
A new nano-on-micro stationary phase based on nanodiamond bonded on silica for hydrophilic interaction chromatography†
Tianpei Caiac,
Haijuan Zhanga,
Zhan Lia,
A. F. M. Mustafizur Rahmana and
Hongdeng Qiu*ab
aKey Laboratory of Chemistry of Northwestern Plant Resources, Key Laboratory for Natural Medicine of Gansu Province, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China. E-mail: hdqiu@licp.cas.cn; Fax: +86 931 8277088; Tel: +86 931 4968877
bState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China
cUniversity of Chinese Academy of Sciences, Chinese Academy of Sciences, Beijing 100049, China
First published on 24th March 2016
Abstract
Nanodiamond particles were covalently bonded on silica microparticles and the resulting material was nicely decorated with a thin layer of oxidized nanodiamonds. This new nano-on-micro hybrid material was packed into a column and successfully evaluated in hydrophilic interaction chromatography.
Carbon nanomaterials (CNMs), thanks to their special qualities such as high thermostability, excellent electrical properties, good biocompatibility and easy derivatization, are acting as vital parts in the development and evolution of many research fields, including chemical, electrical, medical science etc. For analytical chemistry, the large surface area/volume ratio of CNMs make them superior adsorbents, which means they have a vast amount of potential for applications in sample preparation and chromatography separation. Until now, CNMs have been used as extraction material in SPE, SPME or as matrix for liquid–liquid extraction,1 meanwhile they have also been used as chromatographic stationary phase (SP) in monolithic column or stabilized on matrix (e.g. spherical silica) to assemble CNM-grafted packing materials, all of these have been reviewed specifically.2
Compared with other adsorbents, CNMs, especially carbon nanotube (CNT) and graphene (G) show stronger adsorbability.3 While other kind of adsorbents suffering low affinity to the specified sample which hinder effective separation or extraction, CNMs besetting by the same situation caused by their excess adsorbability which make it difficult for sample to be eluted. Speaking of chromatography, good separation performance based on favorable adsorption–desorption equilibrium. However, CNT and G not only have super huge surface area, but also characterized by their large π-conjugated structure, which make it even harder for molecules like aromatic compound to desorb from these packing materials. Besides, CNTs are very easy to twist into bundles4 and G suffers the layered structure,5 which may generate dead volume in column and hinder the mass transfer even coupled with matrix, all of these contribute to the unsatisfactory column efficiency.
Different with CNT and G, nanodiamond (ND) do not contain large π-conjugated system and exist as particulate morphology, meanwhile many effective methods has been used to get perfectly isolate particles,6 so ND is hopeful to become an excellent carbon nanomaterial stationary phase. Thus far, research into nanodiamond-modified stationary phases is relatively scarce. ND was first used as HPLC-SP by Nesterenko's group in 2007, in which porous diamond packing materials of micron size (3–6 μm) was prepared from detonation nanodiamonds (DNDs) by sintering.7 Subsequently, the same group studied the chromatographic performance of this kind of ND-SP in ion chromatography (IC), normal-phase liquid chromatography (NPLC) and hydrophilic interaction liquid chromatography.8 Linford's group synthesized modified core–shell diamond SP with layer-by-layer deposition and studied the chromatographic performance in both reversed-phase liquid chromatography (RPLC) and NPLC mode.9 Then the same group changes the microdiamond core into spherical carbon core and optimized the deposition procedure, the new SP shows improved stability and better separation efficiency.10
In this paper, a new and facile way to prepare nanodiamond coated on silica hybrid material was introduced and this nano-on-micro material was used for HILIC successfully. To be specific, 3-aminopropyltriethoxysilane (APTES) was used as coupling agent to stabilize the oxidized nanodiamond (OND) onto the surface of spherical porous silica through covalent bonding reaction. The resulting packing material coated by a thin layer of OND. For carboxyl groups on OND distinctly enhanced the hydrophilicity of SP,11 so we evaluated the chromatographic performance in HILIC mode with sulfonamides and saccharides as probes.12
