 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The versatility of “click” reactions: molecular recognition at interfaces†
Thomas
Heinrich
ab,
Christoph H.-H.
Traulsen
a,
Erik
Darlatt
b,
Sebastian
Richter
a,
Johannes
Poppenberg
a,
Nora L.
Traulsen
a,
Igor
Linder
a,
Andreas
Lippitz
b,
Paul M.
Dietrich
b,
Baha
Dib
a,
Wolfgang E. S.
Unger
*b and
Christoph A.
Schalley
*a
aInstitut für Chemie und Biochemie der Freien Universität Berlin, Takustraße 3, 14195 Berlin, Germany. E-mail: c.schalley@fu-berlin.de
bBAM Federal Institute for Materials Research and Testing, Unter den Eichen 44-46, 12203 Berlin, Germany. E-mail: wolfgang.unger@bam.de
First published on 31st March 2014
Abstract
In order to investigate molecular recognition on surfaces, an azide-functionalized monolayer was deposited on gold. The monolayer was characterized by X-ray photoelectron spectroscopy (XPS) and angle-resolved near-edge X-ray absorption fine structure (NEXAFS) experiments and the decomposition of the azide upon irradiation with X-ray beams was investigated. Subsequently, various alkyne-functionalized host and guest molecules were attached to the azide by 1,3-dipolar cycloaddition. These modified surfaces and their host–guest chemistry were analysed by XPS and angle-resolved NEXAFS. The reversibility of guest binding was shown for one example as a proof of principle.
Introduction
The controlled modification of interfaces with various functional groups is a main topic in modern physical, materials and bioorganic chemistry.1–9 Therefore, a variety of approaches has been developed. The covalent attachment of organic compounds on surfaces yields highly stable interfaces.10 Among others, the silanization of silica substrates11,12 and the chemisorption of organic thiols13 or liponic acids14 on gold substrates is a far more convenient approach. Numerous examples exist, using the formation of stable self-assembled monolayers (SAMs) on solid supports with different terminal groups such as hydroxides,15 carboxylates,16 pyridines,17 terpyridines,18–22 and amines.23–28 These terminal groups enable the attachment of additional layers on top using for example the formation of thiourea24 or non-covalent complex formation with transition metal ions.17,19,29,30 Further terminal groups which have lately gained attention in this context are 1,3-dipoles such as nitrile oxides and azides which react with alkynes in what has been coined the “click” reaction.31–34 Another approach is the non-covalent physisorption of Langmuir–Blodgett films e.g. the non-covalent attachment of saturated and non-saturated carboxylates.35,36 While covalently attached SAMs are somewhat limited by reduced reversibility of and long reaction times for self-assembly, Langmuir–Blodgett films require amphiphilic molecules and are often less stable against polar solvents and/or reactive species.37The transfer of supramolecular structures and processes from solution to solid supports is still a challenging task in modern physical and biochemistry.38–40 Our approach presented here combines covalent generation of SAMs that can further be functionalized by host or guest molecules which allows the specific non-covalent attachment of another layer through molecular recognition. An azide-functionalized SAM is used here to graft these molecules onto the surface by a copper-catalysed 1,3-dipolar cycloaddition of azides and alkynes forming triazole rings.41 The simple experimental setup, mild reaction conditions and fast conversion of the “click” reaction in solution remain more or less unchanged features of surface-bound triazole formation as shown earlier for the immobilization of terpyridines,31 polymers,42 ferrocene,34 nanoparticle multilayers,43 and metal organic frameworks.44 An advantage of the modification of surfaces via “click” reactions is the orthogonality of the azide and alkyne with most other functional groups. This broadens the variety of molecules significantly, which can be attached. Triazole formation also often gives rise to densely packed consecutive layers.32
Here, we report the formation of an azide-terminated monolayer which is subsequently functionalized with benzo[21]crown-7 ether CE1 and tetralactam macrocycle TLM1 (ref. 45) as specific host molecules and with primary ammonium AM1 and diketopiperazine DP1 as the corresponding guest molecules (Scheme 1). For each surface-attached molecule, a counterpart (CE2, TLM2, AM2 and DP2) exists which does not carry the alkyne and can be used for on-surface host–guest experiments. These molecules were selected as both binding motifs were thoroughly studied in solution. The tetralactam macrocycle nicely binds diketopiperazines in its cavity through four hydrogen bonds between the guest's carbonyl groups and the converging amide NH groups of the macrocycle.46,47 Benzo[21]crown-7 is just large enough to allow pseudorotaxane formation with secondary alkyl ammonium axles.48 Hexyne HEX was used as a reference to investigate triazole formation during the “click” reaction. In former studies, we have demonstrated the utility of copper(I)-catalysed 1,3-dipolar cycloadditions on azide-terminated SAMs assembled from 1,2-bis(11-azidoundecyl)-disulfane (AUD) for the covalent attachment of functionalized alkynes. Since AUD is analogous to alkanethiol SAMs, it should form densely packed and well-ordered SAMs on gold substrates.31,32
