
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Performance evaluation of lithium metal rechargeable batteries with a lithium excess cation-disordered rocksalt based positive electrode under high mass loading and lean electrolyte conditions†
Jittraporn
Saengkaew
a,
Emiko
Mizuki
a and
Shoichi
Matsuda
 *abc
*abc
aResearch Center for Energy and Environmental Materials (GREEN), National Institute for Material Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: matsuda.shoichi@nims.go.jp
bCenter for Advanced Battery Collaboration, Center for Green Research on Energy and Environmental Materials, National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
cNIMS-SoftBank Advanced Technologies Development Center, National Institute for Materials Science, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
First published on 3rd January 2024
Abstract
Although lithium excess cation-disordered rock salt (DRX) metal oxides have been identified as promising candidates for positive-electrode materials, their actual potential remains unclear because previous studies have used inappropriate technological parameters, such as low mass loadings or excessive amounts of electrolyte. In this study, Li2RuO3/Li2SO4 was selected as the model DRX material, and its performance was investigated under cell-level high-energy-density conditions. A highly-mass-loaded positive electrode (30 mg cm−2) with an active material ratio exceeding 96% was fabricated by suppression of the gelation of slurry solution during the electrode preparation process, which is achieved by proper control of the particle size of Li2RuO3/Li2SO4. Notably, using a protected lithium metal electrode setup, superior capacity of the Li2RuO3/Li2SO4 electrode over 180 mA h g−1 was achieved over the 80th cycle under high mass loading and lean electrolyte conditions. The results obtained in the present study reveal the potential of the DRX based positive electrode for realizing superior performance even under practical cell conditions.
Introduction
In recent years, there has been a constantly growing demand for rechargeable energy-storage devices with high energy density. Lithium–metal-based (LMB) rechargeable batteries have attracted researchers’ attention owing to their potential to achieve energy densities considerably higher than those of conventional lithium-ion batteries. In recent studies, LMB rechargeable batteries with cell-level energy densities exceeding 350 W h kg−1 have been developed using NMC811-based positive electrodes.1,2 To achieve high energy density at the cell level, appropriate technological parameters should be adopted. For instance, a high-mass-loading positive electrode can be utilized to achieve high areal capacity, while the use of a lean electrolyte and thin lithium foil can minimize the weight of cell components. In addition, to realize an LMB with a cell-level energy density exceeding 500 W h kg−1, utilizing a high-energy-density positive electrode is crucial.3Among the positive electrode materials, lithium-rich layered oxide materials with the formula Li1+xM1−xO2 (M = transition metal) have gained significant attention as active materials due to their ability to achieve high capacities (over 250 mA h g−1) because of their cumulative cationic redox and anionic lattice oxygen redox reactions.4,5 In particular, there has been growing interest in the class of lithium-excess cation-disordered rock salt (DRX) metal oxides due to their potential for exhibiting superior capacity over 300 mA h g−1.6,7 However, in most studies, battery performance was evaluated under a low electrode mass loading and/or with a cell containing excess electrolyte, thereby limiting the actual cell level energy density. Although there is growing interest for the practical implementation of LMBs equipped with DRX materials, the actual potential of DRX for application in such high-energy-density LMBs at the cell level remains unexplored.
In the present study, we selected Li2RuO3/Li2SO4 as the model DRX system due to the following two reasons. (i) This electrode was recently reported to exhibit a capacity of over 300 mA h g−1,7,8 although the performance was evaluated at low mass loading conditions (<10 mg cm−2). (ii) Ru based materials are known to exhibit superior electrical conductivity compared with Mn based materials. The high electrical conductivity of active materials is beneficial for decreasing the conductive additives in the electrode, which results in the improvement of cell level energy density, especially in the case of high mass loading conditions. Using the Li2RuO3/Li2SO4 positive electrode as the model DRX system, we revealed that proper control of the particle size of active materials is a crucial factor for preparing a high mass loading electrode (>20 mg cm−2). As a result, we successfully prepared a high-mass-loading electrode with an active material ratio of over 96%, while avoiding the undesired gelation of the slurry solution. This resulted in a lithium–metal-based battery cell with a cell-level energy density of more than 500 W h kg−1.
