
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and intramolecular singlet fission properties of ortho-phenylene linked oligomers of diphenylhexatriene†
Oliver
Millington
 ab,
Ashish
Sharma
b,
Stephanie
Montanaro
a,
Anastasia
Leventis
a,
Simon A.
Dowland
b,
Daniel G.
Congrave
a,
Cherie-Anne
Lee
a,
Akshay
Rao
*b and
Hugo
Bronstein
*ab
ab,
Ashish
Sharma
b,
Stephanie
Montanaro
a,
Anastasia
Leventis
a,
Simon A.
Dowland
b,
Daniel G.
Congrave
a,
Cherie-Anne
Lee
a,
Akshay
Rao
*b and
Hugo
Bronstein
*ab
aDepartment of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK. E-mail: hab60@cam.ac.uk
bCavendish Laboratory, University of Cambridge, Cambridge, CB3 0HE, UK. E-mail: ar525@cam.ac.uk
First published on 3rd November 2023
Abstract
In molecular dimers that undergo intramolecular singlet fission (iSF), efficient iSF is typically accompanied by triplet pair annihilation at rates which prohibit effective triplet harvesting. Collisional triplet pair separation and intramolecular separation by hopping to additional sites in extended oligomers are both strategies that have been reported to be effective for acene based iSF materials in the literature. Herein, a family of highly soluble diphenylhexatriene (DPH) oligomers were synthesized and investigated using transient absorption spectroscopy to determine whether these approaches can be applied to the non-acene singlet fission chromophore, DPH. While iSF proceeds rapidly for all three new materials, neither concentration nor oligomer size result in significantly enhanced triplet pair lifetime relative to the dilute dimer case. These null results indicate the fallibility of the collisional separation and oligomerisation strategies.
Introduction
Singlet fission (SF) has attracted significant research attention over the past two decades, as a potential route to enhancing the efficiency of solar power devices beyond the one photon to one electron limit.1,2 This is made possible by the exciton multiplication that occurs when a molecule undergoes SF and a single excited singlet state produces two triplet excitons via interaction with a second chromophore in the ground state.3Singlet fission behaviour is classified into two main categories according to the nature of the interacting chromophore units.4 Intermolecular singlet fission occurs when two discrete molecules interact through space in either the condensed phase or via collisions in sufficiently concentrated solutions. This creates challenges for rational design due to the difficulty to predict solid-state morphologies and the high sensitivity of SF to relatively small changes in molecular packing.5,6 Meanwhile, achieving solution-based intermolecular fission requires very high concentrations that can be challenging to achieve practically with finitely soluble SF materials.7,8
The alternative is intramolecular singlet fission (iSF) in which the SF process occurs between covalently connected chromophore units within a single molecule. iSF enables precise synthetic control over chromophore geometry and provides an ideal platform to study and optimize singlet fission behaviour.9 iSF has been extensively investigated for derivatives of pentacene and tetracene ranging from molecular dimers to polymeric systems.4,9 These materials are challenged by relatively low triplet energies,10,11 which create a barrier toward feasible incorporation with silicon photovoltaic devices.12 In consideration of the limitation of low triplet energy materials, recent work in our group has demonstrated iSF activity in phenylene-bridged dimers based upon the higher triplet energy chromophore diphenylhexatriene (DPH).13 The best-performing material in that study, o-(mDPH)2, successfully achieved fast and efficient iSF. However, fast iSF is accompanied by facile triplet pair annihilation limiting the ability to harvest the formed triplet state.
In this work, we synthesized further phenylene-linked DPH derivatives with the aim of investigating potential strategies for reducing triplet pair annihilation and enhancing free triplet production.
One strategy for separating the triplet pair to avoid annihilation is that of a hybrid intra-intermolecular process, in which triplet pairs are generated by iSF but split to free triplets by intermolecular collisions.14 For the observed triplet pair lifetime of o-(mDPH)2 (τTT = 3.5 ± 0.6 ns),13 accessing a regime where intermolecular collisions can compete effectively with intramolecular triplet pair annihilation should require high concentrations (>10 mM) (ESI Section 4†).
