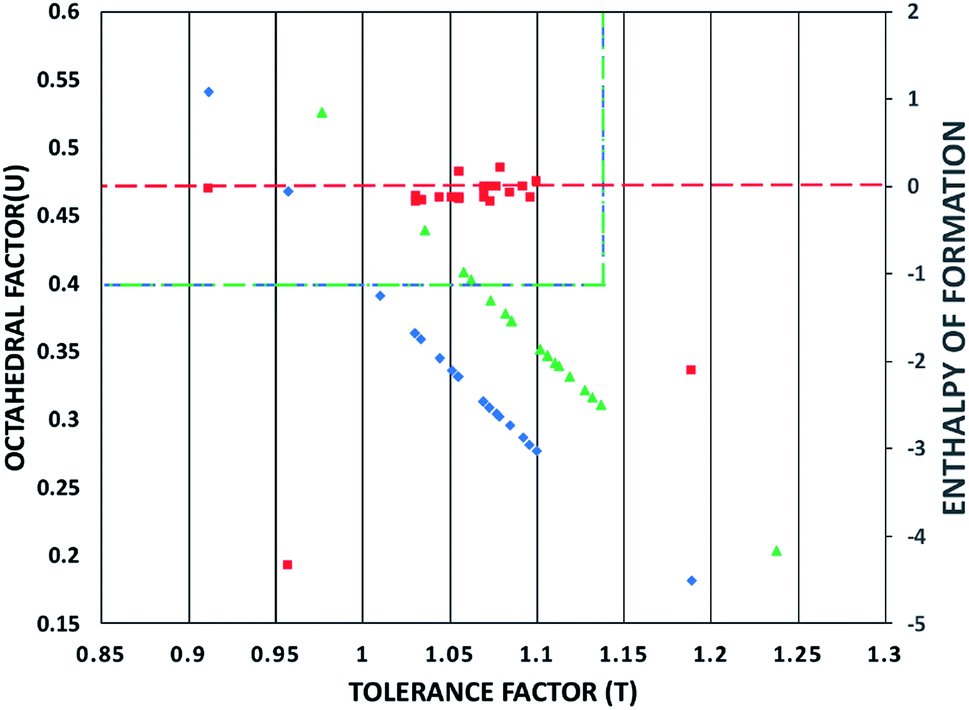 | ||
| Fig. 3 The tolerance factor (t) and the octahedral factor (μ) (blue rhombic symbols) represent the structural stability for the studied triiodide perovskites, while the green triangles represent the structural stability for the studied tribromide perovskites. The red square symbols represent calculated enthalpies of formation in ΔH/f.u. The blue/green dotted lines represent the suitable range for the tolerance and octahedral factors. The red dotted line an enthalpy of formation of 0 eV/f.u. | ||
| Material | me | mh | me (prev.) | mh (prev.) |
|---|---|---|---|---|
| Si | 0.20 | 0.15 | 0.1927 |
0.1627 |
| MAPbI3 | 0.18 | 0.24 | 0.19,27 0.3028 |
0.2527 |
| MAGaI3 | 0.24 | 0.18 | — | — |
| MACuI3 | 0.88 | 0.88 | — | — |
| MAGeI3 | 0.24 | 0.32 | — | — |
| MAInI3 | 0.45 | 0.14 | — | — |
| MASnI3 | 0.17 | 0.14 | — | — |
| MAZnI3 | 0.65 | 0.33 | — | — |
| MAGaBr3 | 0.30 | 0.60 | — | — |
| MACuBr3 | 0.73 | 0.73 | — | — |
| MAGeBr3 | 0.24 | 0.32 | — | — |
| MAInBr3 | 0.15 | 0.30 | — | — |
| MASnBr3 | 0.07 | 0.15 | — | — |
| MABiBr3 | 0.14 | 0.22 | — | — |
| MAZnBr3 | 0.61 | 0.21 | — | — |
| MAZnCl3 | 0.25 | 0.25 | — | — |
3.3 Effective mass
The effective mass of all, with the exception of bismuth, tin, and gallium monoreplacements, had isotropic effective masses of electrons and holes. MABiBr3 has higher effective mass of holes/electrons along one primitive vector. The same effect is seen in MASnI3, with 0.5 effective mass of electron along one primitive vector. Lastly, MAGaBr3 has 1.5 effective mass of electrons along one primitive vector.In general, m* of most monoreplacements are higher than that of MAPbI3 and of crystalline silicon cells. However, the tin monoreplacement performed exceptionally well, consistently achieving lower effective mass of holes and electrons than MAPbI3 with both triiodide and tribromide. As triiodide is replaced with lighter halogens, the m* was reduced. Based on these trends, the lighter the substance at the X-site leads to lower m*.