Experimental
Synthesis of Li2RuO3/Li2SO4 powders
A series of (Li2RuO3)1−x/(Li2SO4)x powders (x = 0.10, 0.16, 0.20, 0.24, and 0.34) were synthesized by a mechanochemistry synthesis, using appropriate amounts of crystalline Li2RuO3 and Li2SO4. Crystalline Li2RuO3 was prepared from a 10% excess of Li2CO3 (99.99%; Kojundo Chemical Laboratory Co., Ltd) and RuO2 (Kojundo Chemical Laboratory Co., Ltd) via a solid-state synthesis. The mixture was preheated in an alumina crucible at 900 °C for 12 h and then calcined at 1100 °C for 12 h under nitrogen and oxygen flows at a heating rate of 5 °C min−1. The obtained sample was ground homogeneously to obtain a well-crystallized Li2RuO3 powder material. Li2SO4·H2O (Kojundo Chemical Laboratory Co., Ltd) was heated at 300 °C for 3 h under an Ar atmosphere to obtain a Li2SO4 crystal powder. For the synthesis of Li2RuO3/Li2SO4 positive electrode materials, stoichiometric amounts of Li2RuO3, and Li2SO4 were mixed and homogenized by using a mortar and pestle. The mixture was ball milled for 50 h in an 80 mL zirconia pot with 5 mm diameter zirconia balls (250 balls) of a planetary ball miller (Pulverisette 6; Fritsch) at different rotating speeds (600 rpm) and then ground well. During sample preparation, the samples were not exposed to the ambient atmosphere.Characterization of the Li2RuO3/Li2SO4 powder
The particle size of the powder-based sample was measured by using a laser scattering particle size distribution analyzer (LA-950V2, HORIBA). Field-emission SEM (FE-SEM, S-4800, Hitachi) and X-ray diffraction (XRD; SmartLab, Rigaku) were used to characterize the powder-based samples.Preparation of the Li2RuO3/Li2SO4 positive electrode
A slurry of Li2RuO3/Li2SO4 active materials (94 wt%), acetylene black (Denka Black HS100; DENKA Co.; 3 wt%), and the polyvinylidene fluoride (PVDF; KUREHA Co.; 3 wt%) binder dissolved in N-methyl-1,2-pyrrolidone (NMP; Super Dehydrated; FUJIFILM Wako Pure Chemical Co.) was coated onto an aluminum (Al) current collector (a thickness of 10 μm). The NMP solvent was removed by heating at 230 °C in a nitrogen atmosphere for 30 minutes, and the electrode sheets were obtained. The loading amount of the active materials was about 30 mg cm−2.Electrochemical measurements
1 M Lithium bis(fluorosulfonyl)imide (LiFSI; Kishida Chemical Co., Ltd, purity > 99.0%) dissolved in sulfolane (Kishida Chemical Co., Ltd, purity > 99%) and 4M LiFSI in 1,2-dimethoxyethane (DME; Kishida Chemical Co., Ltd, purity > 99.5%) were used as electrolytes. A 100 μm thick lithium foil attached with a 12 μm thick copper current collector and a 20 μm thick lithium foil attached with a 2 μm thick copper current collector were used as the negative electrode for cell A and cell B, respectively. For fabrication of cell A, the positive electrode (20 mm × 20 mm), a PO separator (20 μm thickness, 22 mm × 22 mm), a ceramic separator (90 μm thickness, 24 mm × 24 mm), a PO separator (22 mm × 22 mm) and the negative electrode (42 mm × 32 mm) were stacked. In such a configuration, the lithium metal was completely sealed by a ceramic-based solid-state separator and laminated film, which completely isolated the electrolyte on the positive and negative electrode sides. 60 mg of 1M LiFSI in suflolane electrolyte (15 mg cm−2) was injected for the positive electrode side and 10 mg of the 4M LiFSI in the DME electrolyte (2.5 mg cm−2) was injected for the negative electrode side. For fabrication of cell B, the positive electrode (40 mm × 30 mm), separator (46 mm × 36 mm) and the negative electrode (42 mm × 32 mm) were stacked inside a laminated film and sides of the stack were sealed. 120 mg of the electrolyte (10 mg cm−2) was injected before sealing the cell under vacuum. All the cells were fabricated inside a dry room (dew point <−50 °C) and electrolyte injection was carried out inside a fume hood (dew point <−85 °C). Charge and discharge of the cells were carried out with Hokuto Denko HJ1001SD8. All the cells were cycled at a constant current density of 0.6 mA cm−2 in the voltage range of 2–4.2 V.In situ MS analysis