Results and discussion
Molecular design and synthesis
However, o-(mDPH)2, was not sufficiently soluble to study in such a high concentration regime. Here we synthesized a new derivative, which we will refer to simply as (DPH)2, in which the solubilizing alkyl chains are moved from the termini of the DPH chromophores onto the central phenylene linker (Scheme 1). Additionally, the original 2-ethylhexyl chains were replaced by larger 2-butyloctyloxy chains. The net effect of these changes was a solubility enhancement from a maximum of ∼1 mM in toluene to greater than 50 mM. This enabled the investigation of the excited state dynamics across a concentration series to assess the feasibility of a hybrid intra-intermolecular SF mechanism. Larger structures typically exhibit low solubility unless additional solubilizing groups are incorporated and here the highly solubilizing linker additionally facilitated the synthesis of a trimer, (DPH)3, and tetramer, (DPH)4. This enabled study of the impact of increasing the number of chromophore units, a factor which has been reported to influence the dissociation of the bound triplet pair and improve free triplet yield in tetracene and perylene iSF systems.15–17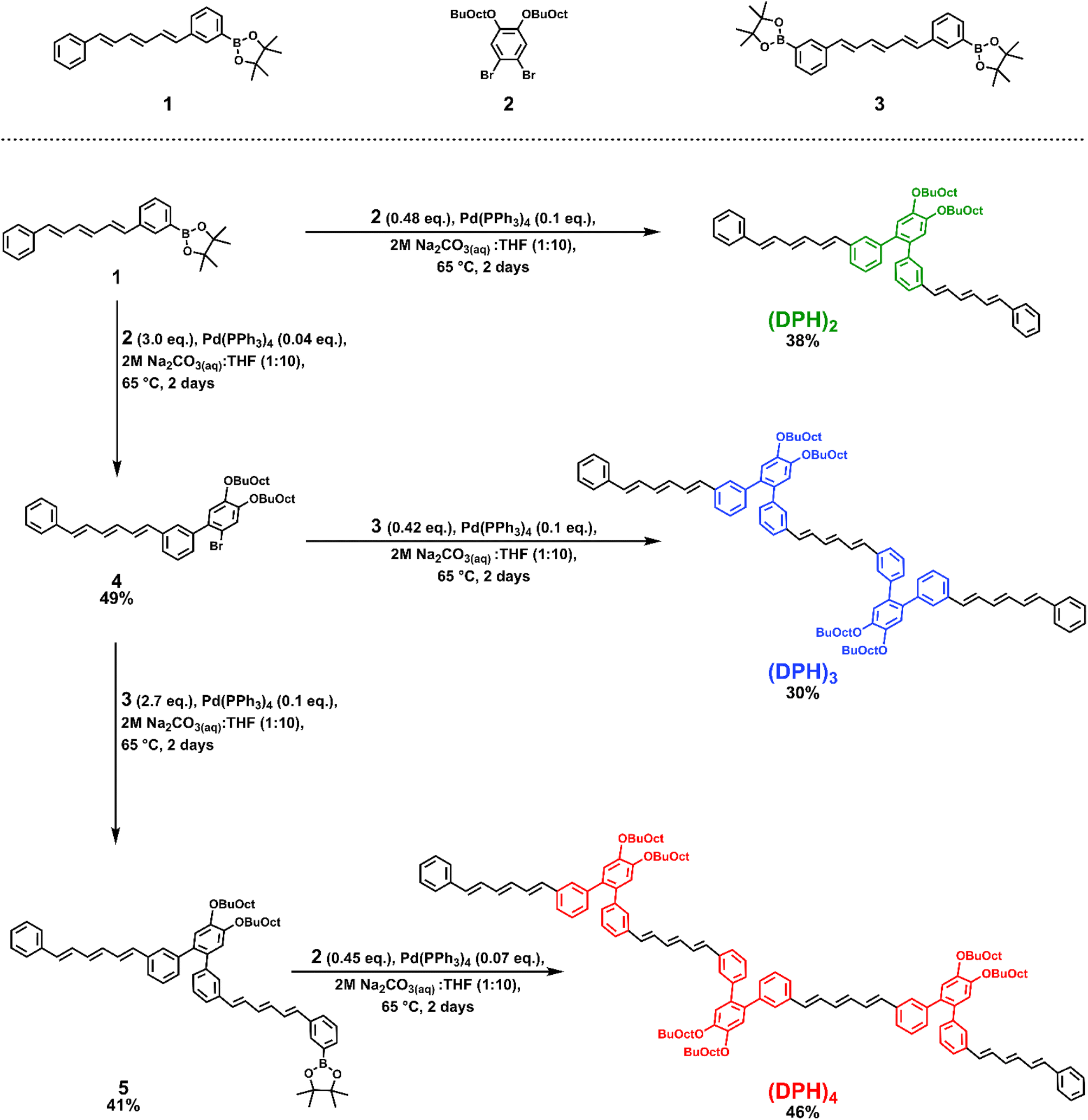 | ||
| Scheme 1 Synthetic route to the diphenylhexatriene oligomers. | ||
The syntheses of (DPH)2, (DPH)3, and (DPH)4 leveraged a diverging sequence of Suzuki coupling reactions using three critical building blocks: an asymmetric boronic acid pinacol ester DPH derivative, 1, a dibrominated linker, 2, and a symmetric bis-boronic acid pinacol ester DPH derivative, 3. Variation of the excess coupling partner enabled selectivity toward the desired Suzuki product at each step of the sequence. Full synthetic details including the syntheses of compounds 1–3 can be found in the ESI.†
Transient absorption spectroscopy
Femtosecond transient absorption spectroscopy (fsTA) was utilized to probe the initial formation of the triplet pair in our oligomer series. The fsTA spectra (Fig. 1a) of all three materials show strong similarity to each other and also that of the predecessor, o-(mDPH)2.13 In all cases, the initial singlet spectra have a broad photoinduced absorption (PIA) feature from 3.1–1.9 eV, which has a slight dip in intensity between 2.6 and 2.2 eV. An additional