
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEPR spin trapping of nucleophilic and radical reactions at colloidal metal chalcogenide quantum dot surfaces†
Caroline J.
Aschendorf
 ,
Mawuli
Degbevi
,
Keaton V.
Prather
and
Emily Y.
Tsui
*
,
Mawuli
Degbevi
,
Keaton V.
Prather
and
Emily Y.
Tsui
*
Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN, USA. E-mail: etsui@nd.edu
First published on 1st November 2023
Abstract
The participation of the surfaces of colloidal semiconductor nanocrystal quantum dots (QDs) in QD-mediated photocatalytic reactions is an important factor that distinguishes QDs from other photosensitizers (e.g. transition metal complexes or organic dyes). Here, we probe nucleophilic and radical reactivity of surface sulfides and selenides of metal chalcogenide (CdSe, CdS, ZnSe, and PbS) QDs using chemical reactions and NMR spectroscopy. Additionally, the high sensitivity of EPR spectroscopy is adapted to study these surface-centered reactions through the use of spin traps like 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) under photoexcitation and thermal conditions. We demonstrate that DMPO likely adds to CdSe QD surfaces under thermal conditions by a nucleophilic mechanism in which the surface chalcogenides add to the double bond, followed by further oxidation of the surface-bound product. In contrast, CdS QDs more readily form surface sulfur-centered radicals that can perform reactions including alkene isomerization. These results indicate that QD surfaces should be an important consideration for the design of photocatalysis beyond simply tuning QD semiconductor band gaps.
Introduction
Colloidal semiconductor nanocrystal quantum dots (QDs) have been used as photosensitizers or photocatalysts for a number of reactions, including small molecule activation (e.g. H2 evolution, N2 or CO2 reduction, or water oxidation)1–3 and organic transformations like C–C bond-forming photoredox reactions.4,5 In most cases, these reactions proceed by charge transfer of photoexcited carriers to substrates or to sacrificial reductants/oxidants. In these systems, the QD surface, which includes supporting ligands, plays a critical role in the reactivity. For example, the QD ligand shell has been shown to influence the reactions by dictating diffusion of reaction components to the QD surface atoms and charge transfer rates.5,6 As such, many efforts have been focused on tuning the ligand shell to facilitate photocatalytic transformations sensitized by QDs.7Beyond the ligand shell, the surface atoms themselves can undergo redox reactions; for example, trapping of photoexcited carriers is common and has been implicated in photolytic QD decomposition.8 Surface traps have also been demonstrated to mediate photoinduced charge transfer from QDs to molecular acceptors, which occurs on much longer timescales (ms–s) than charge trapping (ps).9 The chemical nature and reactivity of these surface atoms are of great interest for QD photocatalytic applications, as they may dictate catalyst degradation, the effectiveness of hole or electron scavengers during photocatalytic reduction reactions, or enhanced electron–hole recombination. One major question that has been less well studied is whether the surface atoms can also participate in inner-sphere reactions. This is particularly salient in commonly studied metal chalcogenide QDs, as both organic and inorganic sulfur- and selenium-containing compounds can undergo many different classes of reactions.
The reactivity of chalcogenide ions outside of nanocrystal contexts is well known, particularly for organic transformations and in biochemical processes. Scheme 1 shows selected examples of chalcogenide reactions that may be hypothesized to occur at metal chalcogenide QD surfaces. For example, reduced chalcogenides (S2− or Se2−) can undergo oxidation to form polysulfide/polyselenide anions or zerovalent oligomeric/polymeric forms (Scheme 1a). While such redox processes are invoked during trapping of photoexcited carriers, they have also been proposed to occur upon treatment of CdS, CdSe, and PbS QDs with chemical reductants and oxidants.10–12 Second, sulfides and selenides (as well as their organic derivatives), are well known to be nucleophilic and to undergo reactions such as alkylation upon treatment with alkyl halides (Scheme 1b). This mode of reactivity has been far less explored for QDs.11 Third, sulfur-centered radicals like thiyl radicals have been demonstrated to participate in a number of different organic transformations, even under photocatalytic conditions, including addition to alkenes (Scheme 1c) and H-atom abstraction.13 Selenium-centered radicals have been less well studied, but have been proposed to participate in some biochemical processes.14 For nanoscale metal chalcogenide semiconductor materials, surface-trapped holes for ZnS and CdS materials have been proposed to undergo radical addition to alkenes to effect cis–trans isomerization and polymerization reactions,15,16 but little has been done to probe the nature of these surface radicals.
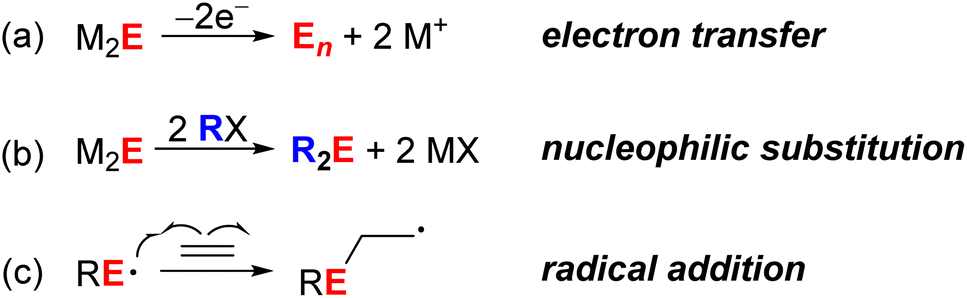 | ||
| Scheme 1 Examples of chalcogenide-centered reactions (E = S, Se). | ||
As research continues to expand the use of QD materials in photocatalysis and chemical reactions, major questions that need to be answered are (1) how relevant such surface reactions can be, (2) how to account for them in experimental design, and (3) how to best study them experimentally. In this work, we study radical and nucleophilic chalcogenide reactions at metal chalcogenide QD surfaces using a number of chemical probes,17,18 as well as spin traps that permit the formation of nitroxide radicals that are readily detected by EPR spectroscopy when used in conjunction with NMR spectroscopy. In particular, the differences between CdSe and CdS QDs in their application toward photocatalytic reactions are discussed.
Results and analysis
Comparison of CdS and CdSe QD alkene photoisomerization
We first examined possible chalcogen radical reactivity at colloidal QD surfaces. Surface-trapped holes (i.e. sulfur-centered radicals) at ZnS and CdS sols have been previously proposed to mediate photocatalytic cis–trans isomerization of internal alkenes in methanol,15 but the effects of ligands or of quantum confinement in these reactions were not studied. Here, irradiation (λ = 370 nm) of a C6D6 suspension of oleate (OA)-capped zinc blende CdS QDs (d ∼ 2.7 nm) and trans-3-hexene (83 equiv. per QD) resulted in photoisomerization to the cis isomer, reaching a 0.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cis:trans ratio over 40 h of irradiation, as quantified through integration of the alkenyl 1H NMR resonances. Irradiation of a mixture of the same CdS QDs and cis-3-hexene also resulted in similar isomerization to trans-3-hexene over many hours. Fig. 1 plots the conversion of both isomers over time, showing the convergence toward a photostationary cis:trans mixture.
:1 cis:trans ratio over 40 h of irradiation, as quantified through integration of the alkenyl 1H NMR resonances. Irradiation of a mixture of the same CdS QDs and cis-3-hexene also resulted in similar isomerization to trans-3-hexene over many hours. Fig. 1 plots the conversion of both isomers over time, showing the convergence toward a photostationary cis:trans mixture.