For on-line MS analysis, the generated gases were directed to the MS detector by the Canon Anelva Quadrupole Mass Spectrometer M-401GA-DM equipped with a capillary tube (internal diameter: 0.05 mm, length: 7 m). After discharge, the test cell was purged with excess He (50 mL min−1) for 1 min to remove the remaining O2. He as a carrier gas was flown at 5 mL min−1 and maintained for 2 h before charge. The measurement was carried out at 100 μV applied voltage, in the m/z range from 11 to 110 under ambient conditions.XCT analysis
X-ray CT analyses of the pouch cells were carried out using a Xradia 520 Versa (ZEISS, Germany) instrument, where the source voltage and power were 140 kV and 10 W, respectively. The cells after certain charge/discharge cycles were mounted on the sample holder, and the cell was rotated 360° for 4501 scans with an exposure time of 10 s. The pixel resolution of XCT imaging was 3.385 μm.Results and discussion
First, we considered how energy density of LMBs is affected by the technological parameters, such as the electrolyte amount and the mass loading of the positive electrode. The parameters of the LMB components used in our simulation are listed in Tables S1 (ESI†), and the simulated energy densities of LMBs are listed in Table S2 (ESI†). Here, we set the capacity of the positive electrode to 300 mA h g−1 and an average discharge voltage of 3.1 V as the model case of the DRX material. Here, the porosity of the positive electrode was set as 33% and the electrolyte amount was estimated for fully filling the pore volume in the positive electrode and separator. When a glass fiber separator (thickness = 100 μm, porosity = 91%) was employed, the cell level energy density was less than 100 W h kg−1 even when the mass loading of the active material in the positive electrode was 10 mg cm−2. Although the amount of electrolyte in the positive electrode is small (1.7 mg cm−2), a large amount of electrolyte (up to 24 mg cm−2) was required to fully fill the pore space in the separator. As a result, the electrolyte accounted for 50% of the total weight (Fig. 1a), which resulted in low energy density. When a polyolefin-based separator (thickness = 20 μm, porosity = 46%) commonly used in commercial lithium-ion batteries was employed, the energy density was 230 W h kg−1, as the electrolyte loading decreased to 0.9 mg cm−2 (Fig. 1b). When the mass loading increases from 10 to 30 mg cm−2, the energy density exceeded over 500 W h kg−1 (Fig. 1c). Under such high energy density conditions, the positive electrode accounted for more than 60% of the total weight. We also performed similar energy density simulations for the NMC811 based positive electrode material under the same conditions as those used for the case of DRX. The results revealed that cell level energy density of the LMB equipped with the NMC based positive electrode is lower than 500 W h kg−1 (Table S3, ESI†), revealing the importance of using high-capacity positive electrode materials for realizing high energy density LMBs.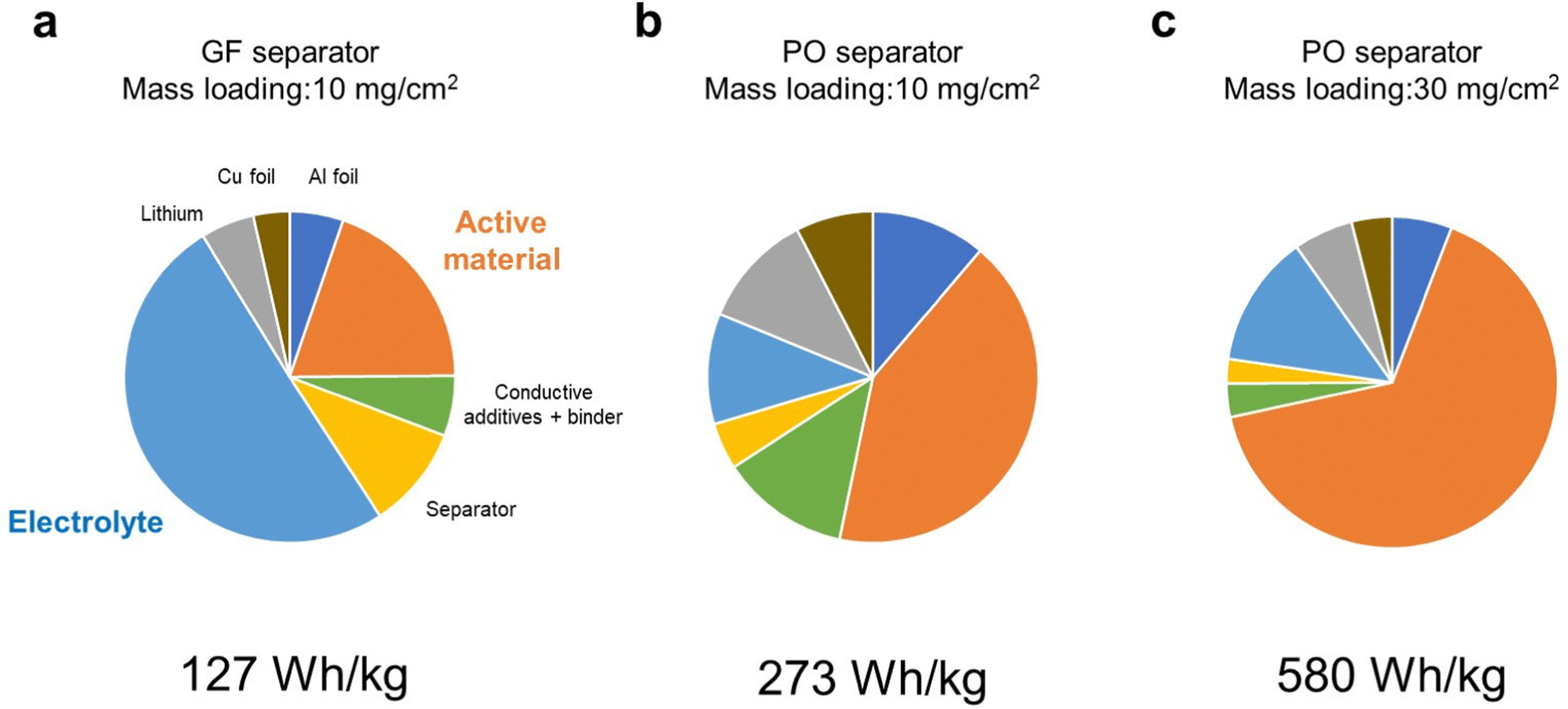 | ||
| Fig. 1 Weight fractions of LMB components calculated using the parameters listed in Table S1 (ESI†). (a) Glass fiber separator, a mass loading of 10 mg cm−2, (b) polyolefin separator, a mass loading of 10 mg cm−2, and (c) polyolefin separator, a mass loading of 30 mg cm−2. | ||
The results of the above simulations revealed the importance of decreasing the electrolyte amount and increasing the mass loading of the positive electrode for maximizing the energy density of LMBs. In particular, preparation of a high mass loading DRX-based positive electrode is crucial.