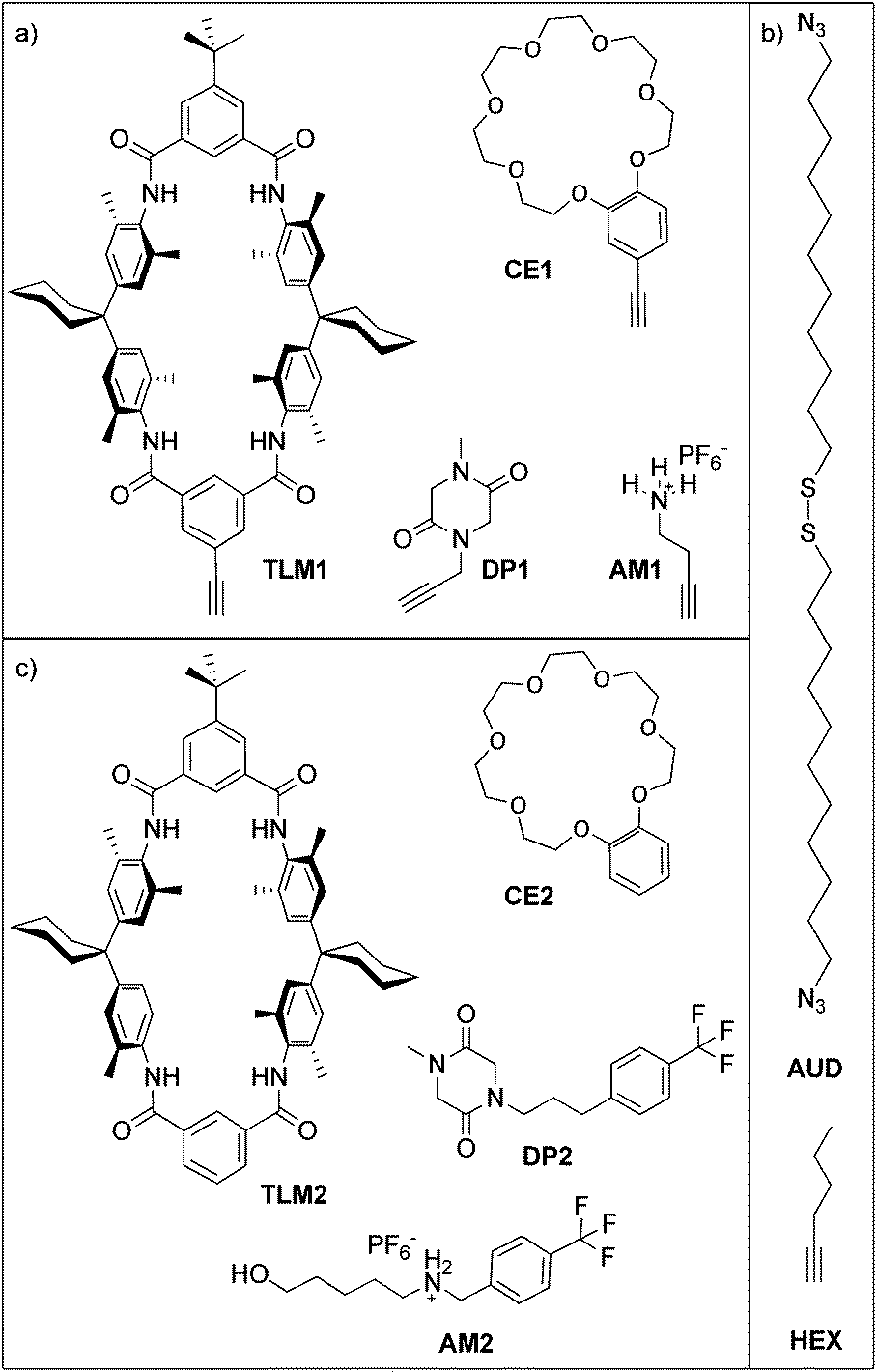 | ||
| Scheme 1 (a) Alkyne-functionalized host and guests, (b) control compound HEX and SAM-forming disulphide AUD, and (c) the corresponding guests and host. The CF3 groups in DP2 and AM2 serve as labels for easy detection by XPS. | ||
Results and discussion
Analysis of azide-decomposition upon irradiation
In the first step, an AUD SAM was prepared which forms the basis for further functionalization with alkyne-substituted organic molecules as shown in Scheme 2. The AUD SAM was characterized by quite surface-sensitive synchrotron radiation X-ray photoelectron spectroscopy (SR-XPS; 500 eV excitation energy, XPS 95% information depth z95% ∼ 2.1 nm) and NEXAFS spectroscopy as described earlier. Angle-resolved NEXAFS experiments provide evidence for a predominant upright orientation of the AUD molecules.31,32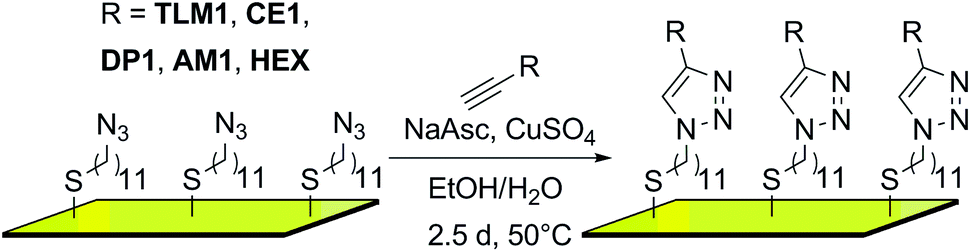 | ||
| Scheme 2 Surface attachment of organic molecules via “click” chemistry (NaAsc = sodium ascorbate). | ||
A major characterization problem of surface-bound azides by XPS is the decomposition of the azides upon X-ray radiation. This irradiation damage yields a new nitrogen component at lower binding energy (BE) in the N 1s XP spectrum of a pristine azide-terminated SAM. Non-damaged SAM molecules should provide three components of equal peak areas diagnostic for azide moieties as displayed in Fig. 1a. Two major components at lower BE (400.9 and 400.3 eV) and a single component at higher BE (404.2 eV) can be assigned to electron-rich and electron-poor azide nitrogen atoms.31,37 During the decomposition process, a new signal appears at a lower BE of 399.0 eV (Fig. 1a). In order to gain more detailed insight into this process and in order to be able to clearly assign all nitrogen species which may arise after the “click” reaction, 35 N 1s spectra were recorded in 4 min intervals at the same spot to follow the time course of the decomposition reaction resulting in a total irradiation time of 142 minutes. In Fig. 1b, the normalized N 1s peak areas of the overall (black line), the azide (green line) and the decomposition products (blue line) are plotted. As expected, the decomposition process is accompanied by an exponential decay of the azide nitrogens. The overall amount of nitrogen does not remain constant, but decreases to about a third of the starting nitrogen peak area at the end of the irradiation time with a half-life of 34 min. Molecular nitrogen is thus released during the decomposition reaction. Nitrogen release follows a first-order rate law (k = 3 × 10−4 s−1) as determined by plotting the natural logarithm of the azide nitrogen mole fraction over the irradiation time resulting in a linear fit (Fig. 1c).
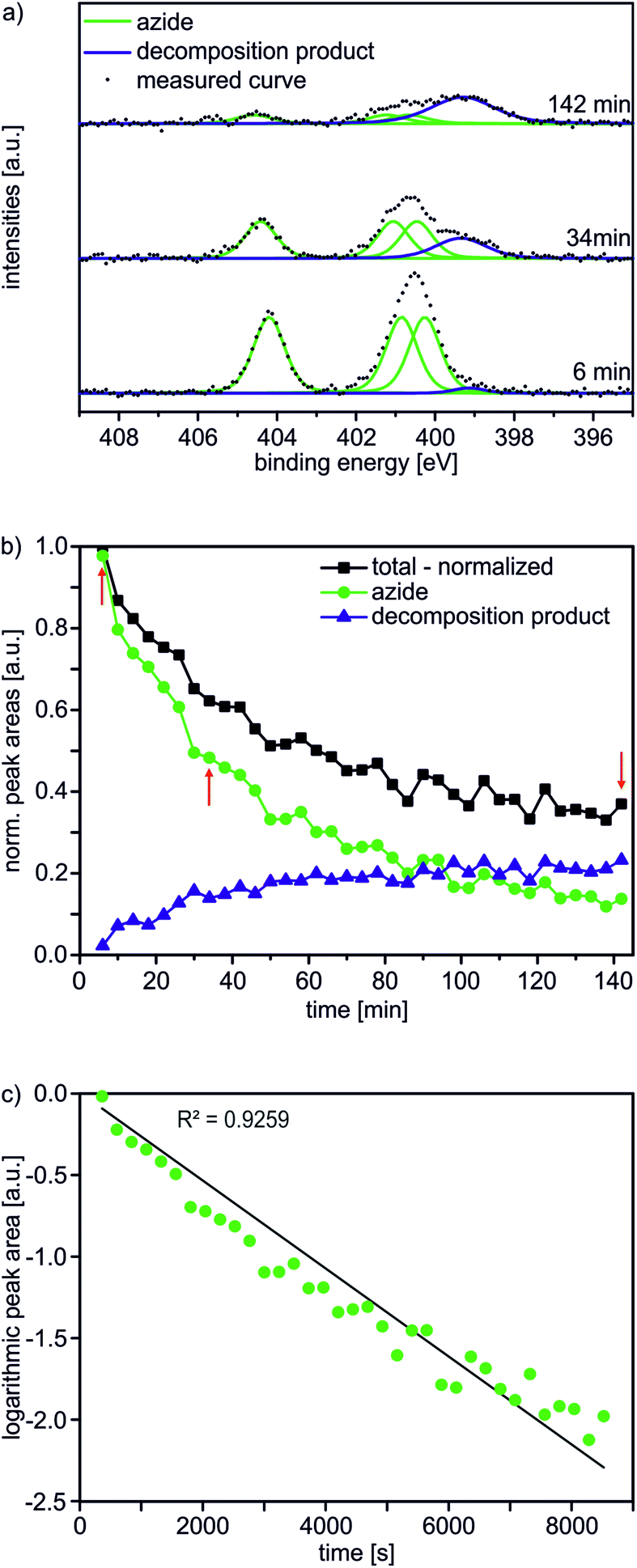 | ||
| Fig. 1 In situ damage of the AUD SAM caused by monochromatic Al Kα radiation (X-ray excitation energy 1486 eV) with (a) peak fits of the N 1s XP spectra, (b) quantitative analysis of the individual peak fits. Red arrows indicate the data points corresponding to the spectra in (a). (c) Logarithmic plot of normalised azide peak areas over time. All peak areas are normalized to the total peak area of the first spectrum after 6 min irradiation time. | ||
Mechanistically, the decomposition can follow different pathways (Scheme 3). The initially formed highly reactive nitrene intermediate can either react by insertion into a C–H bond of a neighbouring AUD molecule. This reaction would give rise to secondary amines, which is however inconsistent with the binding energy of the new nitrogen species.29 Consequently, this reaction does not play a prominent role. The second pathway would be dimerization of two nitrenes to yield an azo-compound, which might react through nitrogen loss forming two alkyl radicals. However, no XPS peak for azo-groups is observed and therefore, we also rule out this pathway as the major decomposition route.28 A third typical reaction of electron sextet compounds such as nitrenes is a 1,2-hydrogen shift from the adjacent carbon to the atom with the electron sextet, here the nitrene nitrogen atom. This hydrogen shift leads to an imine. The binding energy of 399.0 eV for the new component is consistent with the formation of an imine. Also, the π* resonance at 398.3 eV observed in the N K-edge NEXAFS spectroscopy suggests imine formation.28 The 1,2-hydrogen shift pathway is consequently the major reaction following nitrogen loss and nitrene formation. However, it is not possible to determine whether N2 loss and the 1,2-hydrogen shift are subsequent steps or occur in a concerted manner due to the rather long intervals between the spectra.