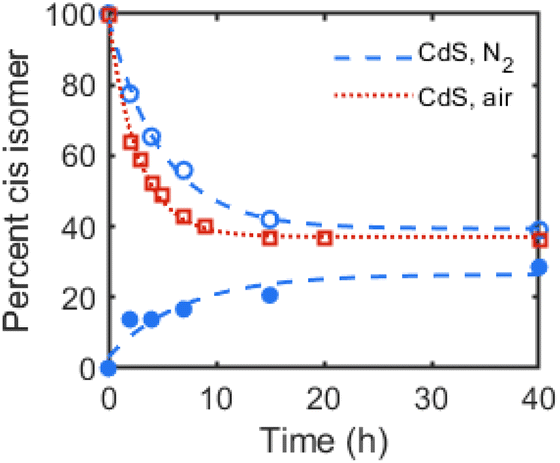 | ||
| Fig. 1 Cis–trans isomerization of 3-hexene in C6D6 with OA-capped CdS QDs (d ∼ 2.7 nm, 1 mol%) during irradiation (λ = 370 nm, 100 mW cm−2) plotted as percentage cis isomer over time. Open markers plot values starting from 100% trans-3-hexene at t = 0, and shaded markers plot values starting from 100% cis-3-hexene at t = 0. Dashed or dotted lines are exponential fits as guides to the eye. | ||
Table 1 summarizes this photocatalytic cis–trans isomerization under different conditions and with different catalysts. A thermal control experiment, in which the CdS/hexene mixture was heated in the dark, showed no detectable isomerization (Table 1, entry 2). Irradiation of the mixture with sub-band-gap light (λ > 515 nm, 100 mW cm−2) also resulted in no observed photoisomerization by 1H NMR spectroscopy (Fig. S14†). To test the involvement of the supporting ligands, the OA ligands were exchanged for octadecylphosphonate (ODPA) by treatment with octadecylphosphonic acid. Similar isomerization activity is observed for octadecylphosphonate (ODPA)-capped CdS QDs (Table 1, entry 3), suggesting that the double bond of the OA ligand is not involved in the reaction. Prolonged irradiation of the reaction mixture using these ODPA-capped CdS QDs showed additional alkenyl resonances, however, possibly indicating some additional double bond migration or other side reactions.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Ligand | d (nm) | t (h) | Conditionsa | % cisb |
|
a Reactions were performed in C6D6 with 1 mol% catalyst loading. Irradiated samples were photoexcited using a 370 nm Kessil lamp (100 mW cm−2).
b Average of duplicate runs. Reported as A/B, where A is the final % cis isomer starting from the trans isomer, while B is the final % cis value starting from the cis isomer.
c Reaction performed in 5:1 THF/C6D6.
d Starting from trans isomer only.
|
||||||
| 1 | CdS | OA | 2.7 | 40 | hν (370 nm) | 31/38 |
| 2 | CdS | OA | 2.7 | 40 | 100 °C | —/0 |
| 3c | CdS | ODPA | 2.7 | 15 | hν (370 nm) | 15/33 |
| 4 | CdS | OA | 3.5 | 40 | hν (370 nm) | 15d |
| 5 | CdS | OA | 3.8 | 40 | hν (370 nm) | 17d |
| 6 | CdSe | OA | 3.6 | 40 | hν (370 or 440 nm) | —/0 |
| 7 | PhSeSePh | — | — | 15 | hν (370 nm) | 15/18 |
| 8 | PhSSPh | — | — | 15 | hν (370 nm) | 19/18 |
To rule out isomerization facilitated by photoinduced charge transfer to the alkene (e.g. reduction to the radical anion), the photoisomerization reaction of cis-3-hexene with OA-capped CdS QDs as the photocatalyst was performed under air rather than under N2. Fig. 1 plots these data over time, in which faster isomerization to the trans isomer was observed under air than under N2. This result contrasts with those of catalytic semiconductor sols, in which photoisomerization of cis-2-octene in methanol under air was shown to inhibit alkene isomerization and was attributed to sulfide oxidation to sulfate, and catalyst degradation.15 While degradation of the CdS QDs is observed by absorption spectroscopy, the same inhibition of isomerization is not observed for our colloidal QD samples. Indeed, the faster rate of isomerization observed under air could be due to the photoinduced formation of hydroxyl or superoxide radicals, which has been previously observed for QD samples irradiated under air.19
Similar photoisomerization of trans-3-hexene was observed for OA-capped CdS QDs of different sizes (d ∼ 3.5, 3.8 nm, Table 1, entries 4 and 5), with no obvious dependence of isomerization activity upon size (or energy of the band gap). The addition of trans-3-hexene (100 equiv. per QD) to a hexanes solution of OA-capped CdS QDs (d ∼ 3.5 nm, 0.8 μM) also does not quench the photoluminescence (PL) emission (see ESI, Fig. S16†). Taken together, these data suggest that photoisomerization does not proceed by charge transfer or energy transfer to the alkene and support the previously proposed surface-radical-mediated mechanism.
Prolonged irradiation of both OA- and ODPA-capped CdS QDs in the presence of alkenes under N2 precipitates a grey solid after several hours of irradiation. While we were unable to characterize this grey solid by powder X-ray diffraction (XRD) due to the small quantities formed, elemental analysis by inductively-coupled plasma optical emission spectroscopy (ICP-OES) confirms the presence of cadmium. The absorption spectra of an irradiated hexanes suspension of OA-capped CdS QDs and cis-3-hexene (Fig. 2A, 80 equiv. per QD) shows slow decrease in the excitonic absorption band intensity (ca. 17% over 24 h) and a slight blue shift (24 meV). In the absence of alkene, bleaching (17%) is observed but no blue shift (Fig. 2B). This bleach is reversed upon exposure to air, and is consistent with previous reports of CdS QD photocharging in the absence of external reductants.20 These results are consistent with a QD-alkene interaction that is surface-mediated and that may result in QD etching over time.
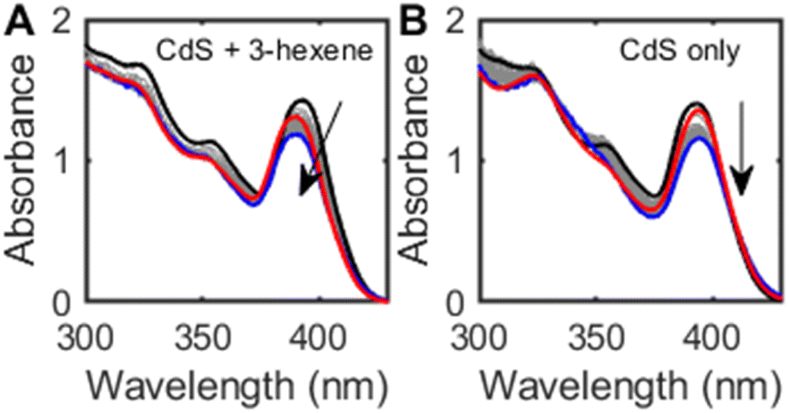 | ||
| Fig. 2 (A) Absorption spectra of a hexanes suspension of OA-capped CdS QDs (d ∼ 2.7 nm, 6.9 μM) and cis-3-hexene (80 equiv. per QD) before (black) and after (blue) irradiation under N2 atmosphere (λ = 370 nm, 24 h). The red trace shows the absorption spectrum of the sample after opening to air. (B) Absorption spectra of a hexanes suspension of CdS QDs under similar conditions, without added cis-3-hexene. | ||
In contrast, irradiation (λ = 440 or 370 nm) of C6D6 mixtures of CdSe QDs (d ∼ 3.8 nm) and either cis- or trans-3-hexene over many hours resulted in no observed cis–trans isomerization, as measured by 1H NMR spectroscopy. Irradiation of these CdSe QD mixtures under air formed new species with downfield 1H NMR resonances (δ ∼ 9–10 ppm). These could be formed upon oxidation of the alkenes to aldehyde-containing products, perhaps due to the photoinduced formation of superoxide or hydroxyl radicals, as has been previously observed for CdSe QDs under O2.19,21 These results suggest that (1) any Se-centered radicals formed from photoinduced hole trapping on CdSe do not appreciably add to alkenes or that (2) no such radicals are formed at all. As a control experiment, the phenylselenyl or phenylthiyl radical formed by photolysis of PhSeSePh or PhSSPh, respectively, (λ = 370 nm) resulted in isomerization of trans-3-hexene to cis-3-hexene over 15 h (Table 1, entries 7 and 8). We note, however, that the phenylthiyl-catalyzed isomerization occurs much more rapidly, consistent with the order of magnitude faster rates of thiyl radical addition to alkenes compared to selenyl radicals.22 This rate discrepancy may also account for the differences in photoisomerization activity for CdS vs. CdSe QDs. CdSe/CdS core–shell QDs prepared from these same CdSe QD cores showed no isomerization activity for trans-3-hexene under irradiation over 15 h. While this result may suggest that surface sulfur radicals are not necessarily involved as the catalytically active species, it could also be explained by inefficient surface hole trapping in this type I core–shell heterostructure.