First, raw ND was oxidized by H2SO4/HNO3 (v/v = 3/1) for 72 h at 90 °C to get a carboxyl terminate surface.13 Then the OND was centrifuged and washed with ultrapure water for many times until neutralized, followed by freeze drying to get grey powder. Second, 150 mg of OND was added into 100 mL dried DMF and sonicated for 1 h to get stable slurry, then 3 mmol of APTES was added into the slurry and 0.50 mL of diethyl cyanophosphonate (DEPC) and 0.65 mL of triethylamine were added as catalyst for the amidation reaction.14 The slurry was heated at 60 °C for 24 h in nitrogen atmosphere, and then washed with DMF and THF (two times for each). At last, the OND–APTES was re-dispersed in 100 mL DMF with 1 h of sonication, followed by adding 3 g of spherical porous silica into the suspension. The system was reacting at 110 °C with mechanical stirring for 24 h under N2 protection (Fig. 1). After reaction, the obtained OND-silica composite was washed and centrifuged with ethanol and water (three times for each) to remove unbounded nanodiamond. The OND-silica packing material was washed one more time with acetone and putted into oven dried at 60 °C. To make a column, the OND modified silica particles were dispersed in acetone/water (v/v = 1/1) to form a slurry,9 which was packed into a stainless steel column (150 × 4.6 mm i.d.) at 55 MPa with acetonitrile as push solvent.
 | ||
| Fig. 1 Synthetic procedure of nanodiamond modified silica stationary phase (Sil-OND). | ||
The modification of silica with ONDs can be clearly seen on scanning electron microscopy (SEM) images, in which the OND particles were uniformly coated on the surface of spherical silica (Fig. 2a–b). From the transmission electron microscopy (TEM) images, the thickness of the OND layer can be identified as around 30 nm, while the diameter of the OND is about 10 nm, that means 2–3 layers of nanodiamonds coated on the silica (Fig. 2c–d). The Fourier transform infrared spectroscopy (FTIR) results show in Fig. S1,† the broad peak at 3400 cm−1 and sharp peak at 1662 cm−1 indicate the existence of carboxyl on oxidized OND. Meanwhile, the C–H stretching of methylene at 2935 cm−1 which was shown in the spectrum of OND–APTES symbolizing the covalent attachment of APTES to OND.
 | ||
| Fig. 2 (a) and (b) are SEM images of Sil-OND. (c) and (d) are TEM images of Sil-OND. | ||
Nine sulfonamides and four saccharides were chosen as probe to evaluate the performance of this OND-silica stationary phase, as shown in Fig. 3c and d. When the mobile phase was composed of acetonitrile/20 mM ammonium acetate (v/v = 89/11) at 30 °C, nine sulfonamides were fully separated as shown in Fig. 3c. Then, the separation performance of OND-silica column (Sil-OND) was compared with a bare silica column and commercial amino column (Sil-NH2) (250 × 4.6 mm i.d.) under the identical chromatographic conditions (Fig. 3a and b). From the comparison image we can observe that the bare silica shows almost no retention for sulfonamides, while the Sil-OND and Sil-NH2 columns demonstrated comparable separation ability (even the Sil-NH2 column is longer) despite of the peaking tailing of Sil-OND, which may be caused by the strong electrostatic attraction between carboxyl on OND and the solutes. As shown in Fig. 3b, sulfadiazine and sulfathiazole are overlapped when using Sil-NH2 column, which can be explained by their similar log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values (logPsulfadiazine = −0.09, logPsulfathiazole = 0.05), for the retention ability of Sil-NH2 is mainly depend on the affinity of solutes to the water-rich layer on stationary phase. In contrast, they can be fully separated with Sil-OND column, which is attributed to the electrostatic attraction between the stationary phase and the solutes, for sulfathiazole (pKasulfathiazole = 7.20) shows stronger electrostatic attraction to the carboxyl than sulfadiazine (pKasulfadiazine = 6.36). That means, electrostatic interaction plays a vital part in the sample separation of Sil-OND. Moreover, in the slightly acidic mobile phase (pH = 6.6), large amount of carboxyl on OND and the sulfonamides exist as ions, the slow sorption–desorption kinetics (“kinetic tailing”) of ionized carboxyl with protonated solutes is regarded as the reason of peak tailing.15 This “kinetic tailing” commonly happen to silica based stationary phase, where ionized silanols and basic samples show strong interactions. As supporting evidence, saccharides, which are non-ionized solutes, show almost no peak tailing when separated with Sil-OND column (Fig. 3d). That means the peak tailing caused by inhomogeneous morphology and strong adsorbability of other CNMs did not happen in this Sil-OND column, which is an evidence for ND to be a better packing material.