A series of (Li2RuO3)1−x/(Li2SO4)x powders (x = 0.10, 0.16, 0.20, 0.24, and 0.34) were synthesized via a mechanochemical method by mixing Li2RuO3 and Li2SO4 in appropriate proportions. Li2RuO3 was synthesized using a solid-state method,9 while Li2SO4 was dried at 300 °C before use. Details of the synthesis procedure can be found in the Experimental section. The XRD patterns of the samples are shown in Fig. S4 (ESI†), which indicate the presence of clear peaks corresponding to the NaCl-type cation-disordered phase, with no clear peaks assignable to the precursor compound of Li2RuO3 or Li2SO4.
Fig. 2 shows the SEM images of a series of Li2RuO3/Li2SO4 samples. For the pristine Li2RuO3 sample, 10 um sized particles can be seen (Fig. 2a). In contrast, the size of mechanically treated Li2RuO3 was hundred nanometer. Notably, SEM observations revealed that an increase in the Li2SO4 concentration was associated with an increase in the Li2RuO3/Li2SO4 particle size (Fig. 2). The particle size distribution was also evaluated, and Fig. S5 (ESI†) shows a histogram of the particle size distributions of pristine Li2RuO3 and a series of Li2RuO3/Li2SO4 samples. The average particle size of the layered Li2RuO3 precursor was found to be 9 μm, whereas the mechanochemically treated Li2RuO3 with a cation disordered rock-salt structure had an average particle size of less than 1 μm. With increasing Li2SO4 concentration, an increase in the average particle size was observed. For samples with Li2SO4 ratios of 0.10 and 0.16, the particle sizes were in the range of 0.1 to 0.3 μm, while in samples with Li2SO4 ratios of 0.20, 0.24, and 0.34, most particles were larger than 0.3 μm.
 | ||
| Fig. 2 SEM-EDS images of (a) and (b) pristine Li2RuO3 and (c)–(h) Li2RuO3/Li2SO4 samples. Scale bars are (a) 10 μm and (b)–(h) 1 μm. | ||
Next, high-mass-loading electrodes were fabricated using prepared Li2RuO3/Li2SO4 samples. A slurry solution was prepared by mixing 94.4 wt% Li2RuO3/Li2SO4 powder, 0.4 wt% CB, 0.2 wt% CNT, and 5 wt% PVDF binders. For the case of Li2RuO3/Li2SO4 with Li2SO4 ratios of 0.2, 0.24, and 0.34, uniform electrodes with mass loadings over 20 mg cm−2 were successfully prepared (Fig. 3a). Fig. S6 (ESI†) displays a representative cross-sectional SEM image of a Li2RuO3/Li2SO4 electrode, showing a uniform distribution of O, F, S, and Ru in the electrode, indicating that the active material, conductive carbon, and binder were well mixed (Fig. 3b). In contrast, slurry gelation occurred in the case of Li2RuO3/Li2SO4 with Li2SO4 ratios of 0, 0.1, and 0.16 (Fig. 3c). Considering the fact that no gelation occurred during the slurry preparation process using Li2RuO3/Li2SO4 with Li2SO4 ratios of 0.2, 0.24, and 0.34, the gelation observed in Li2RuO3/Li2SO4 with Li2SO4 ratios of 0, 0.1, and 0.16 was originated from their high surface area characteristics due to their smaller particle sizes (<0.3 μm).
 | ||
| Fig. 3 (a) Photographic image of the electrode of Li2RuO3/Li2SO4 with a Li2SO4 ratio of 0.24 and with a mass loading of 20 mg cm−2. (b) and (c) Photographic image of slurry solution of Li2RuO3/Li2SO4 with Li2SO4 ratios of (b) 0.24 and (c) 0.1. | ||
To evaluate the battery performance, electrochemical cells were fabricated using Li2RuO3/Li2SO4 electrodes with Li2SO4 ratios of 0.2 and 0.34. To accurately evaluate the performance of the positive electrode, we used a relatively thick 100 μm lithium foil as the negative electrode. In addition, a ceramic-based solid-state separator sandwiched between two pieces of a PO-based separator was used to prevent undesired side reactions at the lithium metal electrode. The lithium metal was completely sealed by a ceramic-based solid-state separator and a laminated film, which completely isolated the electrolyte on the positive and negative electrode sides. A solution of 4 M LiFSI dissolved in DME was used as the electrolyte on the negative electrode side due to its compatibility with the lithium metal electrode.10 Furthermore, 1 M LiFSI in sulfolane was selected as the positive electrolyte because of its high oxidative stability. The details of cell components are described in Table S7 (ESI†). The ratio of the electrolyte weight to areal capacity (E/C, g A−1 h−1) is used as an empirical parameter of the electrolyte amount in the field of LiBs. For the cell A fabricated in the present study, the value of E/C was 2.95 g A−1 h−1. Such a low value of E/C was realized by adopting a high mass loading positive electrode (20 mg cm−2) and a thin separator (20 μm thickness).