MAZnCl3 fits into the above trend well. The zinc triiodide, tribromide, and trichloride perovskites had decreasing effective masses. MAZnCl3 has effective masses of electrons is comparable to that of conventional crystalline silicon PV materials while its effective mass of electrons is comparable to that of MAPbI3.
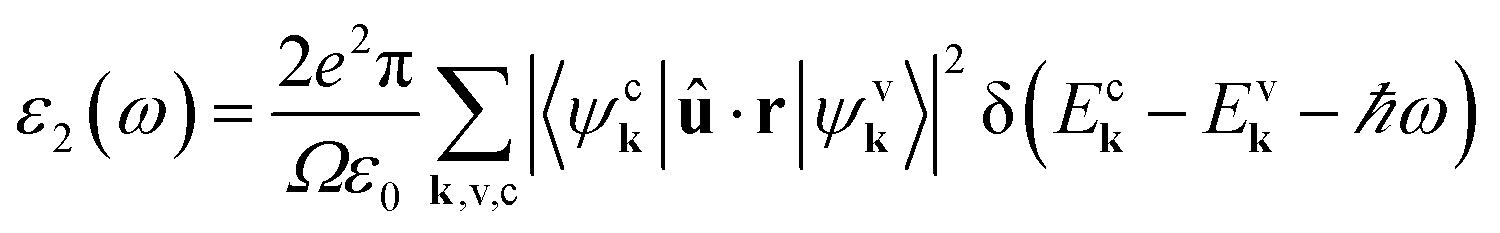 | (3) |
Ninety-eight percent of solar flux is below 3.4 eV,22 concentrated towards infrared light, indicating that such materials with peak absorption below 3.4 eV are strongly efficient for PV applications.
The imaginary part of the dielectric function (ε2) is displayed in Fig. S4† for select monoreplacements, compared to MAPbI3. The MAPbI3 results are comparable to those of previous DFT studies and of experimental results.27,29 All of the selected monoreplacements display absorption peaks at energies lower than that of MAPbI3 (below 2.5 eV). Secondary and higher order peaks appear in lead-free perovskites because there are fewer degenerate bands at critical points S, Q, R, Γ, and T.
Replacement of iodine with bromine shifted the strongest absorption peaks from between 1 eV to 1.8 eV, clustered around 1.75 eV, corresponding to 700 nm red light (in the center of the solar flux concentration). These two trends indicate that lighter halogens at the X-site and lighter monoreplacements at the B-site red-shift absorption towards the bulk of solar concentration, minimizing thermalization loss.
Fig. 4 displays the ε2 of MAPbI3 in comparison to the ε2 of MAZnCl3. MAZnCl3 displayed similar absorption peaks to MAPbI3: one strong absorption peak between 2 eV and 2.5 eV and a second absorption peak around 3 eV. This indicates MAZnCl3 could have high conversion efficiencies for the same energies of light as MAPbI3. Interestingly, MAZnCl3 displayed its strongest absorption peak around 4 eV. Since energy peak is greater than most of the solar flux energy, MAZnCl3 could find use in concentrator solar cells, where the incident energy is much higher.