P values (logPsulfadiazine = −0.09, logPsulfathiazole = 0.05), for the retention ability of Sil-NH2 is mainly depend on the affinity of solutes to the water-rich layer on stationary phase. In contrast, they can be fully separated with Sil-OND column, which is attributed to the electrostatic attraction between the stationary phase and the solutes, for sulfathiazole (pKasulfathiazole = 7.20) shows stronger electrostatic attraction to the carboxyl than sulfadiazine (pKasulfadiazine = 6.36). That means, electrostatic interaction plays a vital part in the sample separation of Sil-OND. Moreover, in the slightly acidic mobile phase (pH = 6.6), large amount of carboxyl on OND and the sulfonamides exist as ions, the slow sorption–desorption kinetics (“kinetic tailing”) of ionized carboxyl with protonated solutes is regarded as the reason of peak tailing.15 This “kinetic tailing” commonly happen to silica based stationary phase, where ionized silanols and basic samples show strong interactions. As supporting evidence, saccharides, which are non-ionized solutes, show almost no peak tailing when separated with Sil-OND column (Fig. 3d). That means the peak tailing caused by inhomogeneous morphology and strong adsorbability of other CNMs did not happen in this Sil-OND column, which is an evidence for ND to be a better packing material.
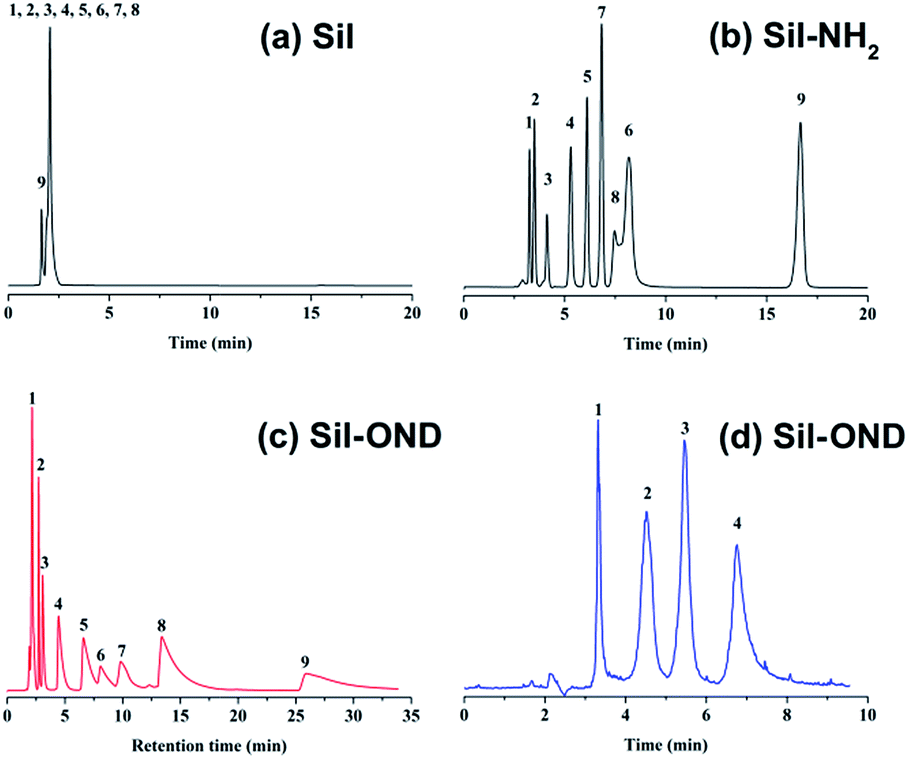 | ||
| Fig. 3 Separation of nine sulfonamides with bare silica column (a), amino column (b), Sil-OND column (c): (1) sulfanilamide, (2) sulfapyridine, (3) sulfamethazine, (4) sulfamerazine, (5) sulfadimethoxine, (6) sulfadiazine, (7) sulfadoxine, (8) sulfathiazole, (9) sulfisoxazole; mobile phase: 89% CH3CN:11% 20 mM NH4AC solution, pH = 6.6, flow rate = 1.0 mL min−1, T = 30 °C. (d) Separation of four saccharides with Sil-OND column: (1) D-ribose; (2) D-galactose; (3) sucrose; (4) lactose; mobile phase: 80% CH3CN:20% 20 mM NH4AC solution, pH = 6.6, flow rate = 1.0 mL min−1, T = 16 °C. | ||