Fig. 4 shows the charge/discharge profile of a cell with Li2RuO3/Li2SO4 (x = 0.20) as the positive electrode. The cell showed a gradual increase in voltage from 3.3 V to 4.0 V (black curve in Fig. 4a) during the first charging process, eventually reaching the cutoff voltage of 4.2 V with a capacity of 260 mA h g−1. Upon switching to discharge, the voltage initially remained around 4.0 V and then decreased gradually, reaching the cutoff voltage of 2.0 V with a capacity of 300 mA h g−1. This voltage profile is consistent with those reported in the literature. In situ MS analysis during the charging process revealed that the oxygen evolution started at a capacity of 225 mA h g−1, corresponding to a charging voltage of 4.0 V (black curve in Fig. 4b). In contrast, no significant CO2 evolution was observed (black curve in Fig. 4c). The cell with Li2RuO3/Li2SO4 (x = 0.34) exhibited a slightly higher charging voltage than the cell with Li2RuO3/Li2SO4 (x = 0.2) (blue curve in Fig. 4a). During discharge, the voltage profile was similar to that observed for Li2RuO3/Li2SO4 (x = 0.2), but the cell exhibited a discharge capacity of 260 mA h g−1. In situ MS analysis of the gas generated during charging revealed that O2 generation started at a capacity of 210 mA h g−1, corresponding to a charging voltage of 4.0 V. The generation of CO2 began at a similar time. After the first charge/discharge process, both cells were subjected to repeated cycling. Fig. 4d shows the voltage profile during the second charge/discharge process, revealing that the cell exhibited a discharge capacity of 270 mA h g−1 for both Li2RuO3/Li2SO4 (x = 0.2) and Li2RuO3/Li2SO4 (x = 0.34). Moreover, no significant O2 or CO2 generation was observed (black curves in Fig. 4b and d), indicating limited side reactions in this system. These results clearly revealed that Li2RuO3/Li2SO4 (x = 0.2) exhibited higher capacity in the charging process and also generated less gas compared with Li2RuO3/Li2SO4 (x = 0.34). Therefore, in the following, we examined the details of Li2RuO3/Li2SO4 (x = 0.2).
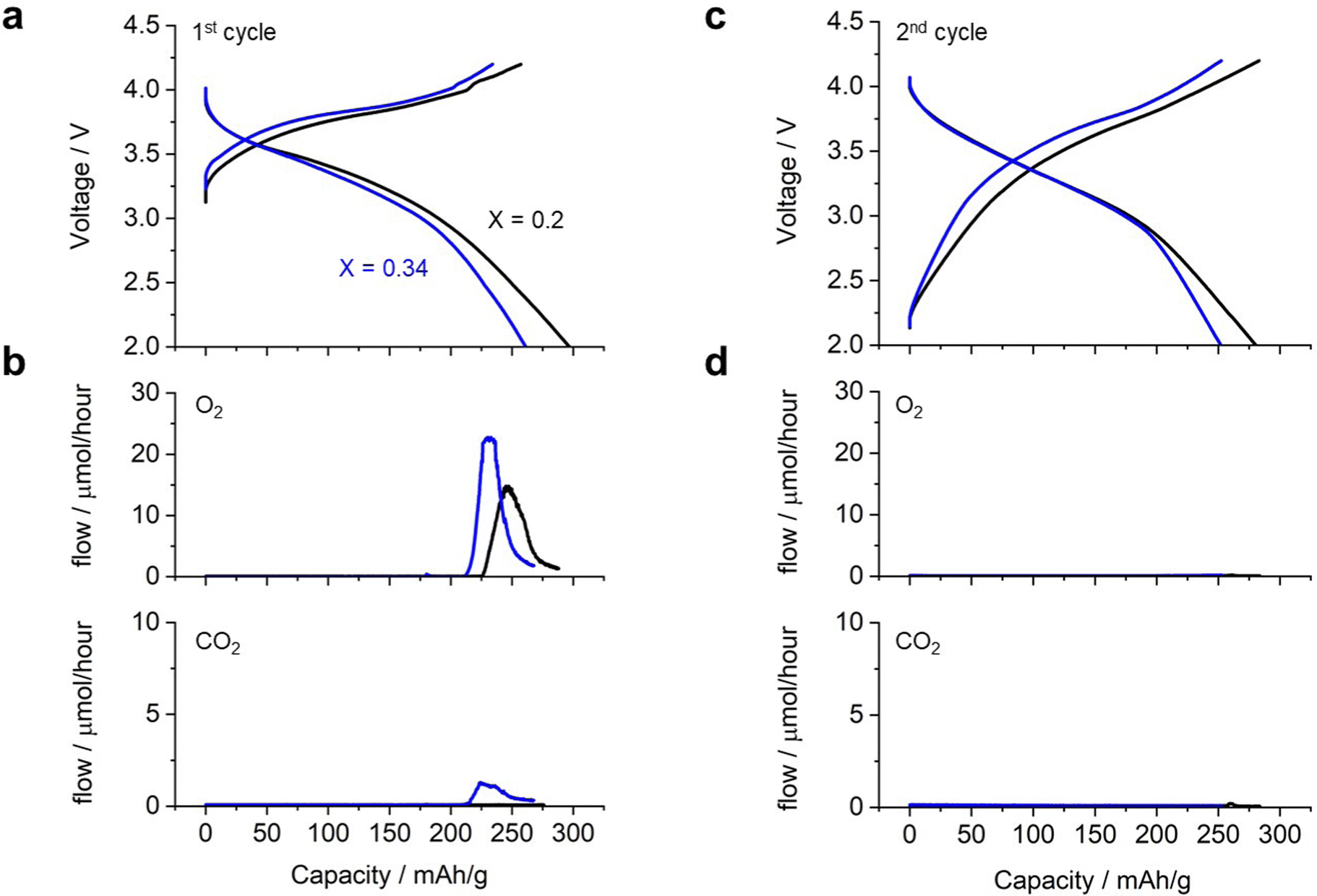 | ||
| Fig. 4 (a) and (c) Charge/discharge profile for 1st and 2nd cycle; (b) and (d) generated gas profile in the (b)1st and (d) 2nd cycles in an electrochemical cell equipped with Li2RuO3/Li2SO4 samples (black curve: x = 0.2 and blue curve: x = 0.34) as positive electrodes. | ||
We conducted an extended long-cycle test on an electrochemical cell (named cell A). As shown in Fig. 5a, the capacity gradually decreased over repeated cycles. Fig. 5c displays the discharge capacity plotted against the cycle life. During the 10th cycle, the capacity decreased rapidly. Subsequently, there was a gradual decrease in the capacity with the progress of the cycles, until it reached 180 mA h g−1 for the 30th cycle, after which the capacity remained stable even up to the 80th cycle. These results indicate that Li2RuO3/Li2SO4 (x = 0.2) exhibited superior performance even under high mass loading and lean electrolyte conditions.