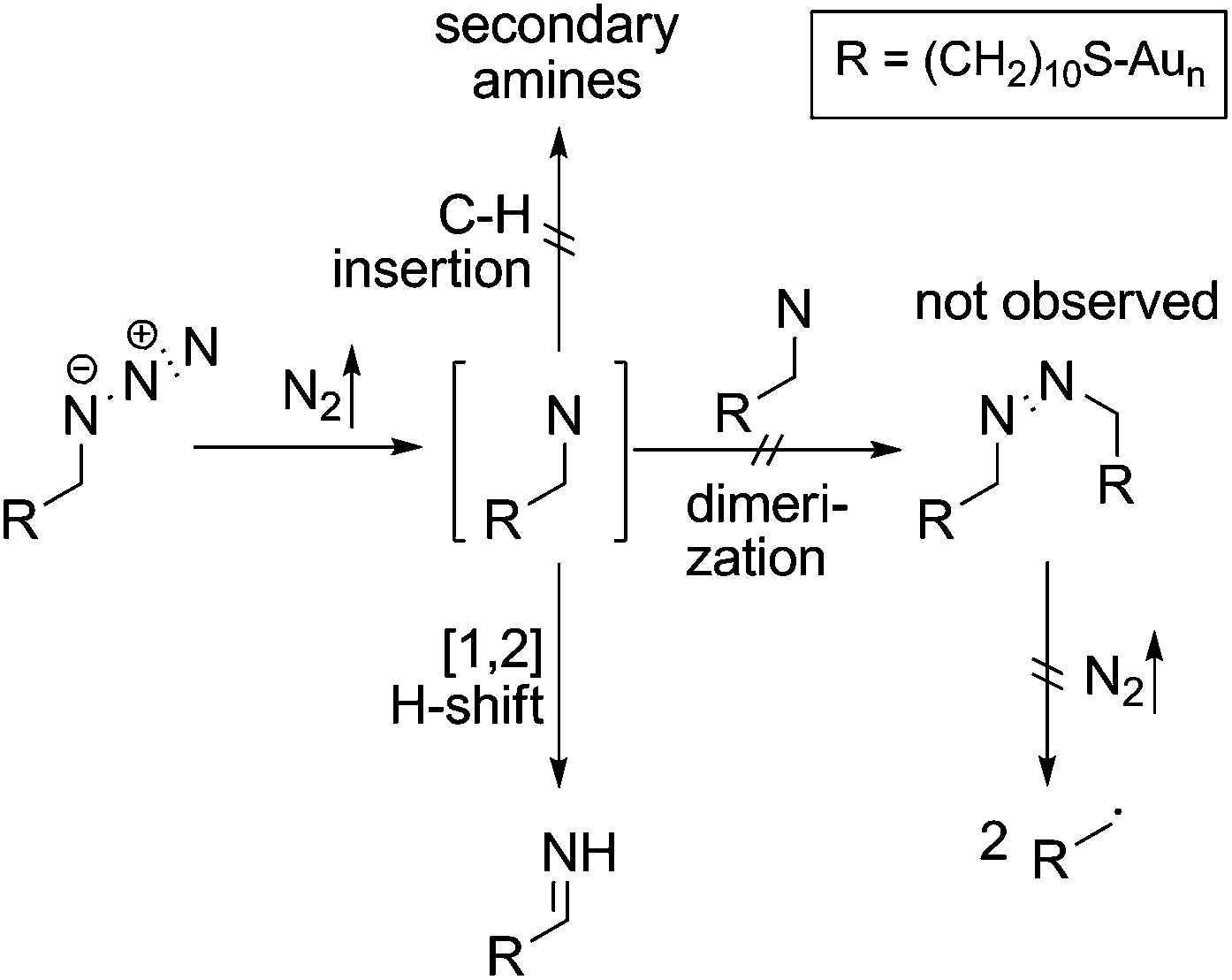 | ||
| Scheme 3 Possible azide decay-mechanisms with a nitrene as the central intermediate. | ||
As a second conclusion, these data reveal azide decomposition to occur rather slowly so that it does not significantly hamper the analysis of azide-functionalized surfaces. This is also important for the following experiments, in which alkynes are clicked to the azides, as for example a sterically demanding molecule such as tetralactam macrocycle TLM1 will like leave azides “unclicked” underneath the macrocycle layer.
Cycloaddition experiments
For all five surfaces, the Au–S interface is stable under the experimental conditions of the “click” reaction as indicated by the main S 2p3/2 species that is detected at ∼162.0 eV (ESI†).32
Fig. 2 (left) summarises the N 1s XP spectra of these five surfaces in comparison to that of the pristine AUD SAM. In order to reduce irradiation damage, irradiation times were kept below three minutes. Clearly, the azide signals of the latter vanish completely, when HEX is added. Instead, three nitrogen signals for the triazole (BE = 399.6, 400.3, and 401.6 eV) with equal peak areas and equal FWHM values can be fitted into the broad N 1s signal. A more or less quantitative “click” reaction was achieved as indicated by the absence of the azide signal at 404.8 eV. A very similar result is obtained for the surface covered with crown ether CE1, which does not bear any additional nitrogen atoms.
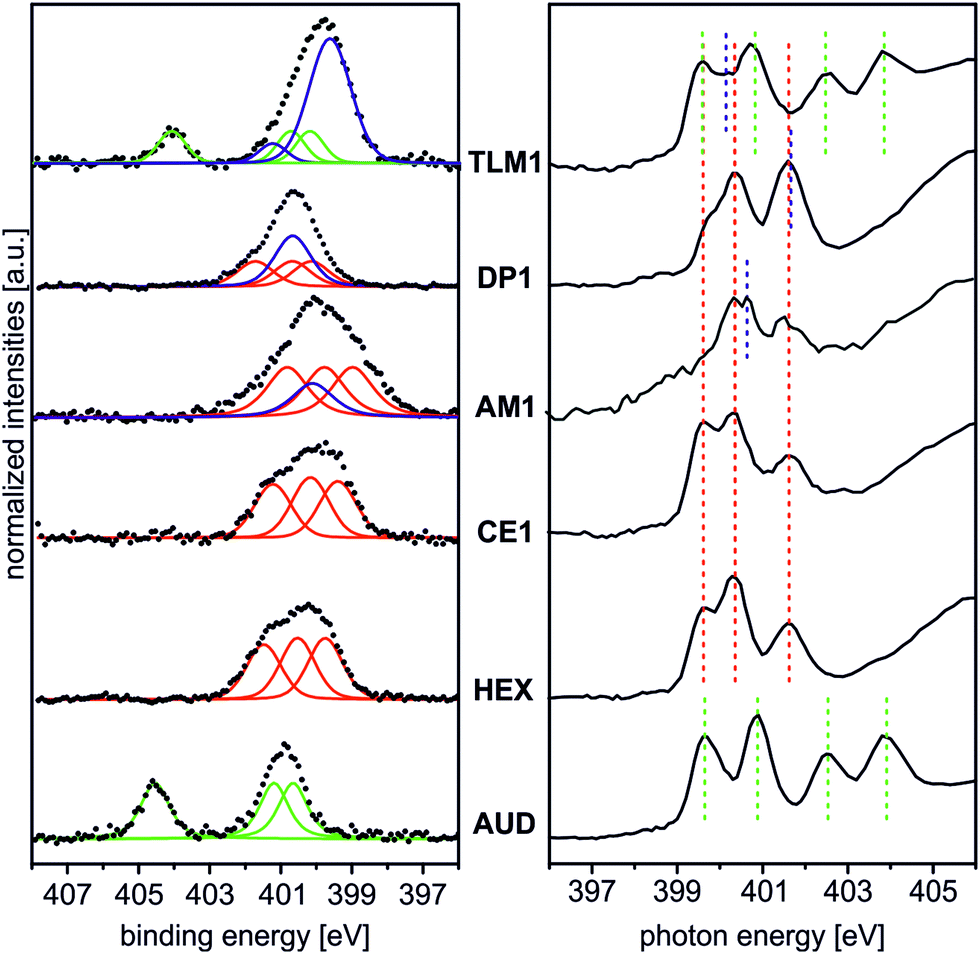 | ||
| Fig. 2 Left: qualitative N 1s XP spectra of the surfaces of HEX, CE1, AM1, DP1 and TLM1 in comparison to that of the pristine AUD SAM (hν = 500 eV and 0° emission angle). Black dotted lines: experimental spectra; green and red curves: contributions of azide and triazole. Blue curves correspond to amine (AM1) and amide nitrogen atoms (DP1, TLM1). To facilitate direct comparison, the backgrounds of the spectra were subtracted followed by normalization to the maximum count rate. Right: π* region of the N K-edge NEXAFS spectra at 55° incident synchrotron light. For qualitative comparison, the spectra were normalized to their maximum count rates. The different photon energies for azide (green), triazole (red), amine and amide nitrogens (blue) are indicated by dotted lines. | ||
The picture changes for AM1, which was “clicked” to the AUD SAM as the free amine followed by treatment with HPF6. This molecule contains an additional ammonium nitrogen atom. Consequently, the triazole signals are superimposed by an additional peak at a BE of 400.3 eV. This binding energy is surprisingly the same as that of free amines as observed for the same surface before protonation, while no new signal for ammonium is observed at the expected BE of 401.8 eV. Thus, only a small protonation yield is obtained. We refrained from using H2SO4 for protonation as reported earlier for surface-bound pyridines,17 as strong ion pairing between the doubly charged sulphate and ammonium ions will likely compete too strongly with crown ether binding in the host–guest studies.