Besides, varies of organic solvent concentrations were used to study the retention behavior of Sil-OND. As the percentage of water increases, the retention factors of four saccharides decrease (Fig. S2†), which is consistent with other references with HILIC mode.16 Both surface adsorption and partitioning mechanisms were proposed to explain the retention behavior of HILIC column.17 Fig. S2a† display the dependence of logk on the logarithm of the mole fraction of water, which fit pretty well to the surface adsorption equation of eqn (1)17 with correlation coefficients of 0.992–0.999. Meanwhile, correlation of logk and the volume fraction of water (Fig. S2b†) shows correlation coefficients of 0.998–0.999 to the partitioning equation of eqn (2).17 In other words, both adsorption and partitioning play important parts in the retention mechanism of Sil-OND column.
 | (1) |
|
logk = Aφ2 + Bφ + C
| (2) |
For further study of the chromatographic properties of Sil-OND, the influences of buffer concentration and buffer pH to the retention of analytes were investigated. Buffer concentration has two effects on the retention ability of HILIC stationary phase: (1) high concentration of organic solvent make salt tend to partition into water-rich layer on stationary phase, the increase of solvated ions in this water-rich layer enlarge its volume, which provide stronger retention for polar solutes; (2) salt in mobile phase would suppress electrostatic interaction (attraction and repulsion) between ionized analytes and stationary phase, which lead to diverse results of enhanced retention or reduced retention.18 According to Fig. 4a, with increasing buffer concentration (10–200 mM) the retention of sulfonamides decreased consistently. It means the retention of sulfonamides is mainly related to the electrostatic attraction with carboxyl on OND, which is getting weaker when higher buffer concentration suppresses the interaction. The influence of buffer pH on analytes retention is realized by controlling the ionization state of analytes and stationary phase. In the