Next, we studied the dependence of CdS-mediated photoisomerization of alkenes on surface ligand density. A single batch of oleate-capped CdS QDs (d ∼ 3.5 nm) was synthesized and either ligand-exchanged with decanethiol or treated with tetramethylethylenediamine (TMEDA) to remove surface-bound Cd(OA)2. The latter treatment is expected to expose additional surface sulfide sites. Ligand coverage of these QDs was quantified by 1H NMR spectroscopy (see Table S1†). Fig. 3 compares the results of photoisomerization of cis-3-hexene using these surface-treated CdS QDs. Faster isomerization was observed for QDs with lower ligand concentrations, consistent with the assignment of the catalytically-active species as surface sulfur radicals. We note that the CdS sample with the highest ligand density (d ∼ 3.5 nm, 105 OA/QD, 2.7 OA per nm2, Fig. 3) exhibited markedly slower photoisomerization rates than the CdS QDs used for the data shown in Fig. 1 (d ∼ 2.7 nm, 49 OA/QD, 2.1 OA per nm2). These data are therefore consistent with a strong dependence on “exposed surface sulfur sites,” although there may be additional batch-to-batch surface differences that are not accounted for.
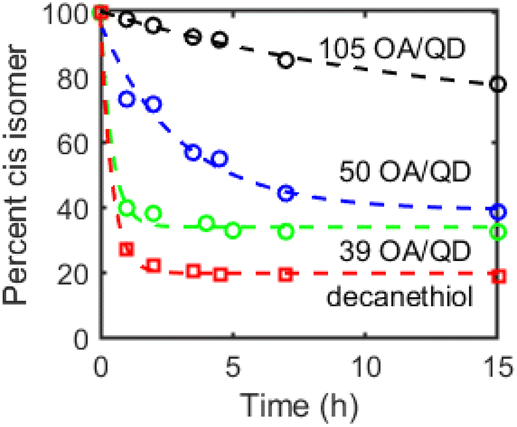 | ||
| Fig. 3 Cis–trans isomerization of cis-3-hexene in C6D6 with OA-capped CdS QDs (d ∼ 3.6 nm, 1 mol%) with 105 (black), 50 (blue), and 39 (red) OA/QD or of decanethiol-capped CdS QDs during irradiation (λ = 370 nm, 100 mW cm−2) plotted as percentage cis isomer over time over 15 h. Dashed lines are exponential fits included as guides to the eye. | ||
The decanethiol-capped QDs exhibited the fastest isomerization activity, possibly due to photooxidation of the coordinated thiol/thiolate ligands to thiyl radicals that can then react with the alkene substrates. Picosecond hole transfer to QD-surface-bound thiolate ligands has previously been reported.23,24 As such, it appears that the concentration of sulfur-centered radicals, whether on the QD surface or on supporting organic ligands, contributes to both the reaction rate as well as the cis–trans ratio of the photostationary state.
Photochemical spin trapping at QD surfaces
In the above studies, the evidence for radical reactivity is predicated on observation of intermolecular chemical reactions and does not conclusively demonstrate reactivity that is localized at the QD surface. However, typical spectroscopic methods of monitoring reactions like solution-phase NMR spectroscopy are challenging to use for these reactions because the NMR resonances of QD-bound functional groups are significantly broadened and can be difficult to observe.25 We hypothesized that the higher sensitivity of EPR spectroscopy may enable more in depth studies of surface bound species through the use of spin traps. While spin trap molecules have been previously used in photochemical experiments with QD samples, these experiments were used primarily in aqueous solutions to detect the formation of hydroxyl or superoxide radicals (or tetrahydrofuran-derived radicals).19,21,26 Other nitroxide radicals with donor moieties like amino groups or thiolate donors have been studied for PL quenching or for ligand dynamics.27,28 Here, we measure the formation and reactions of spin-trap-derived nitroxide radicals that are directly bound to the QD surface without the use of additional donor moieties.Under air-free conditions, a toluene suspension of OA-capped zinc blende CdSe QDs (d ∼ 3.6 nm) was treated with the spin trap 5,5-dimethyl-1-pyrroline N-oxide (DMPO, 1000 equiv. per QD) and photoirradiated for 12 h (λ = 440 nm, 200 mW cm−2). Fig. 4A shows the EPR spectrum of the reaction mixture, which displays an isotropic three-line pattern consistent with a nitroxide radical. Similar spectra are observed for irradiated CdSe QD/DMPO mixtures in hexanes, benzene, and THF, indicating little solvent dependence. This nitroxide radical species (1) can be separated from the QDs by gel permeation chromatography (GPC)29 or by precipitation/centrifugation, indicating that it is a molecular byproduct that is not bound to the QD surface. Interestingly, the EPR signal shows no hyperfine coupling to a hydrogen atom in the β-position of the heterocycle. Similar EPR signals have been previously assigned to structural rearrangement of the DMPO-derived nitroxide radical, including ring-opening reactions, oxidation, or dimerization.30 While there is a background reaction when a solution of DMPO in THF is irradiated (λ = 440 nm) in the absence of QDs, the resulting nitroxide radical product exhibits a different EPR spectrum from that of 1 (see ESI, Fig. S24†). This EPR signal is also distinct from those of DMPO adducts of Cd(OA)2, NaOA, Se, and Ph2Se2 (Fig. S26†).
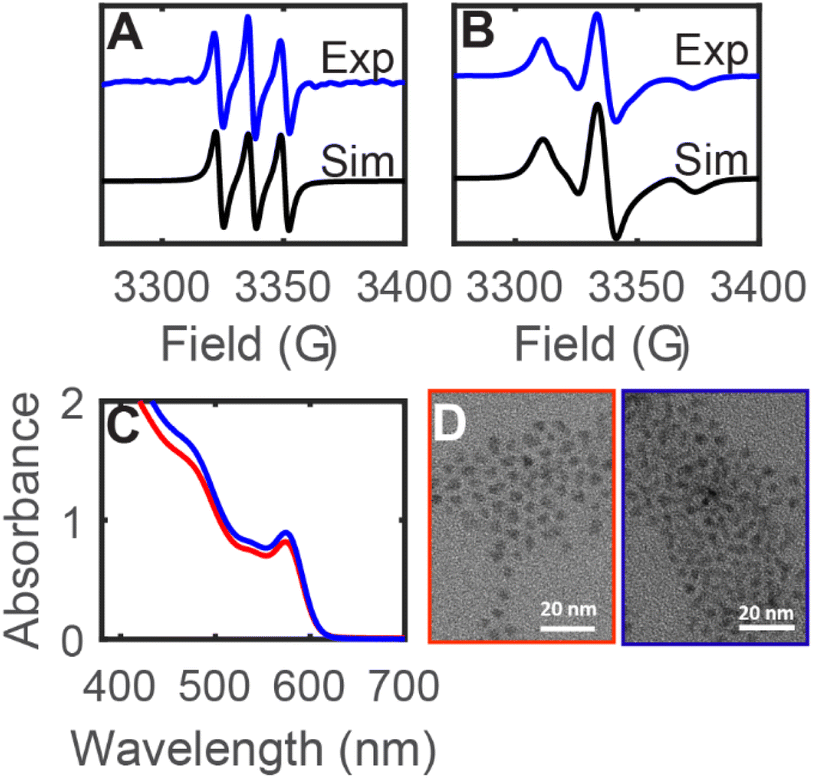 | ||
| Fig. 4 (A and B) Room temperature X-band EPR spectra of a toluene mixture of CdSe QDs (d ∼ 3.6 nm) and DMPO (1000 equiv. per QD) after photoirradiation (λ = 440 nm, 200 mW cm−2, 16 h). (A) Spectrum of the crude reaction mixture (black) and simulation (blue, g = 2.0046, a(N) = 13.5 G), showing the molecular byproduct 1. (B) Spectrum after purification by GPC (black) and simulation (blue, gx = 2.0091, gy = 2.0038, gz = 2.0005, a(N) = 5.4, 3.2, 36.4 G, tcorr = 6.9 ns), showing the QD-bound nitroxide radical (2CdSe). (C) Absorption spectra of toluene solutions of CdSe QDs (d ∼ 3.6 nm) before (red) and after (blue) treatment with DMPO (1000 equiv. per QD). (D) TEM images of CdSe QDs (d ∼ 3.6 nm) before (red) and after (blue) treatment with DMPO, indicating no size differences. | ||
Fig. 4B shows the EPR spectrum of the fraction containing the CdSe QDs after GPC purification of the DMPO/QD reaction mixture (2CdSe). The QD sample 2CdSe shows a broadened asymmetric EPR signal that is consistent with a nitroxide radical in the slow-motion regime, that is, when the rotational motion of the nitroxide radical is on a similar timescale as that of the EPR measurement.31 Similar signals have previously been reported for nitroxide radicals immobilized at polymers or at Au nanoparticle surfaces.32,33 These spectra were simulated using a rotational Brownian diffusion model with long diffusional correlation times (τc ∼ 10−9 s).34 For these reasons, we assign 2CdSe as a nitroxide radical species covalently bound to the QD surface. Due to the broadening and anisotropy of the spectrum, however, only hyperfine coupling to the nitrogen center was used for simulation, and we were unable to simulate additional hyperfine coupling to other nuclei using the EPR spectrum. Absorption spectroscopy and transmission electron microscopy (TEM) show that the QD sample does not undergo any size or morphology changes during this treatment (Fig. 4C and D).