The surface bearing DP1 also exhibits a new signal in addition to the triazole nitrogen atoms which corresponds to the two amide nitrogen atoms of the diketopiperazine and is detected at a binding energy of 400.8 eV.40 Finally, tetralactam macrocycle TLM1 exhibits the triazole signals superimposed by a large peak at BE = 400.3 eV for the four amide nitrogen atoms.28 The azide signal at BE = 404.8 eV is also clearly visible. This indicates the steric bulk of TLM1 to leave unreacted azides underneath the macrocycle layer. Not unexpectedly, the triazole nitrogen atoms exhibit small peak shift changes depending on the exact chemical environment provided by the different molecules attached.
These XPS data are supported by N K-edge NEXAFS experiments (Fig. 2, right). Clearly, azide signals (green dotted lines) appear for the pristine AUD SAM and for the TLM1-functionalized surface. The N K-edge spectrum of the AUD SAM exhibits four pronounced signals in the π* region at 399.6, 400.9, 402.5 and 403.9 eV (Fig. 2, bottom right). Electron transitions from the N 1s energy level of the central azide nitrogen into two different π* orbitals are the reason for the resonances at higher photon energy (PEs). The other two signals at lower PEs can be assigned to the two adjacent nitrogen atoms of the azide moiety.31
Spectral contributions attributed to the triazole group (red lines) are visible for all surfaces except the AUD SAM. The assignment of the signals at 399.6 eV, 400.3 eV and 401.5 eV is in good agreement with DFT calculations reported earlier.31 The additional amine and amide nitrogen atoms of AM1, DP1 and TLM1 lead to the observed changes in the structure of the triazole peaks (e.g. PE = 401.6 eV for the tertiary amide π* resonances in DP1 (ref. 49) or PE = 400.4 eV for the secondary amides in TLM1 (ref. 29)). The presence of remaining azide groups underneath the TLM1 layer are also supported by NEXAFS spectra (Fig. 3): when the surface is irradiated for longer intervals, it clearly reveals the same radiation-induced azide decomposition as the pristine AUD SAM, while similar changes do not occur for the HEX-functionalised surface.
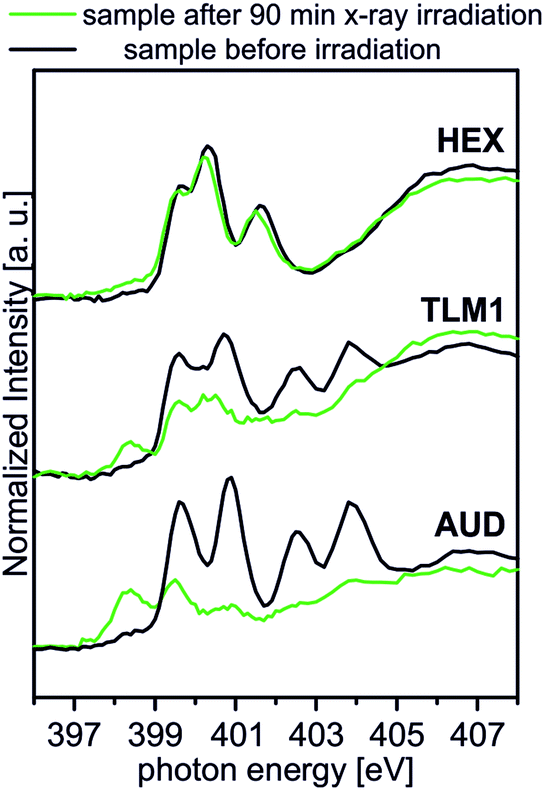 | ||
| Fig. 3 Behaviour of TLM1 attached to the surface upon radiation in comparison to the AUD SAM and the surface attached HEX. | ||
These experiments provide clear evidence for the successful attachment of the alkyne-substituted molecules under study here in copper-catalysed click reactions. The surface coverage is likely high as indicated by the fact that only the largest molecule, TLM1, leaves visible signals for unreacted azide group.
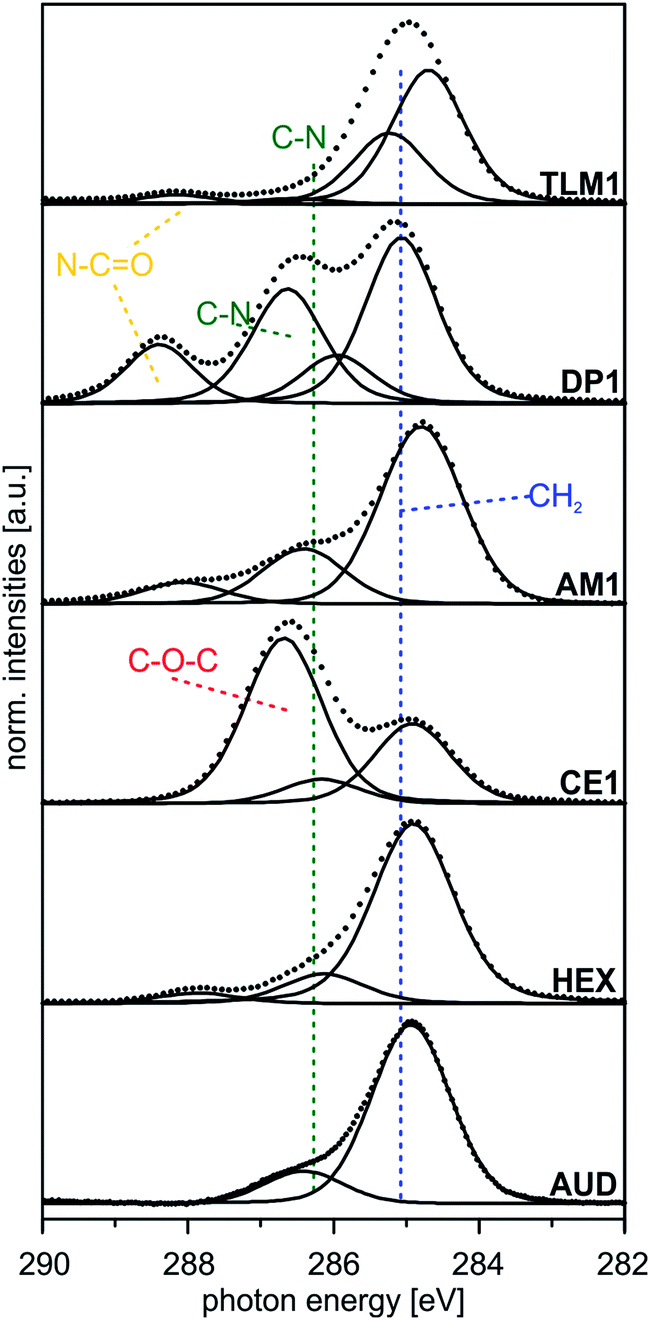 | ||
| Fig. 4 Peak fits of C 1s SR XP spectra (hν = 350 eV, emission angle = 0°) obtained at high surface sensitivity of XPS, 95% information depth (z95% ∼ 2 nm).36 | ||
| Sample | BEa [eV] (rel. peak area) | Theor. ratios | Assigned carbon atoms | FWHM [eV] |
|---|---|---|---|---|
| a Binding energy reference: Au 4f7/2 signal at BE = 84.0 eV. | ||||
| AUD | 284.9 (85) | 82 | –CH2– | 1.3 |
| 286.4 (15) | 8 | –CH2–N3, –CH2–S | ||
| HEX | 284.9 (82) | 82 | –CH2– | 1.3 |
| 286.1 (14) | 18 | Csp2–N, N–CH2– | ||
| CE1 | 284.9 (30) | 34 | –CH2– | 1.3 |
| 286.2 (9) | 10 | Csp2–N, N–CH2–O– | ||
| 286.7 (61) | 56 | CH2–, –O–C(sp2)– | ||
| AM1 | 284.8 (70) | 71 | –CH2– | 1.3 |
| 286.4 (25) | 29 | Csp2–N, N–CH2– | ||
| DP1 | 285.1 (52) | 52 | –CH2– | 1.2 |
| 285.9 (10) | 11 | N–CH2–, N–CH3 | ||
| 286.6 (27) | 26 | OCN–CH2–, Csp2–N | ||
| 288.4 (11) | 11 | O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N– C–N– |
||
| TLM1 | 284.5 (52) | 48 | Csp2 | 1.2 |
| 285.2 (37) | 44 | –CH2–, C–C | ||
| 286.6 (2) | 3 | C–N, Csp2–N | ||
| 288.1 (4) | 5 | OC–N |
||
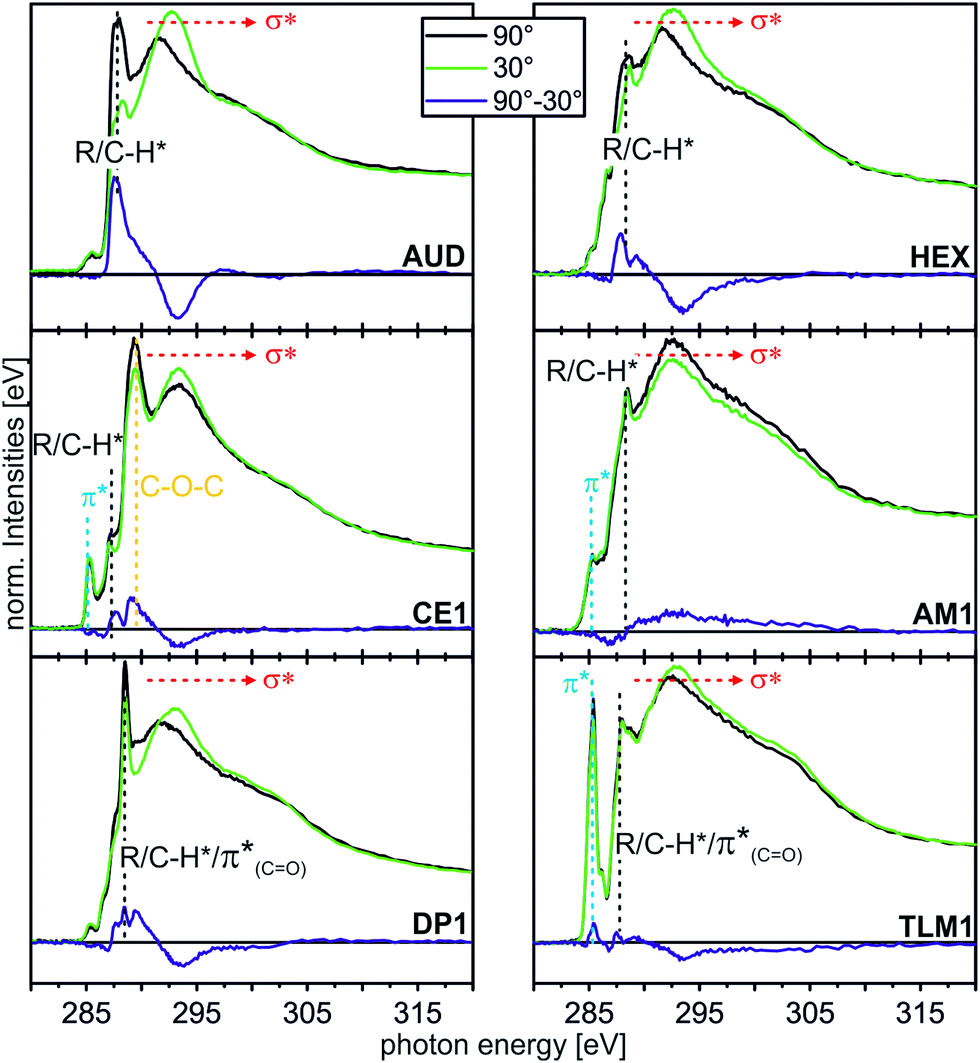 | ||
| Fig. 5 C K-edge NEXAFS spectra (for experimental details, see ESI†) of the AUD SAM and the click-functionalized surfaces with different moieties at incidence angles of 90° (black) and 30° (green). The difference spectra are illustrated by the blue line for every sample. The characteristic resonances are marked by dotted lines. | ||