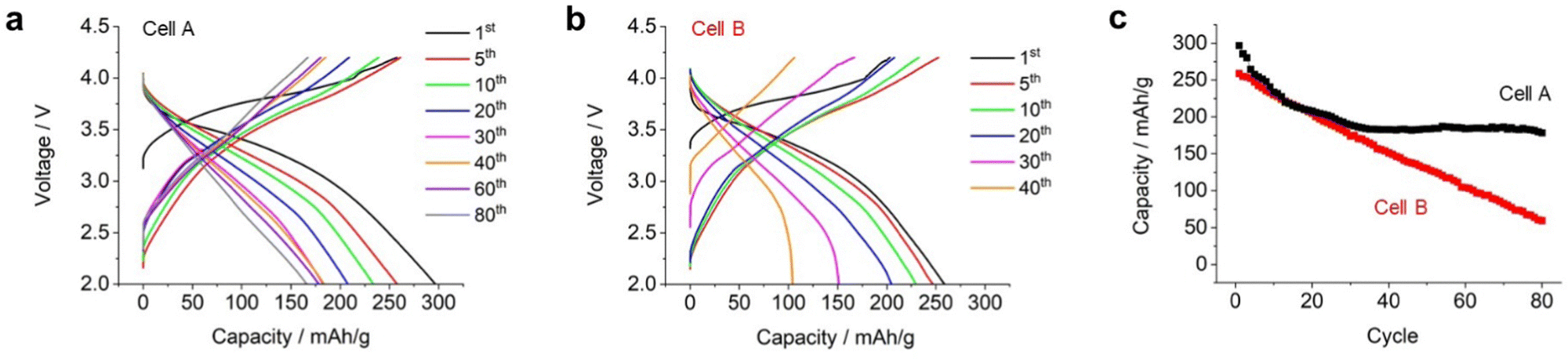 | ||
| Fig. 5 (a) and (b) Charge/discharge profile of the electrochemical cell equipped with the Li2RuO3/Li2SO4 sample (x = 0.2) as the positive electrode: (a) cell A and (b) cell B. (c) Discharge capacity plotted against the cycle number for cell A (black curve) and cell B (red curve). | ||
Subsequently, a high-energy-density battery cell (cell B) was fabricated by utilizing suitable technological parameters, (i) increasing the mass loading of positive electrode (30 mg cm−2), (ii) removing ceramic-based separator, and (iii) replacing thin lithium foil (20 μm thickness). As result, the E/C value of cell B was 1.28 g A−1 h−1. Details of the fabricated cells are listed in Table S1 (ESI†). Fig. 5b shows the repeated charge/discharge profiles. During the initial charging, cell B exhibited a capacity of 220 mA h g−1, which was lower than that of cell A (260 mA h g−1). However, during the overall charging process, cell B exhibited a full capacity of 260 mA h g−1 with an average discharge voltage of 3.1 V. As a result, the energy density at the cell level reached 512 W h kg−1. Cell B exhibited a smaller discharge capacity compared to cell A. It is believed that this could be attributed to the limited amount of electrolyte present in the positive electrode. Upon repeating the charge/discharge cycles, the capacity of cell B further decreased. In the 20th cycle, cell B exhibited a discharge capacity of 230 mA h g−1 with an average discharge voltage of 3.1 V, resulting in a cell-level energy density of 400 W h kg−1. Subsequently, the discharge capacity decreased linearly as the cycles progressed (red plots in Fig. 5c). Although the capacity of cell A was stable at 180 mA h g−1 after the 40th cycle, the capacity of cell B kept decreasing even after 40 cycles. In the voltage profile of cell B, a sudden voltage drop was observed at the end of the 40th discharge cycle, which was significantly different from that of cell A. It should be noted that the average discharge voltage of both cell A and cell B was around 3.1–3.0 V thoughout the cycles.
We performed the SEM analysis of the positive electrodes after the 40th cycle of the cell. No clear difference was observed between the electrodes before and after the cycle test in both cells A and cell B (Fig. S8, ESI†). The results of XRD analysis revealed that intensity of peaks corresponding to the NaCl-type cation-disordered phase decreased with the progress of the cycle (Fig. S9, ESI†), suggesting the deterioration of positive electrode materials.