tested pH range (3.2–6.6), sulfonamides should remain mostly protonated, while the ionization of carboxyl groups on OND would gradually decrease as pH of buffer decrease from 6.6 to 3.2. The decreased ionization of carboxyl groups would provide weaker electrostatic interaction, which is consistent with the experimental results (Fig. 4b). Meanwhile, as described above, the “kinetic tailing” depends on the electrostatic attraction between ionized carboxyl and protonated solutes, so the peak tailing would get more serious when pH increase from 3.2 to 6.6. Take sulfisoxazole for example, the tailing factor increased from 3.36 to 10.65 as the buffer pH changed from 3.2 to 6.6. All the results revealed the importance of ion-exchange sites, which have been emphasized in many works.19
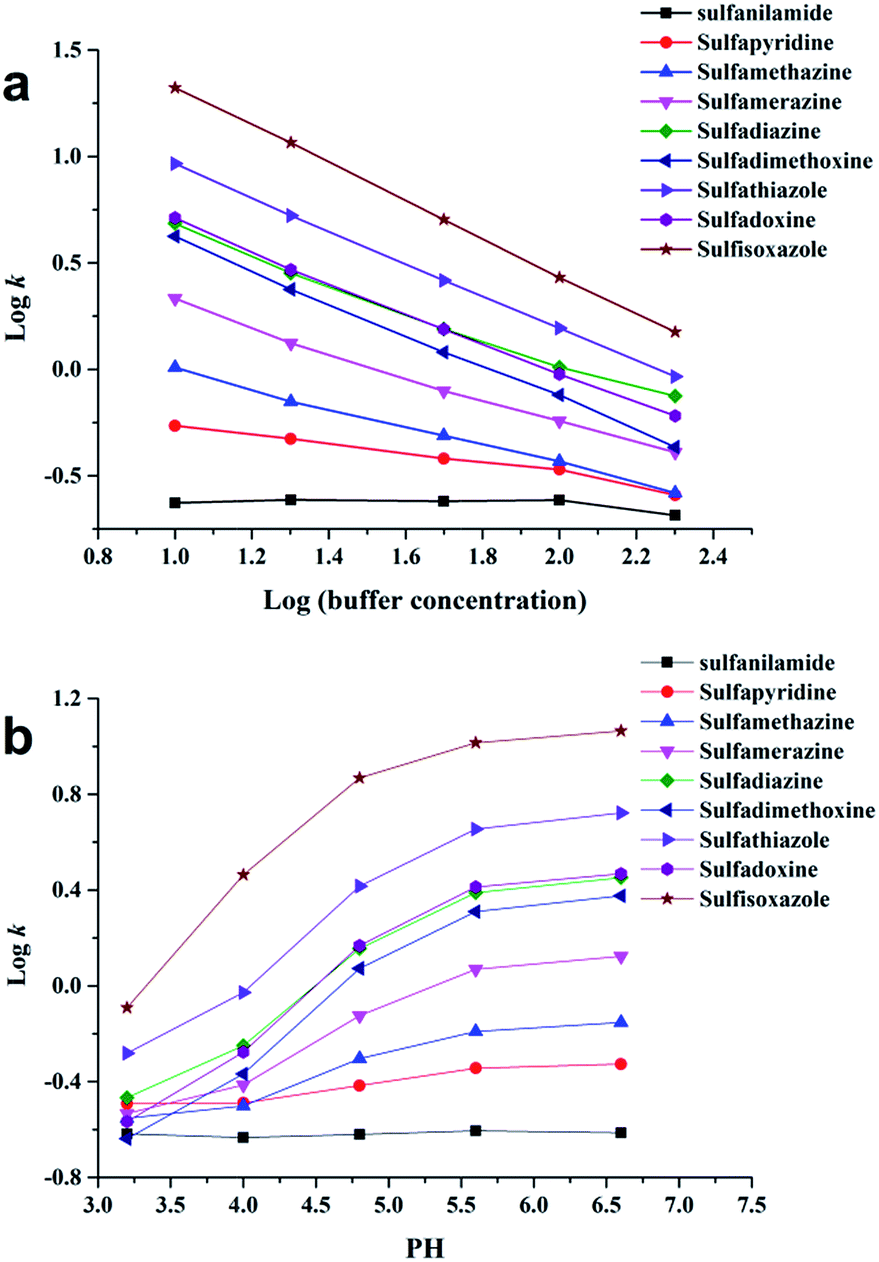 | ||
| Fig. 4 Effect of buffer concentration (a) and pH (b) on the retention factor (k) of sulfonamides; mobile phase: 89% acetonitrile:11% ammonium acetate solution, flow rate = 1.0 mL min−1, T = 30 °C; the buffer pH of (a) were set at 6.6, the buffer concentration of (b) were set at 20 mM. | ||
The advantage of covalent modification is the stability of stationary phase structure, even for long-time using the column pressure remains consistently. Meanwhile, the usage of silica expanded the spaces between ONDs, which significantly reduced the column pressure (only 4.4 MPa under following chromatographic conditions: flow rate = 1.0 mL min−1, T = 30 °C, mobile phase: 89% CH3CN:11% 20 mM NH4AC solution), so it's enabled the stationary phase to be packed into a normal length column instead of shorter ones.10 The reproducibility of the Sil-OND column was tested through the separation of sulfanilamide, sulfapyridine, sulfamethazine and sulfamerazine. The RSD values (n = 10) of the retention factors are within 0.56–1.04%, and the overlapped chromatograms are shown in Fig. S3.† It means the Sil-OND column is competent for the separation of sulfonamides under the present experimental conditions.