In the angle-resolved NEXAFS spectra of the pristine AUD SAM, clear linear dichroism effects are observed. The C–H* resonance decreases significantly when the angle of the incident linear polarized synchrotron light is changed from 90° to 30°. In contrast, the σ* resonance increases. This is typical for well-ordered SAMs of long alkyl chains that are oriented in an upright fashion on the surface.13,50 The same effect is observed for HEX-modified AUD SAMs. Therefore, we conclude the HEX SAM to be highly ordered as well as the AUD precursor SAM.
Linear dichroism effects are also observed for the CE1-functionalized surface. In addition to the effect of the underlying AUD SAM, one observes a linear dichroism effect for the peak corresponding to the σ* resonance of the CH2–O groups, which appears at 289.3 eV.51,52 This effect points to crown ethers with a preferential orientation and thus ordered crown ether layers even though it remains unclear, what their exact packing pattern on the surface is. Furthermore, the DP1-covered surface exhibits preferentially oriented diketopiperazines as indicated by the linear dichroism in the π*(CO) resonance at 288.4 eV.51 In marked contrast, the linear dichroism effects are small for the surfaces loaded with AM1 and TLM1. It is therefore likely that AM1 is not well ordered. The quite complex TLM1 structure with its many aromatic rings that are oriented in different angles relative to the surface, one would expect only a small linear dichroism effect. It is not possible to clearly state whether the small effects observed have their origin in isotropically distributed macrocycles on the surface or in an ordered layer of macrocycles with aromatic rings mutually compensating each other's linear dichroism effects.
As a conclusion, these angle-resolved NEXAFS experiments provide evidence for preferentially ordered molecules in layers of AUD, HEX, CE1 and DP1, while “clicking” AM1 to the surface leads to a more or less isotropic arrangement. The situation after reaction with TLM1 is too complex to be described.
On-surface host–guest experiments
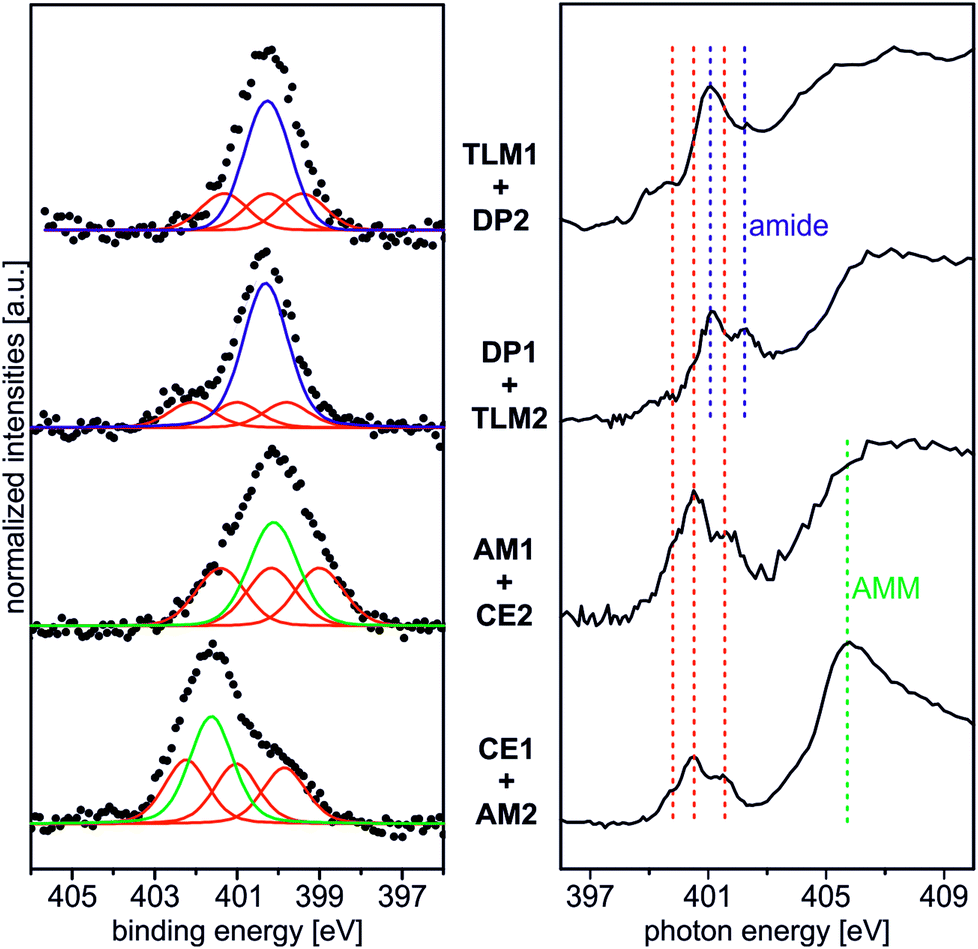 | ||
| Fig. 6 Left: qualitative N 1s XP spectra of the four host–guest pairs (hν = 500 eV and 0° emission angle). Black dotted lines: experimental spectra; red curves: triazoles, blue curves: amide groups (DP1, TLM1), green curves display ammonium (CE1) or amine nitrogen atoms (AM1). To facilitate comparison, the backgrounds of the spectra were subtracted followed by normalization to the maximum count rate. Right: π* region of the N K-edge NEXAFS spectra taken at 55° incident synchrotron light. For qualitative comparison the spectra were normalized to their maximum count rates. The different photon energies for triazole resonances (red), amides (blue), and ammonium nitrogen atoms (green) are marked by dotted lines. | ||
Beside the triazole nitrogen peaks, the CE1–AM2 pair clearly shows additional features at 401.7 eV BE in the N 1s XP spectrum and at 405.8 eV PE in the N K-edge NEXAFS spectrum. These signals can be assigned to the secondary ammonium group of the axle28 and thus provide evidence for its presence even after thoroughly rinsing the surface. We therefore conclude the axle to be bound to surface by threading into the cavity of the crown ether. The opposite pair, AM1 and CE2, exhibits an N 1s XP spectrum, which does not differ much from that of the AM1-covered surface before crown deposition. This is not surprising because no further nitrogen is introduced and the protonation degree is low according to the results discussed above. This becomes also clear, when one compares the binding energies of the nitrogen incorporated in AM1 (BE = 400.3 eV for an amine nitrogen) with that of AM2 (BE = 401.7 eV for the ammonium nitrogen) in the experiment above.