Previous reports of surface-bound nitroxide radicals coordinated to CdSe QD surfaces via amine or thiolate donors have exhibited isotropic EPR signals in the fast rotation regime, perhaps indicating that the surface-bound radical moieties undergo fast exchange with solution-phase nitroxide radicals or due to a greater distance from the QD surface.27,28,35 The anisotropic signal observed here indicates that the nitroxide radicals of 2CdSe are more tightly bound to the QD surface and are an indication of surface-atom-centered reactivity.
Scheme 2 shows a possible route for the photochemical formation of 2CdSe. First, a photogenerated hole is trapped to the surface, forming a surface radical. In previous computational studies, such hole trap sites have been proposed to be two-coordinate surface selenium atoms.36 This selenium-centered surface radical then adds to the spin trap, forming a surface-bound nitroxide radical, 2CdSe. This proposed scheme places an electron in the conduction band that could then decay via slow trapping.37,38 Indeed, the first excitonic feature of the absorption spectrum of a toluene mixture of CdSe QDs and DMPO shows slight bleaching (ca. 8%) upon irradiation with a blue LED, consistent with partial occupation of the 1Se orbital (see Fig. S31†); this bleach reverses upon exposure to air and oxidation. Irradiation of CdSe/DMPO mixtures under air similarly forms 2CdSe, as measured by EPR spectroscopy after GPC purification.
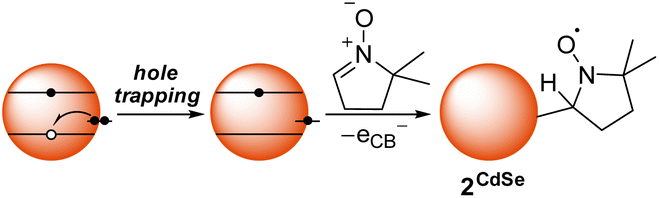 | ||
| Scheme 2 Proposed radical addition pathway of a photogenerated surface-trapped hole to DMPO to form 2CdSe. | ||
We considered the alternative possibility that the reaction could proceed first by photochemical reduction or oxidation of DMPO, followed by addition to the surface. While we cannot entirely rule out this pathway, we disfavor this possibility because DMPO has a relatively wide electrochemical stability window (Ered = –2.35 V vs. SCE, Eox = 1.63 V vs. SCE in MeCN).39 This electrochemical window is wider than the optical band gap of the CdSe QDs (ca. 2.1 eV), although the precise electrochemical potentials of the band edges for the QD samples used for these experiments have not been measured. Analogous surface-bound nitroxide radicals are also observed by EPR spectroscopy when using the spin trap phenyl tert-butylnitrone (PBN) in place of DMPO (see Fig. S27†).
Spin quantification of the surface-bound nitroxide radical of samples of 2CdSe showed low densities, on the order of 0.01–0.1 spins per QD. These low spin concentrations could be due to multiple factors. First, the quantum yield of spin trapping is likely to be low due to competing recombination or detrapping processes, and dictated by access of the spin trap molecule to the CdSe QD surface. Second, although the EPR signal of solutions of 2CdSe persists for days under inert atmosphere at room temperature, photoirradiation results in a decrease of the nitroxide EPR signal within hours (Fig. S29†). This decay may arise from photoinduced oxidation of the surface-bound radical by a photogenerated hole from CdSe. From these processes, the surface-trapped nitroxide radical species could then be converted to an EPR-silent diamagnetic product. Nitroxide radicals, for example of trapped thiyl radicals, can be oxidized to the oxoammonium cation or can undergo disproportionation or other decomposition processes.30,40,41 Due to this competing photooxidation of 2CdSe, accumulation of the surface-bound nitroxide radical under irradiation is likely to be low.
To study the fate of DMPO, the reactions were monitored by 1H NMR spectroscopy. Fig. 5A shows the 1H NMR spectra of C6D6 mixture of OA-capped CdSe QDs (d ∼ 3.6 nm), DMPO (300 equiv. per QD), and a 1,3,5-trimethoxybenzene internal standard during irradiation (λ = 440 nm, 200 mW cm−2). Upon addition of DMPO to the CdSe QDs, the resonance of the alkenyl C–H proton shifts and broadens (δ ∼ 6.2 ppm). The other 1H NMR resonances corresponding to DMPO are also shifted and broadened, indicating some fast exchange reaction with the QDs, possibly related to association of DMPO with the QD surface.
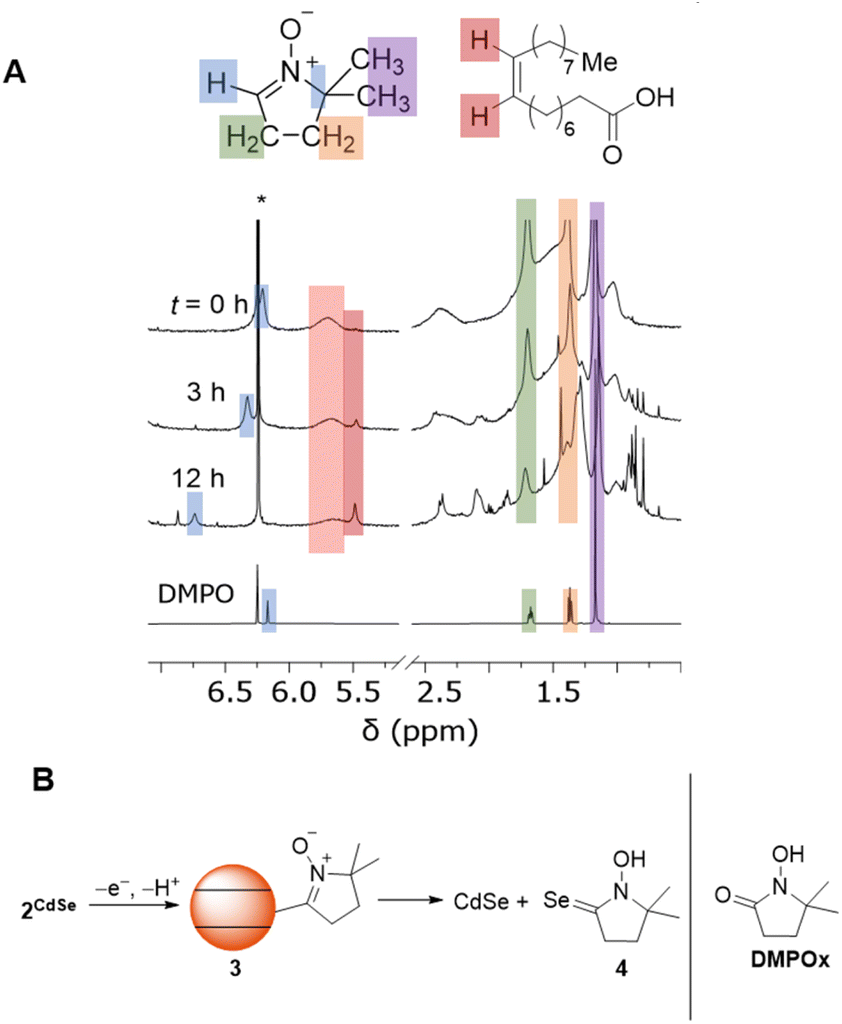 | ||
| Fig. 5 (A) 1H NMR spectra of a C6D6 mixture of zinc blende OA-capped CdSe QDs (d ∼ 3.6 nm) and DMPO (300 equiv. per QD) during irradiation (λ = 440 nm, 200 mW cm−2). (B) Proposed oxidative formation of DMPO-derived diamagnetic products from 2CdSe. | ||
Upon irradiation at room temperature (λ = 440 nm), the alkenyl proton resonance shifts downfield and decreases in intensity over 24 h. The resonances corresponding to the methylene and methyl moieties of DMPO also decrease, indicating conversion to new DMPO-derived products. The 1H NMR resonances of these species are broadened and obscured by those of the oleate ligands, so we are unable to identify whether these species are coordinated to the QD or not during the reaction. Similar spectra are observed upon irradiation of a C6D6 mixture of OA-capped CdS QDs and DMPO (λ = 370 nm) or upon irradiation of a C6D6/THF mixture of ODPA-capped CdSe QDs and DMPO (λ = 440 nm). Irradiation of a C6D6 solution of DMPO under the same conditions in the absence of QDs does not result in similar conversion of DMPO to other products.