In order to explain the poor capacity retention of cell B, the difference in the cell configuration between cells A and B must be considered. The observed severe capacity fading phenomenon in cell B could be attributed to the degradation of the lithium metal electrode. Thus, we conducted an analysis of the lithium metal electrode post cycling. However, during the cell disassembly process, the electrode easily collapsed and could not be analyzed using conventional ex situ techniques such as SEM and XRD. Hence, we performed XCT analysis of the cell, which is a representative non-destructive analytical technique for monitoring the structural changes in the electrode.11Fig. 6a shows the side-view XCT image of cell B under the prepared conditions. There can be seen the Li2RuO3/Li2SO4-based positive electrode with a thickness of 150 μm and a lithium metal negative electrode with a thickness of 20 μm. Fig. 6b shows the XCT image of the cell after the 10th cycle, revealing a large volume change in the lithium metal electrode. Although the thickness of the Li2RuO3/Li2SO4-based positive electrode did not change, that of the lithium–metal-based negative electrode exceeded 120 μm, which is six times greater than the initial thickness (20 μm). After the 20th cycle, the thickness of the lithium–metal electrode reached 150 μm. Fig. 6c shows the cross-sectional XCT image of the lithium electrode after the 20th cycle. Dense lithium metal remained in the region close to the copper foil, which is shown in Fig. 6d and e. On the other hand, a porous structure was detected in the central area and in the region close to the separator side (Fig. 6f and g). A similar porous Li electrode has been reported in the literature.11 However, such porous characteristics can accelerate the formation of electrochemically isolated dead lithium, resulting in a poor reaction efficiency at the lithium negative electrode. Moreover, the electrolyte is absorbed into the porous lithium electrode, resulting in a shortage of electrolytes in the entire cell. Further detailed analyses of the electrodes in high-energy-density lithium–metal-based rechargeable battery cells are currently underway in our laboratory.
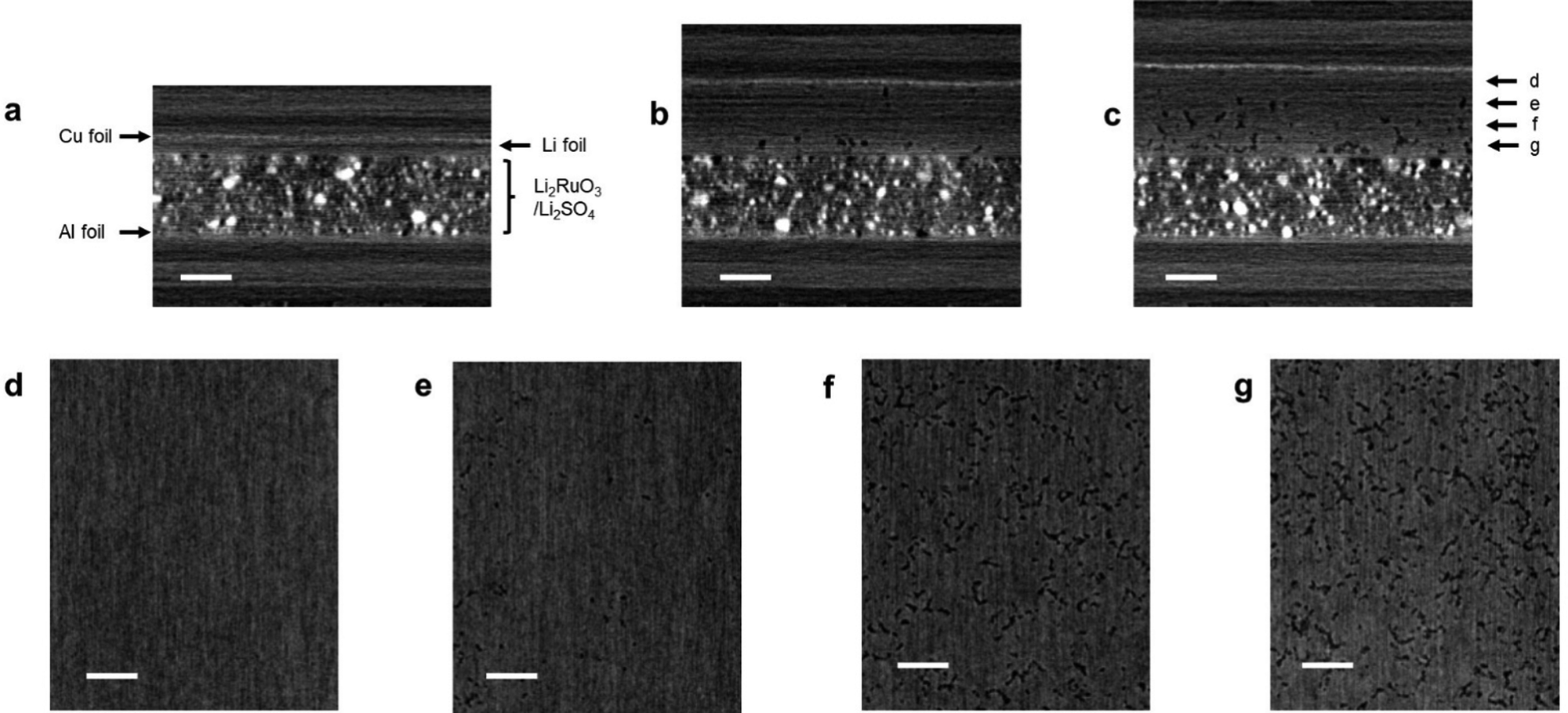 | ||
| Fig. 6 (a)–(c) X-ray CT images of cell B; (a) as prepared, (b) after the 10th cycle, and (c) after the 20th cycle in the cross-sectional direction; (d)–(g) X-ray CT images of cell B after the 20th cycle in the surface direction. Scale bars are 100 μm. | ||
Conclusions
In this study, we used a Li2RuO3/Li2SO4-based positive electrode as the model material for lithium–metal-based rechargeable batteries. By controlling the particle size of Li2RuO3/Li2SO4 by varying the Li2SO4 ratio, a high-mass-loading electrode with an active material ratio of over 96% was successfully prepared by suppressing undesired gelation of the slurry solution. Notably, using a protected lithium metal electrode setup, superior capacity of the Li2RuO3/Li2SO4 electrode over 180 mA h g−1 was achieved over the 80th cycle under high mass loading and lean electrolyte conditions. We also fabricated a high-energy-density battery cell, exhibiting energy density over 500 W h kg−1 and a stable charge/discharge reaction. However, as cycling progressed, the cell capacity decreased rapidly, causing the energy density of the cell to drop to 400 W h kg−1 by the 20th cycle. Non-destructive XCT analysis revealed a significant volume expansion of the lithium electrode, which is considered to be the primary reason for the capacity fading. Importantly, not the reaction at the DRX positive