Deposition of the tetralactam macrocycle in the DP1–TLM2 host–guest combination can already be clearly observed in the N 1s XP spectra. The intensity of the peak at BE = 400.8 eV representing the amides in DP1 as well as TLM2 is significantly higher compared to the triazole peaks of the DP1-covered surface before TLM1 deposition (Fig. 2). For the opposite combination of TLM1 and DP2, the situation is not as clear, and C 1s XP spectra will provide more detailed insight.
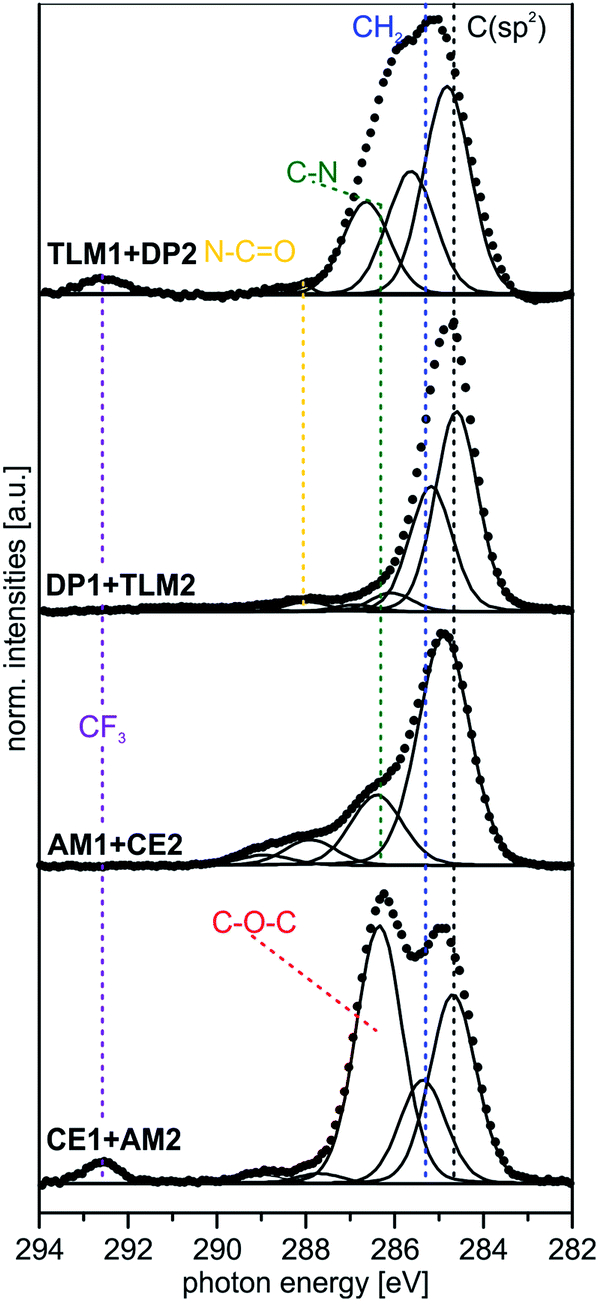 | ||
| Fig. 7 Peak fits of C 1s SR-XP spectra (hν = 350 eV, emission angle = 0°) for the four different host–guest pairs. | ||
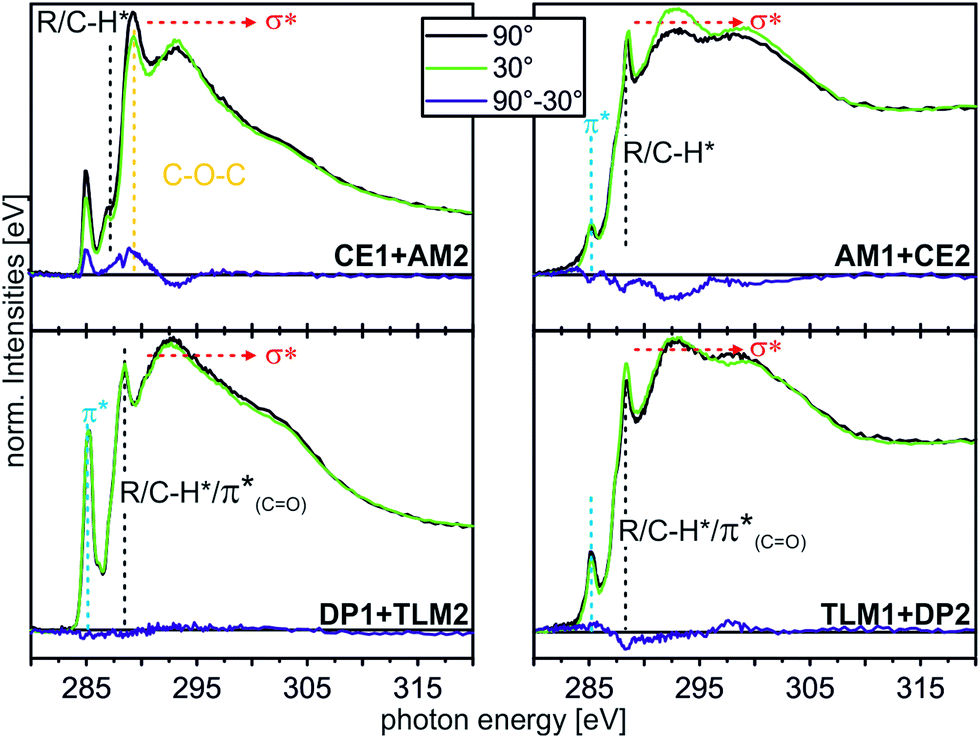 | ||
| Fig. 8 C K-edge NEXAFS spectra (for experimental details, see ESI†) of the host–guest experiments with different moieties at incident angles of 90° (black) and 30° (green). The differences between the spectra are illustrated by the blue lines. Characteristic resonances are marked by dotted lines. | ||
The CE1–AM2 pair clearly exhibits linear dichroism effects on the σ*(CH2O) resonance of the crown ether methylene groups. Consequently, the crown ethers still have a preferred orientation. In addition, a new linear dichroism effect is observed for the π* resonance of the aromatic rings. As this was not present in the corresponding spectra of surface-attached CE1 prior to AM2 deposition (Fig. 5), we attribute this effect to the aromatic ring incorporated in the ammonium axle. Consequently, CE1 on the surface does not only retain its orientational preference, but also induces a preferred orientation into the deposited guest molecule AM2.