We currently propose that oxidation and deprotonation of the surface-bound nitroxide radical of 2CdSe photocatalytically forms diamagnetic DMPO-derived products (for example, 3 or 4, Fig. 5B), but we cannot presently rule out secondary pathways in which free radical species are formed by QD irradiation that then further react with DMPO. For the reaction mixtures containing OA-capped QDs (both CdSe and CdS), a sharper resonance (δ ∼ 5.5 ppm) increases in intensity, corresponding to an unbound oleate-containing species. Fig. 6 tracks these concentrations over the course of the experiment. As this rise corresponds to the decrease in the intensity of the DMPO-derived alkenyl proton, these data are consistent with deprotonation of the DMPO β-H by OA− anion to form oleic acid. Additionally, the 1H NMR spectrum of a CDCl3 solution of the DMPO-derived product after irradiation is complete after precipitation and removal of the QDs shows two triplet resonances corresponding to the methylene moieties of the heterocycle but no resonance corresponding to the β-H. The spectrum is similar to that of 1-hydroxy-5,5-dimethylpyrrolidinone (DMPOx, Fig. 5B), a previously characterized product of hydroxyl radical addition to DMPO that results in similar removal of the β-H.42 This deprotonation reaction is also consistent with the observed three-line hyperfine coupling pattern of 1.
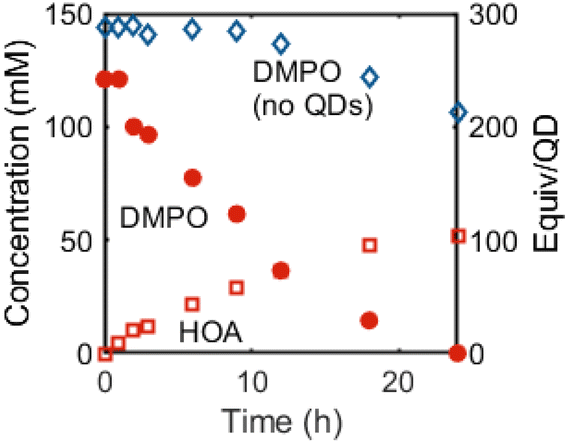 | ||
| Fig. 6 Concentrations of DMPO (red circles) and oleic acid (red squares) in a C6D6 mixture of OA-capped CdSe QDs (d ∼ 3.6 nm, 0.5 mM) and DMPO (300 equiv. per QD) over time during irradiation (λ = 440 nm, 200 mW cm−2). The concentration of a C6D6 solution of DMPO during irradiation (λ = 370 nm, 100 mW cm−2) in the absence of QDs is included for comparison (blue diamonds). | ||
Nucleophilic reactions of surface chalcogenides
Next, we studied the nucleophilic reactivity of surface chalcogenides in CdSe and CdS QDs. As discussed above, it has previously been demonstrated that CdSe QDs can undergo selenium-centered alkylation upon treatment with alkyl halides, resulting in the formation of a C–Se bond and the observation of alkyl diselenide compounds by GC-MS.11 Here, heating a C6D6 mixture of OA-capped CdSe QDs (d ∼ 3.6 nm) and benzyl bromide (BnBr, 30–50 equiv. per QD) at 90 °C in the dark resulted in consumption of BnBr (1H NMR spectroscopy) over several hours and formation of new benzyl-containing products that were identified as benzyl oleate (BnOA), dibenzyl selenide (Bn2Se), and dibenzyl diselenide (Bn2Se2) in a 1:0.3:0.05 ratio, respectively (Fig. 7). While these data show that carboxylate alkylation is the major pathway, selenide alkylation is competitive. OA-capped CdS QDs (d ∼ 3.5 nm) similarly undergo alkylation to form Bn2S2 and Bn2S, along with BnOA.
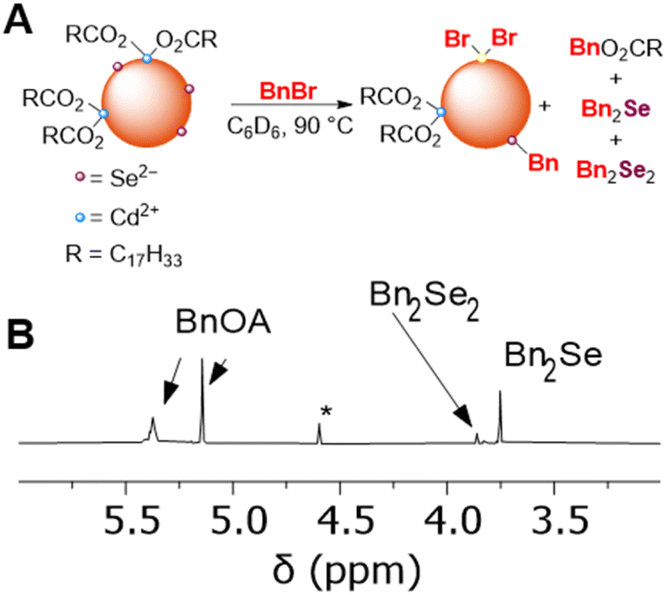 | ||
| Fig. 7 (A) Scheme showing that benzylation of carboxylate ligands and of surface selenide ions of CdSe QDs upon treatment with benzyl bromide. (B) Truncated 1H NMR spectrum showing benzylic resonances of a CDCl3 solution of molecular products formed upon heating a benzene mixture of OA-capped CdSe QDs (d ∼ 3.6 nm, 0.5 mM) and BnBr (50 equiv. per QD) at 90 °C for 15 h. (*) indicates a Bn2O impurity. | ||
The formation of BnOA likely occurs by dissociation of surface-bound OA− ligands followed by alkylation with benzyl bromide. This reaction appears to be facile – BnOA is observed by 1H NMR spectroscopy even at room temperature upon addition of BnBr to OA-capped CdSe QDs. Selenide alkylation to form Bn2Se and Bn2Se2 requires higher temperatures of activation (>60 °C) and could be envisioned to proceed by two possible pathways: first, Se2− could dissociate from the QD surface to then participate in alkylation to form the benzylselenolate anion (BnSe−) that could then further undergo benzylation with a second equivalent of BnBr to form Bn2Se or could undergo oxidation to form Bn2Se2. Alternatively, benzylation could proceed directly at the QD surface to form QD-bound BnSe− that then dissociates from the surface. While we are unable to distinguish these possibilities from our experiments, we consider the second pathway more likely, as free S2− or Se2− anions are relatively basic and would have low equilibrium dissociation constants.
Integration of the 1H NMR resonances of OA-capped CdSe QDs treated with BnBr (50 equiv. per QD) against an internal standard was performed to quantify the nucleophilic selenide sites at the CdSe QD. The 1H NMR spectrum shows that the molecular benzyl-containing byproducts (BnOA, Bn2Se, etc.) account for only of ca. 60% BnBr added; the remainder may be QD-bound, with broadened 1H NMR signals. Additionally, absorption spectroscopy of the same mixture shows minimal blue-shifting of the excitonic absorption, meaning that not much etching is occurring (as might be expected if significant formation of Bn2Se occurs). Brutchey and co-workers have previously prepared CdSe QDs supported by BnSeH or PhSeH ligands by the reductive addition of the corresponding organic diselenide compounds to stearate-capped CdSe QDs in the presence of Ph2PH.43 Unlike our samples, the absorption spectra of those samples displayed a red shift of the first exciton peak due to QD growth upon addition of the organoselenol ligands.