Conclusions
In summary, a convenient and effective method to prepare ND covalent modified silica stationary phase was developed. The spherical silica coated with homogeneous and thin layer of oxidized ND was easily prepared by simply coupling reaction. The presence of OND not just enlarged the surface area of packing material, but introduced functional groups to the column after functionalization. The good adsorbability combines with electrostatic interaction give the Sil-OND stationary phase excellent retention ability for polar analytes in HILIC mode. Even in the initial modification of ND surface with carboxyl groups provide tailing peaks which hinder a better column efficiency, it is reasonable to believe with proper modification, this kind of covalent modified Sil-ND stationary phase would demonstrate much better chromatographic performance. Besides, the new prepared nano-on-micro particles would be having a broad application in other fields of material science.Acknowledgements
Financial supports from the “Hundred Talents Program” of Chinese Academy of Sciences and the National Natural Science Foundation of China (21475142) and the funds for Distinguished Young Scientists of Gansu (1506RJDA281) and the top priority program of “One-Three-Five” Strategic Planning of Lanzhou Institute of Chemical Physics, CAS, are gratefully acknowledged.Notes and references
- R. Lucena, B. M. Simonet, S. Cardenas and M. Valcarcel, J. Chromatogr. A, 2011, 1218, 620–637 CrossRef CAS PubMed.
- M. Zhang and H. Qiu, Trends Anal. Chem., 2015, 65, 107–121 CrossRef CAS.
- (a) W. Chen, L. Duan and D. Zhu, Environ. Sci. Technol., 2007, 41, 8295–8300 CrossRef CAS PubMed; (b) S. Zhang, Z. Du and G. Li, Anal. Chem., 2011, 83, 7531–7541 CrossRef CAS PubMed; (c) X. Zhang, S. Chen, Q. Han and M. Ding, J. Chromatogr. A, 2013, 1307, 135–143 CrossRef CAS PubMed; (d) H. Wan, J. Huang, Z. Liu, J. Li, W. Zhang and H. Zou, Chem. Commun., 2015, 51, 9391–9394 RSC.
- A. V. Herrera-Herrera, M. Á. González-Curbelo, J. Hernández-Borges and M. Á. Rodríguez-Delgado, Anal. Chim. Acta, 2012, 734, 1–30 CrossRef CAS PubMed.
- Q. Qu, C. Gu and X. Hu, Anal. Chem., 2012, 84, 8880–8890 CrossRef CAS PubMed.
- (a) Y. Liang, M. Ozawa and A. Krueger, ACS Nano, 2009, 3, 2288–2296 CrossRef CAS PubMed; (b) Y. L. Hsin, H.-Y. Chu, Y.-R. Jeng, Y.-H. Huang, M. H. Wang and C. K. Chang, J. Mater. Chem., 2011, 21, 13213 RSC; (c) K. Y. Niu, H. M. Zheng, Z. Q. Li, J. Yang, J. Sun and X. W. Du, Angew. Chem., Int. Ed., 2011, 50, 4099–4102 CrossRef CAS PubMed.