The other two pairs, DP1–TLM2 and TLM1–DP2, do not exhibit any significant angle-dependence. These two host–guest pairs thus either remain unordered (TLM1–DP2) or even lose their orientational preference upon complex formation (DP1–TLM2). In particular the latter host–guest pair shows that DP1 is capable of adapting its position to the steric requirements of its complex with TLM2 because the dichroism effect of the CO band at 288.4 eV vanishes upon addition of TLM2.
Reversibility of the on-surface molecular recognition events
In order to provide a proof of principle that the host–guest complexation is reversible, the DP1–TLM2 pair was chosen. First of all, the hydrogen bonding between these two molecules is strongly affected in solution by competitive solvents. Second, it is not associated with a similarly high dissociation barrier as the benzo[21]crown-7/secondary ammonium ion motif.Consequently, a sample surface covered with DP1 and subsequently treated with TLM2 for complex formation was rinsed vigorously with dimethylformamide which is a quite strong hydrogen-bond competitor. The XP N 1s and C 1s spectra of the surface after this treatment are displayed in Fig. 9. Both, the N 1s and the C 1s spectra are almost identical to those of the untreated DP1-functionalized surface (Fig. 2 and 4). The characteristic features for DP1 “clicked” to the surface can be observed and furthermore, the relative intensities are similar to those of the pristine DP1. Consequently, the host–guest complex can be dissociated with competitive solvents providing evidence for its reversible formation.
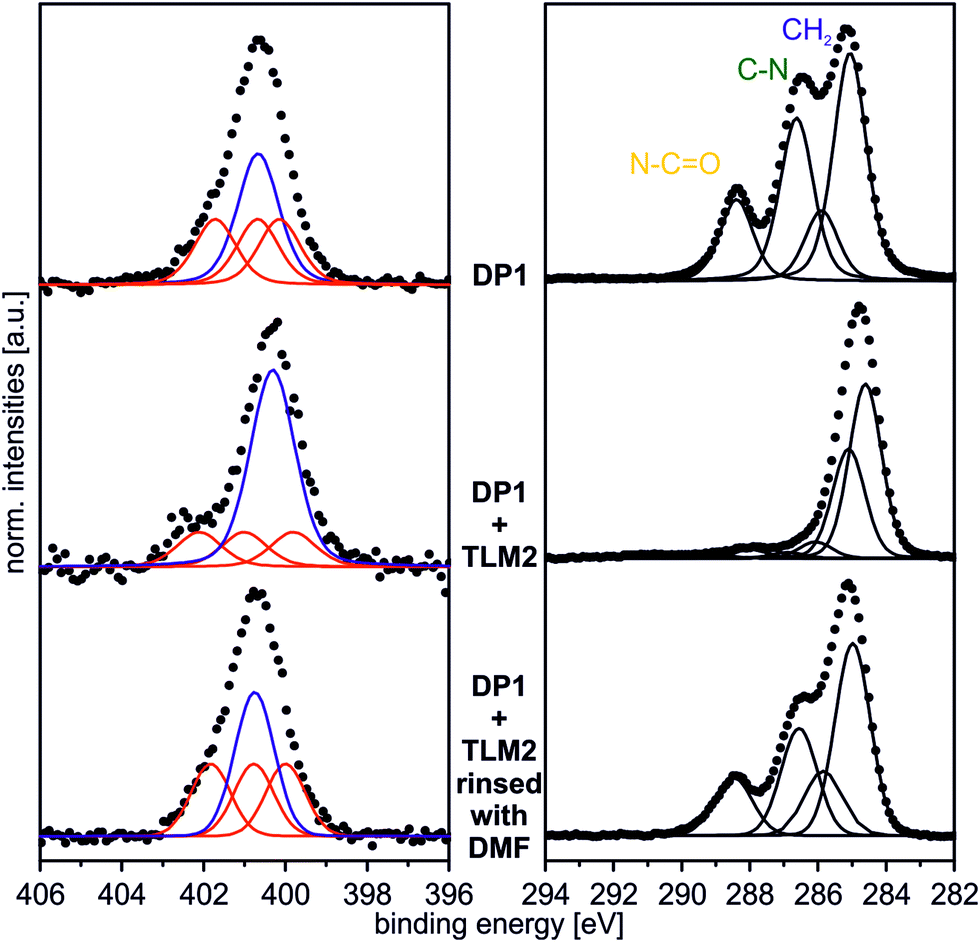 | ||
| Fig. 9 N 1s (hν = 500 eV; 0°) and C 1s (hν = 350 eV; 0°) spectrum of DP1 + TLM2 rinsed vigorously with DMF. | ||
Conclusions
In this contribution, click chemistry was utilized to attach host and guest molecules to Au surfaces covered with a self-assembled monolayer with azide head groups. Prior to the cycloaddition reactions, the X-ray irradiation-induced decomposition of the azide groups was investigated in detail by N 1s XPS in order to ensure that XP and NEXAFS analysis delivers valid data for the characterization of the “clicked” surfaces. X-ray damage experiments clearly suggest that molecular nitrogen is lost from the azide to the nitrene intermediate followed by a 1,2-hydrogen shift to the resulting imine.Covalent anchoring of four host or guest molecules to the above mentioned azide monolayer was obtained by copper-catalysed 1,3-dipolar cycloaddition. The resulting samples were investigated via XPS and NEXAFS which confirmed the cycloadditions to be successful for all functional molecules. Surface-attached crown ether CE1 and diketopiperazine DP1 exhibit a preferred orientation within the monolayer.
Finally, the attached molecules – except for the primary amine – are found to be able to build host–guest complexes with their corresponding counterpart on the surface as again shown by XPS and NEXAFS experiments. The reversibility of complex formation was shown for the DP1–TLM2 complex.
Acknowledgements
The authors gratefully acknowledge the Deutsche Forschungsgemeinschaft (SCHA 893/9-1, UN 80/8-1), the Freie Universität Berlin and the Fonds der Chemischen Industrie (FCI) for financial support. S. R. thanks the FCI for a Chemiefonds Ph.D. fellowship. We are grateful to D. Treu and Dr T. Gross for operating the XPS instrument at BAM 6.8. Support by the staff at BESSY II and the HE-SGM CRG (Dr A. Nefedov, KIT) during our activities at the HE-SGM beamline is gratefully acknowledged.Notes and references
- C. Nicosia and J. Huskens, Mater. Horiz., 2014, 1, 32–45 RSC
.