While both sulfide and selenide anions are nucleophilic, as shown by their reactions with BnBr, selenides should be expected to be more nucleophilic than sulfide due to their greater size and polarizability. To compare the reactivities of the surface chalcogenide anions in CdS and CdSe QDs, we investigated CdS or CdSe QD-mediated scrambling of alkyl disulfide compounds. The S–S bonds of disulfides are known to cleave upon the addition of soft nucleophiles like thiolates and phosphines,44 and QDs have been previously demonstrated to promote alkyl disulfide cleavage.28
Table 2 reports the yields of the mixed disulfide product, BuSSBn, formed from 1:1 mixtures of BuSSBu and BnSSBn in C6D6 after heating or irradiation with or without added QD catalyst, as measured by 1H NMR spectroscopy (see Fig. S21†). In these experiments, while heating the disulfide mixtures in the dark without QDs did not form appreciable amounts of the BuSSBn (Table 2, entry 1), heating the mixture with added OA-capped CdSe QDs (d ∼ 3.6 nm, 1 mol%) formed the mixed disulfide product (Table 2, entry 6). In contrast, heating the disulfide mixture with OA-capped CdS QDs (d ∼ 2.7 nm, 1 mol%) resulted in minimal disulfide exchange over 17 h (Table 2, entry 4). These results are consistent with higher selenide nucleophilicity compared to sulfide.
|
|
|||
|---|---|---|---|
| Entry | Cat. | Conditions | Yielda (%) |
|
a Reactions were performed in C6D6 and heated or irradiated for 17 h. Yields were calculated by integrating the benzylic 1H resonance of BnSSBn (δ 3.34 ppm, 4H) against that of BnSSBu (δ 3.60 ppm, 2H), where a statistical 1:2:1 BnSSBn/BuSSBn/BuSSBu product distribution would be achieved at a yield of 33% of the mixed product.
b Other unidentified products were observed by 1H NMR spectroscopy.
c Complete consumption of BnSSBn and BuSSBu was observed with the formation of additional unidentified products.
|
|||
| 1 | None | Dark, 100 °C | 0 |
| 2 | None | 370 nm | 33b |
| 3 | None | 440 nm | 3.5 |
| 4 | CdS QDs | Dark, 100 °C | <5 |
| 5 | CdS QDs | 370 nm | N/Ac |
| 6 | CdSe QDs | Dark, 100 °C | 26 |
| 7 | CdSe QDs | 440 nm | 23 |
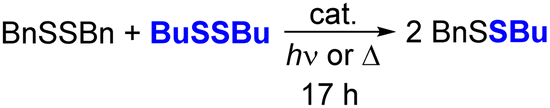
We note that disulfide exchange upon photoexcitation proceeds differently. Since direct photoexcitation of disulfides has been shown to homolytically cleave the disulfide S–S bond, resulting in disulfide exchange or other radical reactions,45,46 irradiation of the mixture of BuSSBu and BnSSBn (λ = 370 nm) forms BuSSBn, along with other unidentified products that may arise from H-atom abstraction or other radical processes (Table 2, entry 2). Irradiation with lower energy light (λ = 440 nm) results in minimal scrambling, as neither disulfide strongly absorbs visible light (Table 2, entry 3). However, irradiation of a mixture of the disulfide compounds and CdSe QDs (λ = 440 nm) results in disulfide scrambling to form the mixed disulfide product (Table 2, entry 7). While we cannot rule out nucleophile-induced disulfide exchange due to some slight heating of the sample, this reaction could proceed from reduction of the disulfide compounds by photoexcited conduction band electrons resulting in disulfide radical anions that dissociate to form thiolate anions that are capable of catalyzing disulfide exchange.
Thermal “spin trapping” at QD surfaces
We hypothesized that DMPO could also be used to test nucleophilic 2-electron activity at QD surfaces, as DMPO has been previously demonstrated to undergo nucleophilic addition at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond, followed by oxidation to form a nitroxide radical (the Forrester–Hepburn mechanism).47 This pathway can compete with and obscure radical trapping reactions. Scheme 3 shows the proposed nucleophilic addition pathway at QD surfaces. Here, DMPO undergoes nucleophilic addition by a QD surface anion (followed by protonation from residual acid) to form a bound hydroxylamine species (5) that can then be oxidized to 2CdSe. In this manner, this route could enable the use of EPR spectroscopy to observe a two-electron reaction with high sensitivity.
N double bond, followed by oxidation to form a nitroxide radical (the Forrester–Hepburn mechanism).47 This pathway can compete with and obscure radical trapping reactions. Scheme 3 shows the proposed nucleophilic addition pathway at QD surfaces. Here, DMPO undergoes nucleophilic addition by a QD surface anion (followed by protonation from residual acid) to form a bound hydroxylamine species (5) that can then be oxidized to 2CdSe. In this manner, this route could enable the use of EPR spectroscopy to observe a two-electron reaction with high sensitivity.
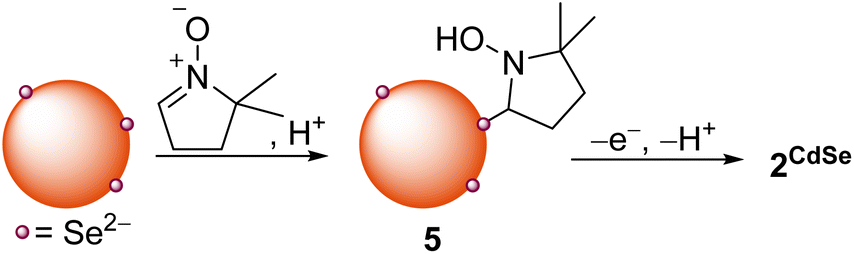 | ||
| Scheme 3 Nucleophilic addition of QD surface anions to DMPO forms a hydroxylamine product (5) that can undergo oxidation to the bound nitroxide radical. | ||
In the absence of light, heating a toluene mixture of OA-capped CdSe QDs and DMPO (1000 equiv. per QD) under N2 at 90–100 °C for 12 h forms the same surface-bound nitroxide radical species (2CdSe), as measured by EPR spectroscopy after GPC purification. This thermal reaction proceeds without the generation of photoexcited carriers. Unlike the photochemical reaction between CdSe QDs and DMPO, the thermal reaction is not catalytic. Heating a C6D6 mixture of CdSe QDs and DMPO (500 equiv. per QD) over multiple days does not result in appreciable decay of the DMPO-derived 1H resonances. Similarly, heating a solution of 2CdSe over 12 h does not result in a decrease in the EPR signal corresponding to the nitroxide radical. This suggests that while nitroxide radical formation can be thermally mediated, the resulting oxidation/deprotonation steps to form 1 are likely photoinduced.
Spin quantification of thermally generated 2CdSe is still low, on the order of 0.1 spins per QD. To study whether there are greater quantities of EPR-silent 5, as suggested by the proposed mechanism in Scheme 3, we performed two experiments. First, a C6D6 suspension of OA-capped CdSe QDs (d ∼ 3.8 nm) was treated with DMPO (200 equiv. per QD). Heating this sample at 90 °C in the dark for 24 h under N2 did not show any consumption of DMPO by 1H NMR spectroscopy. Addition of benzoic acid as a proton source followed by heating in the dark resulted in complete consumption of DMPO within hours. The QD sample was purified by GPC. The EPR spectrum of this sample showed minimal nitroxide signal, suggesting that any DMPO-derived QD-bound products exist as an EPR-silent form, possibly as 5. Treatment of this sample with the oxidant ferrocenium triflate (FcOTf) resulted in the formation of free HOA (1H NMR spectroscopy) as well as an increase in the 2CdSe EPR signal, consistent with the mechanism in Scheme 3.
We considered the possibility of an alternative pathway that proceeds via thermal homolytic cleavage of diselenide or disulfide moieties at the QD surfaces to form the corresponding sulfur- and selenium-centered radicals that then add to DMPO. Such oxidized chalcogen species have previously been proposed as reducible species in as-prepared CdSe or related QDs.11 Similarly, surface selenide oxidation upon treatment with chemical oxidants has previously been reported to form interparticle Se–Se bonds.10 However, Se–Se and S–S bond cleavage would be expected to require higher temperatures than those applied here (ca. 80–100 °C). For example, homolytic cleavage of the S–S bond in the S8 allotrope of elemental sulfur to form the corresponding biradical has been demonstrated to occur at temperatures higher than 430 K.48 Additionally, this pathway would not be expected to be affected by addition of acid.
