
Wide-range work-function tuning of active graphene transparent electrodes via hole doping†
Abstract
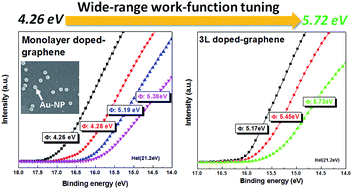
Graphene is regarded as a potential candidate to replace the transparent conductive (TC) electrodes that are currently used in various optoelectronic applications. However, there is still a lack of methods by which to achieve low sheet resistance (Rs) with stable doping and work functions with a wide range of tunability, which is significant for band alignment at the interface to enhance charge transport and thus to achieve higher device performance. We developed a novel strategy for preparing a TC electrode by doping layer-by-layer (LBL)-stacked graphene with AuCl3, by which means an excellent TC performance (an Rs of 40 ohm sq−1 at a transmittance (T) of 89.5%) and an extremely wide range of work-function tunability (∼1.5 eV) were successfully achieved. Moreover, a hybrid electrode prepared by transferring doped graphene onto a pre-patterned Cu metal mesh exhibited a low resistance of ∼4.9 ohm sq−1. In addition, we monitored the long-term stability of AuCl3-doped graphene for 6 months and also constructed a model for accelerated degradation testing. The relevant mechanism of charge transfer between the graphene and the dopants was characterized based on X-ray photoelectron spectroscopy (XPS) spectra to elucidate degradation observed after long-term testing. This work contributes a novel type of “active electrode”; the doped graphene film not only serves as a high-performance TC electrode but also provides a wide range of tunable work functions. The proposed active electrode is prepared using a scalable and facile doping process, which paves the way for its usage in applications such as optoelectronic devices.
 Please wait while we load your content...
Please wait while we load your content...