electrode, but the reaction at the lithium metal negative electrode is the bottleneck process for achieving a prolonged cycle life. Similar problems can exist for the case in which Mn-based DRX electrode materials were used, which is more attractive from a practical point of view. The results obtained in the present study highlight the importance of suppressing the significant volume change of lithium–metal electrodes, as well as the development of DRX materials, in order to achieve a prolonged cycle life in high energy density rechargeable batteries.Author contributions
S. M. supervised the project. J. S. and E. M. performed experiments. All authors contributed for data analysis and discussion. J. S. and S. M. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A part of this work was carried out at the SoftBank-NIMS Advanced Technologies Development Center as a joint research between NIMS and SoftBank Corp. This work also received support from the National Institute for Materials Science (NIMS) Battery Research Platform.Notes and references
- C. Niu, H. Lee, S. Chen, Q. Li, J. Du, W. Xu, J.-G. Zhang, M. S. Whittingham, J. Xiao and J. Liu, High-energy lithium metal pouch cells with limited anode swelling and long stable cycles, Nat. Energy, 2019, 4(7), 551–559 CrossRef CAS
.
- C. Niu, D. Liu, J. A. Lochala, C. S. Anderson, X. Cao, M. E. Gross, W. Xu, J. G. Zhang, M. S. Whittingham, J. Xiao and J. Liu, Balancing interfacial reactions to achieve long cycle life in high-energy lithium metal batteries, Nat. Energy, 2021, 6(7), 723–732 CrossRef CAS
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Pathways for practical high-energy long-cycling lithium metal batteries, Nat. Energy, 2019, 4(3), 180–186 CrossRef CAS
- M. Sathiya, G. Rousse, K. Ramesha, C. P. Laisa, H. Vezin, M. T. Sougrati, M.-L. Doublet, D. Foix, D. Gonbeau, W. Walker, A. S. Prakash, M. Ben Hassine, L. Dupont and J.-M. Tarascon, Reversible anionic redox chemistry in high-capacity layered-oxide electrodes, Nat. Mater., 2013, 12(9), 827–835 CrossRef CAS
- H. Koga, L. Croguennec, M. Ménétrier, K. Douhil, S. Belin, L. Bourgeois, E. Suard, F. Weill and C. Delmas, Reversible oxygen participation to the redox processes revealed for Li1.20Mn0.54Co0.13Ni0.13O2, J. Electrochem. Soc., 2013, 160(6), A786 CrossRef CAS
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Unlocking the potential of cation-disordered oxides for rechargeable lithium batteries, Science, 2014, 343(6170), 519–522 CrossRef CAS PubMed
- R. J. Clément, Z. Lun and G. Ceder, Cation-disordered rocksalt transition metal oxides and oxyfluorides for high energy lithium-ion cathodes, Energy Environ. Sci., 2020, 13(2), 345–373 RSC
- K. Nagao, A. Sakuda, W. Nakamura, A. Hayashi and M. Tatsumisago, Fast cationic and anionic redox reactions in Li2RuO3-Li2SO4 positive electrode materials, ACS Appl. Energy Mater., 2019, 2(3), 1594–1599 CrossRef CAS
- G. J. Moore, C. S. Johnson and M. M. Thackeray, The electrochemical behavior of xLiNiO2·(1 − x)Li2RuO3 and Li2Ru1−yZryO3 electrodes in lithium cells, J. Power Sources, 2003, 119–121, 216–220 CrossRef CAS
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, High rate and stable cycling of lithium metal anode, Nat. Commun., 2015, 6 Search PubMed
- R. Tamate and S. Matsuda, Asymmetric volume expansion of the lithium metal electrode in symmetric lithium/lithium cells under lean electrolyte and high areal capacity conditions, ACS Appl. Energy Mater., 2023, 6(1), 573–579 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ya00281k |
| This journal is © The Royal Society of Chemistry 2024 |