- P. N. Nesterenko, O. N. Fedyanina and Y. V. Volgin, Analyst, 2007, 132, 403–405 RSC.
- (a) P. N. Nesterenko, O. N. Fedyanina, Y. V. Volgin and P. Jones, J. Chromatogr. A, 2007, 1155, 2–7 CrossRef CAS PubMed; (b) P. N. Nesterenko and O. N. Fedyanina, J. Chromatogr. A, 2010, 1217, 498–505 CrossRef CAS PubMed; (c) O. N. Fedyanina and P. N. Nesterenko, Russ. J. Phys. Chem. A, 2011, 85, 1773–1777 CrossRef CAS.
- G. Saini, D. S. Jensen, L. A. Wiest, M. A. Vail, A. Dadson, M. L. Lee, V. Shutthanandan and M. R. Linford, Anal. Chem., 2010, 82, 4448–4456 CrossRef CAS PubMed.
- L. A. Wiest, D. S. Jensen, C. H. Hung, R. E. Olsen, R. C. Davis, M. A. Vail, A. E. Dadson, P. N. Nesterenko and M. R. Linford, Anal. Chem., 2011, 83, 5488–5501 CrossRef CAS PubMed.
- L. Yu, X. Li, J. Dong, X. Zhang, Z. Guo and X. Liang, Anal. Methods, 2010, 2, 1667 RSC.
- (a) T. Gu, M. Zhang, J. Chen and H. Qiu, Chem. Commun., 2015, 51, 9825–9828 RSC; (b) Z. Guo, A. Lei, Y. Zhang, Q. Xu, X. Xue, F. Zhang and X. Liang, Chem. Commun., 2007, 2491–2493 RSC.
- (a) J. Cheng, J. He, C. Li and Y. Yang, Chem. Mater., 2008, 20, 4224–4230 CrossRef CAS; (b) A. Krueger and D. Lang, Adv. Funct. Mater., 2012, 22, 890–906 CrossRef CAS.
- S. Ray, M. Takafuji and H. Ihara, J. Chromatogr. A, 2012, 1266, 43–52 CrossRef CAS PubMed.
- (a) D. V. McCalley, J. Chromatogr. A, 2010, 1217, 858–880 CrossRef CAS PubMed; (b) D. H. Marchand, L. R. Snyder and J. W. Dolan, J. Chromatogr. A, 2008, 1191, 2–20 CrossRef CAS PubMed.
- (a) H. Qiu, A. K. Mallik, T. Sawada, M. Takafuji and H. Ihara, Chem. Commun., 2012, 48, 1299–1301 RSC; (b) H. Qiu, A. K. Mallik, M. Takafuji, X. Liu, S. Jiang and H. Ihara, J. Chromatogr. A, 2012, 1232, 116–122 CrossRef CAS PubMed.
- P. Hemström and K. Irgum, J. Sep. Sci., 2006, 29, 1784–1821 CrossRef.
- (a) A. J. Alpert, Anal. Chem., 2008, 80, 62–76 CrossRef CAS PubMed; (b) R. I. Chirita, C. West, S. Zubrzycki, A. L. Finaru and C. Elfakir, J. Chromatogr. A, 2011, 1218, 5939–5963 CrossRef CAS PubMed; (c) X. Xiong and Y. Liu, Talanta, 2016, 150, 493–502 CrossRef CAS PubMed.
- (a) B. Chen, J. Xu, Q. Fu, X. Dong, Z. Guo, Y. Jin and X. Liang, Analyst, 2015, 140, 4676–4686 RSC; (b) L. Chen, X. Dong, L. Cao, Z. Guo, L. Yu, L. Zou and X. Liang, Anal. Methods, 2013, 5, 6919 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra04824b |
| This journal is © The Royal Society of Chemistry 2016 |