near-infrared (NIR) PIA below 1.9 eV is the most characteristic feature of the singlet. On timescales faster than 100 ps, the spectra evolve with a significant reduction in the intensity of the NIR PIA and the emergence of a new strong PIA between 3.2 and 2.7 eV. This PIA feature is assigned to the triplet pair state, TT, that is formed by intramolecular singlet fission.13 The TT feature has a primary peak at ∼2.9 eV and a higher energy shoulder at ∼3.1 eV. As the TT feature evolves there is a small decrease in the relative intensity of the higher energy shoulder relative to the main peak (ESI Fig. S2†). The shoulder may arise from a binding energy associated with the strongly correlated1(TT) pair that is initially formed by fission. Since the shoulder diminishes relative to the main peak, this suggests that weakly coupled triplet pairs are gradually formed from the strongly coupled state.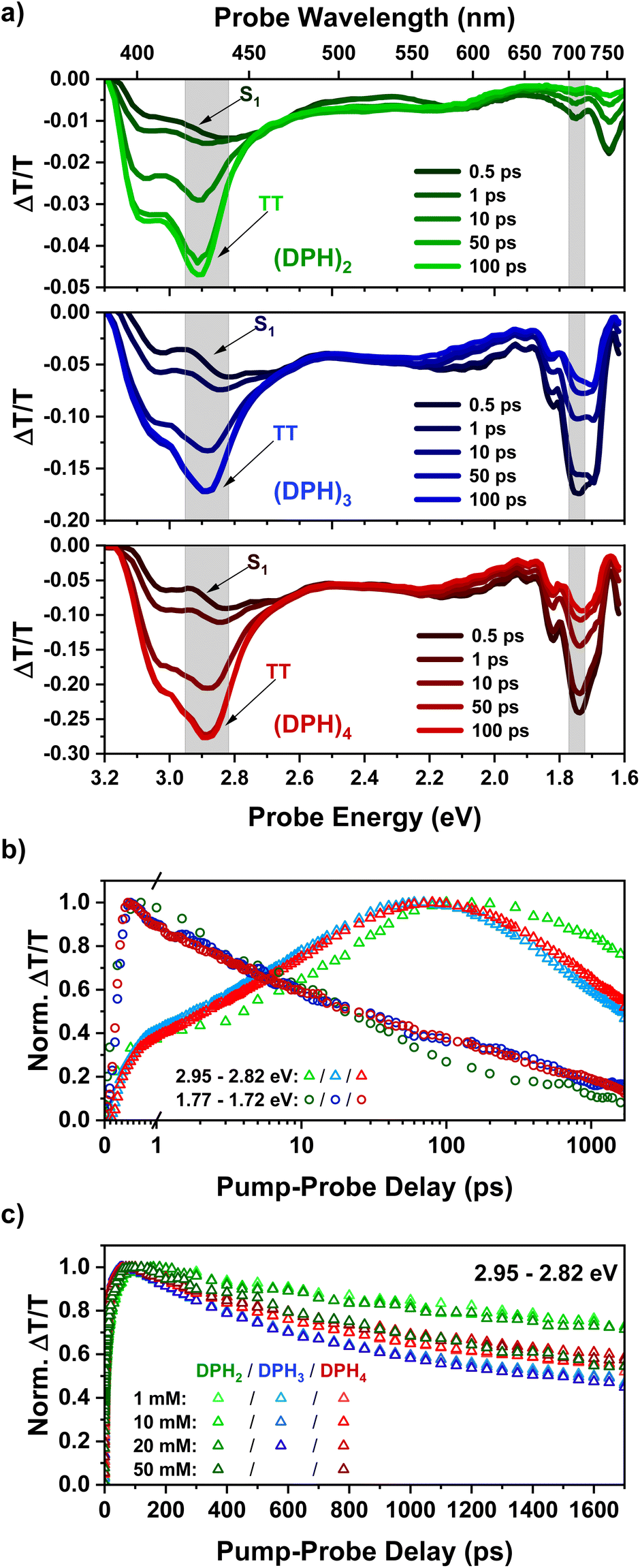 | ||
| Fig. 1 (a) fsTA spectra for solutions (1 mM) of the DPH oligomer series excited at 400 nm, showing spectral slices up to 100 ps. Spectra at later time intervals during which the TT PIA starts to decay may be found in the ESI (Fig. S2–S4).† (b) Kinetics corresponding to the shaded regions above with (DPH)2, (DPH)3 and (DPH)4 indicated by the colours green, blue and red respectively. (c) Kinetics in the region of the TT peak plotted for each measured concentration utilizing a linear temporal axis and the same colour scheme as above. | ||
The similarity of the singlet fission dynamics in these materials is made further evident by the kinetics in key wavelength regions (Fig. 1b). In the NIR singlet region, indicated by a spectral slice from 1.77–1.72 eV (700–720 nm), the decay profiles of the spectra of all three materials overlap significantly. Meanwhile, the rise of the TT feature is near-identical for (DPH)3 and (DPH)4 and only slightly different for (DPH)2. The slightly faster rise of the TT PIA in the larger oligomers potentially suggests that the rate of iSF is enhanced by the increased number of chromophore units. Of greater significance and contrary to the design aim, the triplet pair lifetime appears reduced rather than improved in (DPH)3 and (DPH)4 compared to (DPH)2. One possibility is that the greater bulk reduces torsional flexibility in the larger oligomers making it more difficult for triplet pair interactions to be reduced by conformational changes and thus enhancing the triplet pair annihilation rate relative to (DPH)2.