- J. J. Gooding and S. Ciampi, Chem. Soc. Rev., 2011, 40, 2704–2718 RSC
- D. Samanta and A. Sarkar, Chem. Soc. Rev., 2011, 40, 2567–2592 RSC
- C. Haensch, S. Hoeppener and U. S. Schubert, Chem. Soc. Rev., 2010, 39, 2323–2334 RSC
- G. Fioravanti, N. Haraszkiewicz, E. R. Kay, S. M. Mendoza, C. Bruno, M. Marcaccio, P. G. Wiering, F. Paolucci, P. Rudolf, A. M. Brouwer and D. A. Leigh, J. Am. Chem. Soc., 2008, 130, 2593–2601 CrossRef CAS PubMed
- A. F. M. Altelaar, I. Klinkert, K. Jalink, R. P. J. de Lange, R. A. H. Adan, R. M. A. Heeren and S. R. Piersma, Anal. Chem., 2005, 78, 734–742 CrossRef PubMed
- D. Wasserberg, C. Nicosia, E. E. Tromp, V. Subramaniam, J. Huskens and P. Jonkheijm, J. Am. Chem. Soc., 2013, 135, 3104–3111 CrossRef CAS PubMed
- T. Chang, D. I. Rozkiewicz, B. J. Ravoo, E. W. Meijer and D. N. Reinhoudt, Nano Lett., 2007, 7, 978–980 CrossRef CAS PubMed
- L. Tauk, A. P. Schröder, G. Decher and N. Giuseppone, Nat. Chem., 2009, 1, 649–656 CrossRef CAS PubMed
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS PubMed
- M. Li, Y. Wang, V. Pinon III and M. Weck, Chem. Commun., 2011, 47, 2802–2804 RSC
- Y. Liu, S. Yamazaki and S. Izuhara, J. Organomet. Chem., 2006, 691, 5809–5824 CrossRef CAS PubMed
- C. Vericat, M. E. Vela, G. Benitez, P. Carro and R. C. Salvarezza, Chem. Soc. Rev., 2010, 39, 1805–1834 RSC
- K. Kim, W. S. Jeon, J.-K. Kang, J. W. Lee, S. Y. Jon, T. Kim and K. Kim, Angew. Chem., Int. Ed., 2003, 42, 2293–2296 CrossRef CAS PubMed
- A. Pulsipher, N. P. Westcott, W. Luo and M. N. Yousaf, Adv. Mater., 2009, 21, 3082–3086 CrossRef CAS
- M. J. Shuster, A. Vaish, M. L. Gilbert, M. Martinez-Rivera, R. M. Nezarati, P. S. Weiss and A. M. Andrews, J. Phys. Chem. C, 2011, 115, 24778–24787 CAS
- J. Poppenberg, S. Richter, E. Darlatt, C. H.-H. Traulsen, H. Min, W. E. S. Unger and C. A. Schalley, Surf. Sci., 2012, 606, 367–377 CrossRef CAS PubMed
- A. Auditore, N. Tuccitto, G. Marzanni, S. Quici, F. Puntoriero, S. Campagna and A. Licciardello, Chem. Commun., 2003, 2494–2495 RSC
- C. H.-H. Traulsen, E. Darlatt, S. Richter, J. Poppenberg, S. Hoof, W. E. S. Unger and C. A. Schalley, Langmuir, 2012, 28, 10755–10763 CrossRef CAS PubMed
- N. Tuccitto, V. Ferri, M. Cavazzini, S. Quici, G. Zhavnerko, A. Licciardello and M. A. Rampi, Nat. Mater., 2009, 8, 41–46 CrossRef CAS PubMed
- N. Tuccitto, I. Delfanti, V. Torrisi, F. Scandola, C. Chiorboli, V. Stepanenko, F. Wurthner and A. Licciardello, Phys. Chem. Chem. Phys., 2009, 11, 4033–4038 RSC
- C. Battocchio, G. Polzonetti, L. Gambino, N. Tuccitto, A. Licciardello and G. Marletta, Nucl. Instrum. Methods Phys. Res., Sect. B, 2006, 246, 145–150 CrossRef CAS PubMed
- P. M. Dietrich, N. Graf, T. Gross, A. Lippitz, S. Krakert, B. Schüpbach, A. Terfort and W. E. S. Unger, Surf. Interface Anal., 2010, 42, 1184–1187 CrossRef CAS
- P. M. Dietrich, N. Graf, T. Gross, A. Lippitz, B. Schüpbach, A. Bashir, C. Wöll, A. Terfort and W. E. S. Unger, Langmuir, 2010, 26, 3949–3954 CrossRef CAS PubMed
- N. Graf, E. Yegen, T. Gross, A. Lippitz, W. Weigel, S. Krakert, A. Terfort and W. E. S. Unger, Surf. Sci., 2009, 603, 2849–2860 CrossRef CAS PubMed
- M. R. Lockett, M. R. Shortreed and L. M. Smith, Langmuir, 2008, 24, 9198–9203 CrossRef CAS PubMed
- Y. Luo, M. Piantek, J. Miguel, M. Bernien, W. Kuch and R. Haag, Appl. Phys. A, 2008, 93, 293–301 CrossRef CAS PubMed
- X. Song, Y. Ma, C. Wang, P. M. Dietrich, W. E. S. Unger and Y. Luo, J. Phys. Chem. C, 2012, 116, 12649–12654 CAS
- S. Richter, J. Poppenberg, C. H.-H. Traulsen, E. Darlatt, A. Sokolowski, D. Sattler, W. E. S. Unger and C. A. Schalley, J. Am. Chem. Soc., 2012, 134, 16289–16297 CrossRef CAS PubMed
- C. H.-H. Traulsen, V. Kunz, T. Heinrich, S. Richter, M. Holzweber, A. Schulz, L. K. S. von Krbek, U. T. J. Scheuschner, J. Poppenberg, W. E. S. Unger and C. A. Schalley, Langmuir, 2013, 29, 14284–14292 CrossRef CAS PubMed
- E. Darlatt, A. Nefedov, C. H.-H. Traulsen, J. Poppenberg, S. Richter, P. M. Dietrich, A. Lippitz, R. Illgen, J. Kühn, C. A. Schalley, C. Wöll and W. E. S. Unger, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 621–624 CrossRef CAS PubMed
- E. Darlatt, C. H.-H. Traulsen, J. Poppenberg, S. Richter, J. Kühn, C. A. Schalley and W. E. S. Unger, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 85–89 CrossRef CAS PubMed
- I. F. Gallardo and L. J. Webb, Langmuir, 2010, 26, 18959–18966 CrossRef CAS PubMed
- J. P. Collman, N. K. Devaraj and C. E. D. Chidsey, Langmuir, 2004, 20, 1051–1053 CrossRef CAS
- D. Zacher, R. Schmid, C. Wöll and R. A. Fischer, Angew. Chem., Int. Ed., 2011, 50, 176–199 CrossRef CAS PubMed
- J. Berná, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Pérez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nat. Mater., 2005, 4, 704–710 CrossRef PubMed
-
A. Ulman, An Introduction to Ultrathin Organic Films-From Langmuir–Blodgett
to Self-Assembly, Academic Press, London, UK, 1991 Search PubMed
- A. C. Fahrenbach, S. C. Warren, J. T. Incorvati, A.-J. Avestro, J. C. Barnes, J. F. Stoddart and B. A. Grzybowski, Adv. Mater., 2013, 25, 331–348 CrossRef CAS PubMed
- J. Poppenberg, S. Richter, C. H.-H. Traulsen, E. Darlatt, B. Baytekin, T. Heinrich, P. M. Deutinger, K. Huth, W. E. S. Unger and C. A. Schalley, Chem. Sci., 2013, 4, 3131–3139 RSC
- S. Richter, C. H.-H. Traulsen, T. Heinrich, J. Poppenberg, C. Leppich, M. Holzweber, W. E. S. Unger and C. A. Schalley, J. Phys. Chem. C, 2013, 117, 18980–18985 CAS
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS
- G. K. Such, J. F. Quinn, A. Quinn, E. Tjipto and F. Caruso, J. Am. Chem. Soc., 2006, 128, 9318–9319 CrossRef CAS PubMed
- A. P. Upadhyay, D. K. Behara, G. P. Sharma, A. Bajpai, N. Sharac, R. Ragan, R. G. S. Pala and S. Sivakumar, ACS Appl. Mater. Interfaces, 2013, 5, 9554–9562 CAS
- Z. Wang, J. Liu, H. K. Arslan, S. Grosjean, T. Hagendorn, H. Gliemann, S. Bräse and C. Wöll, Langmuir, 2013, 29, 15958–15964 CrossRef CAS PubMed
- B. Baytekin, S. S. Zhu, B. Brusilowskij, J. Illigen, J. Ranta, J. Huuskonen, L. Russo, K. Rissanen, L. Kaufmann and C. A. Schalley, Chem.–Eur. J., 2008, 14, 10012–10028 CrossRef CAS PubMed
- C. Allott, H. Adams, C. A. Hunter, J. A. Thomas, P. L. Bernad Jr and C. Rotger, Chem. Commun., 1998, 2449–2450 RSC
- E. V. Dzyuba, L. Kaufmann, N. L. Löw, A. K. Meyer, H. D. F. Winkler, K. Rissanen and C. A. Schalley, Org. Lett., 2011, 13, 4838–4841 CrossRef CAS PubMed
- W. Jiang, H. D. F. Winkler and C. A. Schalley, J. Am. Chem. Soc., 2008, 130, 13852–13853 CrossRef CAS PubMed
- Y. Zubavichus, M. Zharnikov, A. Schaporenko and M. Grunze, J. Electron Spectrosc. Relat. Phenom., 2004, 134, 25–33 CrossRef CAS PubMed
- L. Strong and G. M. Whitesides, Langmuir, 1988, 4, 546–558 CrossRef CAS
- K. C. Prince, R. Richter, M. de Simone, M. Alagia and M. Coreno, J. Phys. Chem. A, 2003, 107, 1955–1963 CrossRef CAS
- D. J. Menzies, B. Cowie, C. Fong, J. S. Forsythe, T. R. Gengenbach, K. M. McLean, L. Puskar, M. Textor, L. Thomsen, M. Tobin and B. W. Muir, Langmuir, 2010, 26, 13987–13994 CrossRef CAS PubMed
- D. Brete, D. Przyrembel, C. Eickhoff, R. Carley, W. Freyer, K. Reuter, C. Gahl and M. Weinelt, J. Phys.: Condens. Matter, 2012, 24, 394015 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures and characterization of new compounds, copies of 1H, 13C and 19F NMR spectra, multilayer preparation, instrumentation and data processing used for the described experiments, additional transmission, XPS and NEXAFS data for the characterization of samples. See DOI: 10.1039/c4ra01730g |
| This journal is © The Royal Society of Chemistry 2014 |