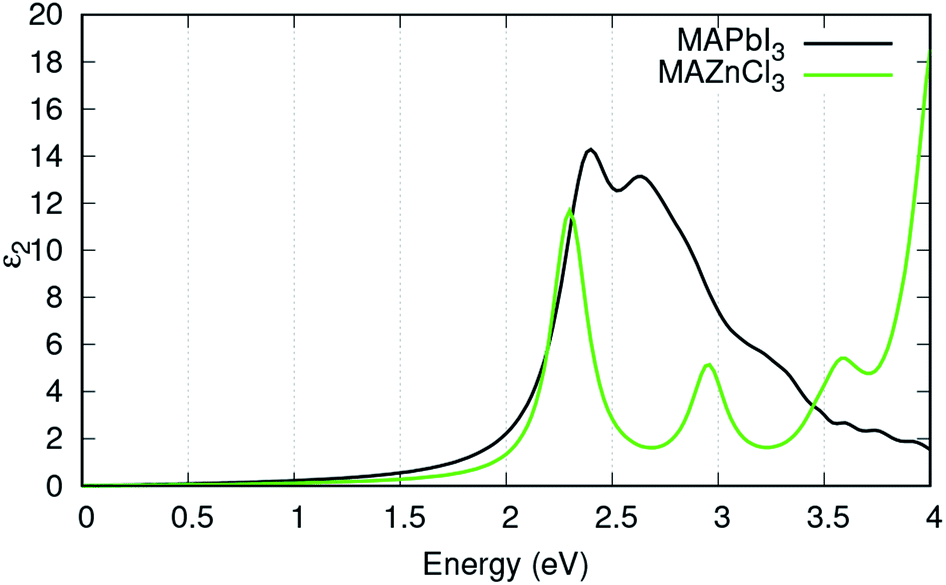 | ||
| Fig. 4 Imaginary part of the calculated dielectric function, ε2 of MAPbI3 in black, MAZnCl3 in green. | ||
4 Conclusions
This study presents a systematic DFT study exploring the possibility of monoreplacements of lead in MAPbI3 with non-toxic metallic elements. For homogeneous single-junction solar cells, it can be determined that Ge, Sn, Si, and Zn are the best replacements of lead in MAPbI3. However, incorporation of other transition metals is feasible with light anions at the X-site, due to the tunability of the band gap. Based on discussed trends, a perovskite with a relatively lighter replacement of lead at the B-site and a lighter anion at the X-site would maximize structural stability, carrier and hole mobility, and absorption efficiency. MAZnCl3 fits this description well; it has a close-to-ideal band gap (1.69 eV), low effective mass (0.25 m*), high structural stability (t = 1.06, μ = 0.52), high thermal stability (ΔH −49.5 kJ mol−1), and absorption similar to MAPbI3. Zinc is also a highly abundant element in the Earth's crust, reducing production cost. MAZnCl3, as well as the data and trends in this study, establishes a new foundation for experimental investigations in low cost, non-toxic, lead-free perovskites for photovoltaic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was conducted in the National Graphene Research and Development Center under the supervision of Dr Xuan Luo. The author would like to thank Dr GeFei Qian for technical support and fruitful discussion.Notes and references
- R. Ali, G.-J. Hou, Q.-B. Zhu, Z.-G. Yan, Q.-R. Zheng and G. Su, Chem. Mater., 2018, 30, 718–728 CrossRef CAS
.