The fsTA response of all three oligomers was investigated over a concentration series ranging from 1 mM up to 50 mM in the case of (DPH)2 and (DPH)4 and up to 20 mM for (DPH)3. At higher concentrations, the spectra were observed to be qualitatively similar to the dilute cases (Fig. S2–S4†), barring a red shift in the observed spectral edge due to increasing probe absorption beyond the onset of the ground state absorption. At higher concentrations, there was no significant improvement in the decay profile of the triplet pair feature (Fig. 1c). In fact, for (DPH)2 the triplet pair decays markedly more quickly at 50 mM than at lower concentrations. This suggests that intermolecular interactions can even promote annihilation of the triplet pair.
The fsTA studies were supported by nanosecond transient absorption spectroscopy (nsTA), using a setup that is able to probe decay of the excited state populations up to 90 μs but is limited by an instrument response time of ∼2 ns. First, sensitization experiments were performed in order to characterize the triplet excited states of the DPH oligomers (Fig. S5d†). These confirmed that the triplet has a characteristic PIA feature peaking at ∼2.8–2.9 eV, which is similar for all three materials and matches the triplet feature observed in the original dimer series.13 Additionally, these experiments indicated the expected triplet lifetime for isolated triplets on the oligomers to be ∼50 μs for all three materials.
The nsTA spectra of the DPH oligomers are qualitatively similar (Fig. S5a–c†). All three materials exhibit a clear triplet PIA that is strongest at the instrument response limited peak of ∼2 ns. This finding is in agreement with the sub-nanosecond rise time of the TT feature in the fsTA data. In all three materials and at all studied concentrations, the majority of the PIA intensity decays in under ten nanoseconds (Fig. S5e and f†). We assign this decay to intramolecular triplet pair annihilation. The similarity of the decay supports the fsTA results and is further evidence that (DPH)3 and (DPH)4 do not achieve the design aim of enhanced triplet pair lifetime relative to (DPH)2.