This thermal method of synthesizing 2CdSe was applied toward zinc blende CdSe QDs of different diameters (3.0–6.1 nm). For each sample, toluene suspensions of the CdSe QDs were treated with DMPO (1000 equiv. per QD), and the mixture was heated at 100 °C for 12 h. The reaction mixtures were purified by GPC, and the resulting EPR spectra are shown in Fig. 8A. While these spectra all show the broadened signal corresponding to a nitroxide radical in the slow rotation regime, as discussed above, the features shift and sharpen with increasing QD size. The spectrum of 2CdSe for the largest size of CdSe QDs (d ∼ 6.1 nm) also exhibits more features; this may indicate multiple EPR-active nitroxide components that are bound to the QD surface.
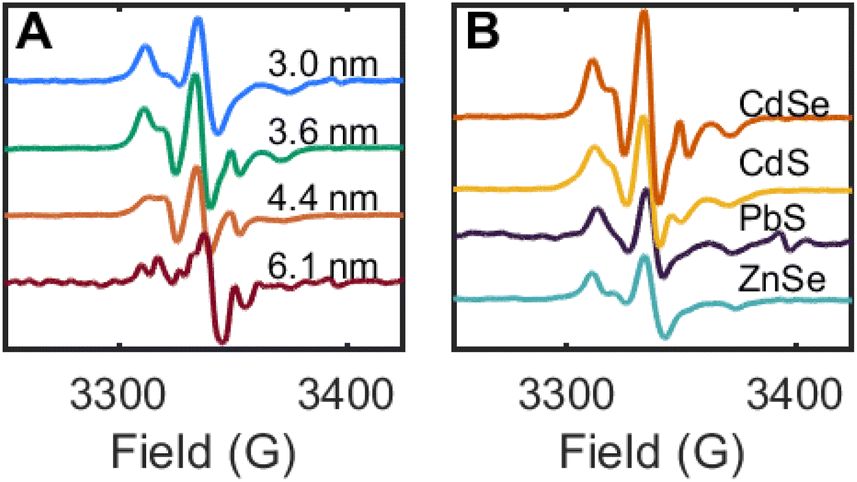 | ||
| Fig. 8 (A) Room temperature X-band EPR spectra of toluene suspensions of CdSe QDs of different sizes after thermal treatment with DMPO (1000 equiv. per QD) and GPC purification. (B) Room temperature X-band EPR spectra of toluene suspensions of different QD materials after thermal treatment with DMPO and GPC purification. | ||
This spin trapping reaction and formation of surface-bound nitroxide radicals is readily reproduced for other QD materials of different band edge potentials, including CdS and ZnSe (both photochemical and thermal), and PbS QDs (thermal only). In each of these cases, the QD samples were purified by GPC and show similar EPR signals corresponding to surface-bound nitroxide radicals in the slow rotation regime (Fig. 8B). These data further support the assignment of chalcogenide-centered DMPO reactions in these experiments.
Discussion
Implications for nucleophilic surface chalcogenides
The observation that heating different QDs of different band gaps and structures (e.g. zinc blende and rock salt lattices) with DMPO all form the same QD-bound product (2) suggests that broadly speaking, surface chalcogenides of different materials and lattices can perform similar nucleophilic addition reactions. This is important, particularly as much current research is focused on the exploration of new chalcogenide-based nanomaterials, including ternary lattices, chalcogenide-derived perovskites, etc.49 These reactions could present a general way of changing the surface chemistry of new QD materials, even for those supported by different types of ligands. For example, alkylation of S2− terminated QDs using long-chain alkyl halides should form the corresponding thiolate-terminated QDs, perhaps presenting a way to readily control QD solubility and hydrophobicity.The nucleophilic reactions described above occur at relatively low temperatures (80–100 °C). As such, nucleophilic substitution/addition may be competitive with photoinduced processes for reactions currently considered to be photocatalytic. For example, a recent report of CdSe-photocatalyzed aldehyde olefination with benzyl bromide may undergo selenide-centered benzylation in addition to the proposed photoinduced reaction steps.50 These would be particularly important in reactions illuminated under high flux (with localized heating) or for reactions with low quantum yields. Such reactions could result in lower selectivity or lower product conversion or may even, in some cases, be the operative pathway for the photocatalytic products.
The examples discussed above, which include addition to DMPO, substitution of benzyl bromide, and thermal catalysis of alkyl disulfide exchange, demonstrate that both sulfide- and selenide-derived QDs can perform nucleophilic additions. The higher activity toward disulfide exchange of the CdSe QDs indicates that selenides are more nucleophilic than sulfides, as expected, due to their softness and higher polarizability.51,52 Nevertheless, the reactions described here demonstrate broad application and high nucleophilicity for both anions. While for the OA-capped CdS and CdSe QDs, alkylation of the carboxylate ligands was competitive, this may still indicate a higher than expected surface reactivity due to (1) the carboxylate ligands should be more accessible and (2) higher in number, due to typical cadmium-enriched QD surfaces.53 We note that phosphonate-capped QDs may not exhibit the same alkylation chemistry due to stronger binding to cadmium, supporting a dissociative mechanism for ligand alkylation.
EPR spectroscopy and spin trapping as a tool for studying QD surfaces
The high sensitivity of EPR spectroscopy makes it an attractive tool for studying species that may be at low concentrations at QD surfaces. From the above results, we have demonstrated that it is possible (1) to quantify surface-bound radicals, (2) to distinguish the resulting surface-bound nitroxide radicals from unbound, molecular products by the differences in the EPR spectral features. This method may present a useful way to monitor intramolecular and intermolecular reactions that may both occur under photocatalytic conditions.This method yet presents some limitations, however. As discussed above, in our experiments we routinely observed low concentrations of the surface-bound nitroxide radicals, and demonstrated the formation of other diamagnetic DMPO-derived products that were formed by photooxidation and proton-transfer events. While protonation/deprotonation is readily explained by reactions with OA− and residual oleic acid that is present in the QD sample and that can act as a sort of pH buffer, we have not identified any additional external oxidants or reductants in these samples. However, these results may point to an additional role of other redox-active surface trap states that may serve as the final electron sink in these transformations. Future work could include studying these transformations in the presence of redox buffers.54
Conclusions
The results above demonstrate that the nature of the QD surface and its atoms can be an important consideration in designing photocatalytic reactions. The nucleophilicity of the ligands and surface chalcogenides, as well as possible radical reactions with substrates or other reaction components should be taken into account, as these reactions can occur under conditions that are relevant to catalysis. However, this reactivity may also suggest possibilities for the design of QD-photocatalyzed reactions that operate by different mechanisms and form different products compared to more commonly-used outer-sphere photosensitizers. The use of spin trap molecules and EPR spectroscopy as a way to interrogate such reactions is also expected to be a useful tool that can be applied in tandem with other, less sensitive methods such as NMR and IR spectroscopy.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
C. J. A., M. D., K. V. P., and E. Y. T. conceived the research and designed the experiments. C. J. A., K. V. P., and M. D. synthesized QDs. C. J. A., K. V. P., and E. Y. T. carried out spin trapping experiments and EPR and NMR spectroscopy. M. D. carried out photoisomerization experiments. All authors analyzed data and participated in writing the manuscript. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF (CHE-2154948). Financial support from the German Academic Exchange Service (fellowship for C. J. A. through the ISAP program) is gratefully acknowledged. We thank Maksym Zhukovskyi for assistance with TEM imaging.Notes and references
- L. Amirav and A. P. Alivisatos, J. Phys. Chem. Lett., 2010, 1, 1051–1054 CrossRef CAS.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters, L. C. Seefeldt and P. W. King, Science, 2016, 352, 448–450 CrossRef CAS PubMed.
- H. B. Yang, J. Miao, S.-F. Hung, F. Huo, H. M. Chen and B. Liu, ACS Nano, 2014, 8, 10403–10413 CrossRef CAS PubMed.
- J. A. Caputo, L. C. Frenette, N. Zhao, K. L. Sowers, T. D. Krauss and D. J. Weix, J. Am. Chem. Soc., 2017, 139, 4250–4253 CrossRef CAS PubMed.