- M. Johnston and L. Herz, Acc. Chem. Res., 2016, 49, 146–154 CrossRef CAS PubMed
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS
- N. Rathore, N. L. Panwar, F. Yettou and A. Gama, Int. J. Ambient Energy, 2019, 1–18 Search PubMed
- A. Babayigit, A. Ethirajan, M. Muller and B. Conings, Nat. Mater., 2016, 15, 247–251 CrossRef CAS PubMed
- J. Gong, S. B. Darling and F.-Q. You, Energy Environ. Sci., 2015, 8, 1953 RSC
- X. Gonze, B. Amadon, P.-M. Anglade, J.-M. Beuken, F. Bottin, P. Boulanger, F. Bruneval, D. Caliste, R. Caracas, M. Cote, T. Deutsch, L. Genovese, P. Ghosez, M. Giantomassi, S. Goedecker, D. Hamann, P. Hermet, F. Jollet, G. Jomard, S. Leroux, M. Mancini, S. Mazevet, M. Oliveira, G. Onida, Y. Pouillon, T. Rangel, G.-M. Rignanese, D. Sangalli, R. Shaltaf, M. Torrent, M. Verstraete, G. Zerah and J. W. Zwanziger, Comput. Phys. Commun., 2009, 180, 2582–2615 CrossRef CAS
- P. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed
- C. G. Broyden, J. Inst. Math. Its Appl., 1970, 6, 76 CrossRef
- R. Fletcher, Comput. J., 1970, 13, 317 CrossRef
- D. F. Shanno, Math. Comput., 1970, 24, 647 CrossRef
- D. Yang, X. Lv, J. Zhao, Q. Xu, Y. Fu, Y. Zhan, A. Zunger and L. Zhang, Chem. Mater., 2017, 29, 524–538 CrossRef CAS
- J. Laflamme Janssen, Y. Gillet, S. PoncÃľ, A. Martin, M. Torrent and X. Gonze, Phys. Rev. B, 2016, 93, 205147 CrossRef
- M. Giantomassi, M. Stankovski, R. Shaltaf, M. GrÃijning, F. Bruneval, P. Rinke and G.-M. Rignanese, Phys. Status Solidi B, 2011, 248, 275–289 CrossRef CAS
- W. G. Aulbur, L. Jönsson and J. W. Wilkins, Solid State Phys., 2000, 54, 1–218 CAS
- I. C. for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, 2019 Search PubMed.
- G. Giorgi and K. Yamashita, Nanotechnology, 2015, 26, 442001 CrossRef PubMed
- G. E. Eperon, S. D. Stranks, C. Menelaou, M. B. Johnston, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 982–988 RSC
- J. Jin, H. Li, C. Chen, B. Zhang, L. Xu, B. Dong, H. Song and Q. Dai, ACS Appl. Mater. Interfaces, 2017, 9, 42875–42882 CrossRef CAS PubMed
- P. P. Boix, S. Agarwala, T. M. Koh, N. Mathews and S. G. Mhaisalkar, J. Phys. Chem. Lett., 2015, 6, 898–907 CrossRef CAS PubMed
- S. Bremner, C. Yi, A. Almansouri, I. Ho-Baillie and M. Green, Sol. Energy, 2015, 135, 750–757 CrossRef
- C. Henry, J. Appl. Phys., 1980, 51, 4494 CrossRef CAS
- C. Li, X. Lu, W. Ding, L. Feng, Y. Gao and Z. Guo, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 702–707 CrossRef CAS
- H. KronmÃijller and S. Parkin, Handbook of magnetism and advanced magnetic materials: Spintronics and magnetoelectronics., John Wiley & Sons, 2007 Search PubMed
- J. A. Wasastjerna, Soc. Sci. Fenn., Commentat. Phys.-Math., 1923, 1, 1–25 Search PubMed
- Q. Wang, Q. Dong, T. Li, A. Gruverman and J. Huang, Adv. Mater., 2016, 28, 6734–6739 CrossRef CAS PubMed
- R. Ali, G.-J. Hou, Q.-B. Zhu, Z.-G. Yan, Q.-R. Zheng and G. Su, J. Mater. Chem. A, 2018, 6, 9220–9227 RSC
- K. Wang, X. Liang, K. L. Anokhin, N. Gerasimov and P. A. Dremova, Adv. Electron. Mater., 2015, 1, 150089 Search PubMed
- Y. Jiao, F. Ma, G. Gao, H. Wang, J. Bell, T. Frauenheim and A. Du, RSC Adv., 2015, 5, 82346–82350 RSC
Footnote |
| † Electronic supplementary information (ESI) available: PDOS and TDOS of MAPbI3, studied band structures, and highlights in transition metal perovskite band gaps. See DOI: 10.1039/d0ra03034a |
| This journal is © The Royal Society of Chemistry 2020 |