- Z. Zhang, K. Edme, S. Lian and E. A. Weiss, J. Am. Chem. Soc., 2017, 139, 4246–4249 CrossRef CAS PubMed.
- A. B. Nepomnyashchii, R. D. Harris and E. A. Weiss, Anal. Chem., 2016, 88, 3310–3316 CrossRef CAS PubMed.
- F. Y. Dou, S. M. Harvey, K. G. Mason, M. K. Homer, D. R. Gamelin and B. M. Cossairt, J. Chem. Phys., 2023, 158, 184705 CrossRef CAS PubMed.
- K. V. Prather, J. T. Stoffel and E. Y. Tsui, Chem. Mater., 2023, 35, 3386–3403 CrossRef CAS.
- M. K. Homer, D.-Y. Kuo, F. Y. Dou and B. M. Cossairt, J. Am. Chem. Soc., 2022, 144, 14226–14234 CrossRef CAS PubMed.
- I. R. Pala, I. U. Arachchige, D. G. Georgiev and S. L. Brock, Angew. Chem., Int. Ed., 2010, 49, 3661–3665 CrossRef CAS PubMed.
- E. Y. Tsui, K. H. Hartstein and D. R. Gamelin, J. Am. Chem. Soc., 2016, 138, 11105–11108 CrossRef CAS PubMed.
- M. D. Reichert, C.-C. Lin and J. Vela, Chem. Mater., 2014, 26, 3900–3908 CrossRef CAS.
- F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef PubMed.
- S. Nehzati, N. V. Dolgova, D. Sokaras, T. Kroll, J. J. H. Cotelesage, I. J. Pickering and G. N. George, Inorg. Chem., 2018, 57, 10867–10872 CrossRef CAS PubMed.
- S. Yanagida, K. Mizumoto and C. Pac, J. Am. Chem. Soc., 1986, 108, 647–654 CrossRef CAS.
- N. C. Strandwitz, A. Khan, S. W. Boettcher, A. A. Mikhailovsky, C. J. Hawker, T.-Q. Nguyen and G. D. Stucky, J. Am. Chem. Soc., 2008, 130, 8280–8288 CrossRef CAS PubMed.
- C. L. Hartley and J. L. Dempsey, Chem. Mater., 2021, 33, 2655–2665 CrossRef CAS.
- C. Y. Dones Lassalle, J. E. Kelm and J. L. Dempsey, Acc. Chem. Res., 2023, 56, 1744–1755 CrossRef CAS PubMed.
- B. I. Ipe, M. Lehnig and C. M. Niemeyer, Small, 2005, 1, 706–709 CrossRef CAS PubMed.
- K. E. Shulenberger, H. R. Keller, L. M. Pellows, N. L. Brown and G. Dukovic, J. Phys. Chem. C, 2021, 125, 22650–22659 CrossRef CAS.
- Z. Hu, S. Liu, H. Qin, J. Zhou and X. Peng, J. Am. Chem. Soc., 2020, 142, 4254–4264 CrossRef CAS PubMed.
- O. Ito, J. Am. Chem. Soc., 1983, 105, 850–853 CrossRef CAS.
- M. Abdellah, S. Zhang, M. Wang and L. Hammarström, ACS Energy Lett., 2017, 2, 2576–2580 CrossRef CAS.
- L. Lystrom, A. Roberts, N. Dandu and S. Kilina, Chem. Mater., 2021, 33, 892–901 CrossRef CAS.
- Z. Hens and J. C. Martins, Chem. Mater., 2013, 25, 1211–1221 CrossRef CAS.
- J. Qiao, Z.-Q. Song, C. Huang, R.-N. Ci, Z. Liu, B. Chen, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2021, 60, 27201–27205 CrossRef CAS PubMed.
- P. Dutta, Y. Tang, C. Mi, M. Saniepay, J. A. McGuire and R. Beaulac, J. Chem. Phys., 2019, 151, 174706 CrossRef PubMed.
- P. S. Billone, L. Maretti, V. Maurel and J. C. Scaiano, J. Am. Chem. Soc., 2007, 129, 14150–14151 CrossRef CAS PubMed.
- Y. Shen, M. Y. Gee, R. Tan, P. J. Pellechia and A. B. Greytak, Chem. Mater., 2013, 25, 2838–2848 CrossRef CAS.
- K. Makino, A. Hagi, H. Ide, A. Murakami and M. Nishi, Can. J. Chem., 1992, 70, 2818–2827 CrossRef CAS.
- K. A. Earle, D. E. Budil and J. H. Freed, J. Phys. Chem., 1993, 97, 13289–13297 CrossRef CAS.
- V. Chechik, H. J. Wellsted, A. Korte, B. C. Gilbert, H. Caldararu, P. Ionita and A. Caragheorgheopol, Faraday Discuss., 2004, 125, 279–291 RSC.
- P. Ionita, A. Caragheorgheopol, B. C. Gilbert and V. Chechik, J. Phys. Chem. B, 2005, 109, 3734–3742 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- C. Tansakul, E. Lilie, E. D. Walter, F. Rivera III, A. Wolcott, J. Z. Zhang, G. L. Millhauser and R. Braslau, J. Phys. Chem. C, 2010, 114, 7793–7805 CrossRef CAS PubMed.
- A. J. Houtepen, Z. Hens, J. S. Owen and I. Infante, Chem. Mater., 2017, 29, 752–761 CrossRef CAS.
- J. D. Rinehart, A. M. Schimpf, A. L. Weaver, A. W. Cohn and D. R. Gamelin, J. Am. Chem. Soc., 2013, 135, 18782–18785 CrossRef CAS PubMed.
- E. Y. Tsui, G. M. Carroll, B. Miller, A. Marchioro and D. R. Gamelin, Chem. Mater., 2017, 29, 3754–3762 CrossRef CAS PubMed.
- G. L. McIntire, H. N. Blount, H. J. Stronks, R. V. Shetty and E. G. Janzen, J. Phys. Chem., 1980, 84, 916–921 CrossRef CAS.
- D. I. Potapenko, E. G. Bagryanskaya, Y. P. Tsentalovich, V. A. Reznikov, T. L. Clanton and V. V. Khramtsov, J. Phys. Chem. B, 2004, 108, 9315–9324 CrossRef CAS.
- K. Ranguelova and R. P. Mason, Magn. Reson. Chem., 2011, 49, 152–158 CrossRef CAS PubMed.
- A. Thomas and S. Rajappa, Tetrahedron, 1995, 51, 10571–10580 CrossRef CAS.
- R. L. Brutchey, Acc. Chem. Res., 2015, 48, 2918–2926 CrossRef CAS PubMed.
- R. Caraballo, M. Rahm, P. Vongvilai, T. Brinck and O. Ramström, Chem. Commun., 2008, 6603–6605 RSC.
- E. Banchereau, S. Lacombe and J. Ollivier, Tetrahedron Lett., 1995, 36, 8197–8200 CAS.
- H. Otsuka, S. Nagano, Y. Kobashi, T. Maeda and A. Takahara, Chem. Commun., 2010, 46, 1150–1152 RSC.
- D. I. Potapenko, E. G. Bagryanskaya, V. V. Reznikov, T. L. Clanton and V. V. Khramtsov, Magn. Reson. Chem., 2003, 41, 603–608 CrossRef CAS.
- N. P. Tarasova, A. A. Zanin, E. G. Krivoborodov and Y. O. Mezhuev, RSC Adv., 2021, 11, 9008–9020 RSC.
- D. Zilevu and S. E. Creutz, Chem. Commun., 2023, 59, 8779–8798 RSC.
- I. N. Chakraborty, P. Roy and P. P. Pillai, ACS Catal., 2023, 13, 7331–7338 CrossRef CAS.
- R. G. Pearson, H. R. Sobel and J. Songstad, J. Am. Chem. Soc., 1968, 90, 319–326 CrossRef CAS.
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821–841 CrossRef CAS PubMed.
- A. J. Morris-Cohen, M. D. Donakowski, K. E. Knowles and E. A. Weiss, J. Phys. Chem. C, 2010, 114, 897–906 CrossRef CAS.
- J. H. Engel, Y. Surendranath and A. P. Alivisatos, J. Am. Chem. Soc., 2012, 134, 13200–13203 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional sample characterization and spectra. See DOI: https://doi.org/10.1039/d3sc04724e |
| This journal is © The Royal Society of Chemistry 2023 |