In each case, a residual long-lived triplet population is produced, which we assign to molecules on which triplet-pair annihilation results in the generation of an isolated triplet. The height of this plateau relative to the peak does exhibit a small increase in the larger oligomers but this does not necessarily indicate a greater yield of long-lived triplets. In consideration of the fsTA data, the shift in relative intensity of the IRF limited peak and the long-lived signal may be attributed to increased triplet-pair decay occurring within the instrument response time for the larger oligomers and therefore suppressing the observed peak.
Conclusions
A new series of DPH oligomers ranging in size from dimer to tetramer have been synthesized and their intramolecular singlet fission properties evaluated. These materials exhibit significantly enhanced solubility relative to their parent material that was studied in our previous work. All three materials undergo fast iSF with the triplet pair signature PIA peaking on timescales faster than 100 ps. The iSF rate in the trimer and tetramer is slightly enhanced relative to the dimer, suggesting that iSF can be favoured by having more chromophore sites between which singlet fission can occur.The main aim of this work was to investigate the role of concentration and oligomer size on triplet pair dissociation in DPH iSF materials. At high concentrations, up to 50 mM, (DPH)2 exhibited no evidence of intermolecular triplet harvesting. Instead, the decay of triplet-containing states is faster at 50 mM suggesting that intermolecular interactions only result in quenching. Furthermore, triplet pair lifetime showed no significant enhancement in (DPH)3 and (DPH)4. This may indicate that triplets formed by singlet fission are unable to migrate between the different DPH units in these oligomers and as such the triplet recombination is governed by the interaction of triplets localized on adjacent DPH chromophores, regardless of oligomer size. The formation of bound triplet states that cannot be separated by hopping to additional sites in larger oligomers has precedent in the reported behaviour of pentacene oligomers.18 However, in this case the explanation for why triplets cannot hop to adjacent sites appears more nuanced than the existence of a significant triplet pair binding energy, given that signal indicating a bound state evolves to that consistent with weakly coupled triplets well before the triplet pair population starts to decay significantly. Overall, this study of ortho-phenylene linked DPH oligomers provides further evidence that while oligomerization may reduce triplet pair annihilation and enhance free triplet yield for some iSF systems,15–17 it is not a universal strategy.
Author contributions
O. M. designed the oligomer series and the synthetic route in discussion with S. M., A. L., D. G. C. and H. B. O. M. carried out the synthetic steps except for intermediate 2, which was undertaken by C.-A. L. O. M. carried out the transient absorption spectroscopy and discussed the TA data with A. S. and A. R. PLQE measurements were performed by S. A. D. The manuscript was drafted by O. M. under the supervision of H. B. and A. R.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Winton Programme for the Physics of Sustainability and the Engineering and Physical Sciences Research Council (EP/S003126/1, EP/V055127/1, EP/P007767/1) for funding.References
- A. Rao and R. H. Friend, Nat. Rev. Mater., 2017, 2, 17063 CrossRef CAS.
- M. C. Hanna and A. J. Nozik, J. Appl. Phys., 2006, 100, 074510 CrossRef.
- M. B. Smith, J. Michl, M. B. Smith, J. Michl, M. B. Smith, J. Michl, M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- R. Casillas, I. Papadopoulos, T. Ullrich, D. Thiel, A. Kunzmann and D. M. Guldi, Energy Environ. Sci., 2020, 13, 2741–2804 RSC.
- K. J. Fallon, N. Sawhney, D. T. W. Toolan, A. Sharma, W. Zeng, S. Montanaro, A. Leventis, S. Dowland, O. Millington, D. Congrave, A. Bond, R. Friend, A. Rao and H. Bronstein, J. Am. Chem. Soc., 2022, 144, 23516–23521 CrossRef CAS PubMed.
- X. Feng, A. B. Kolomeisky and A. I. Krylov, J. Phys. Chem. C, 2014, 118, 19608–19617 CrossRef CAS.
- B. J. Walker, A. J. Musser, D. Beljonne and R. H. Friend, Nat. Chem., 2013, 5, 1019–1024 CrossRef CAS.
- H. L. Stern, A. J. Musser, S. Gelinas, P. Parkinson, L. M. Herz, M. J. Bruzek, J. Anthony, R. H. Friend and B. J. Walker, Proc. Natl. Acad. Sci., 2015, 112, 7656–7661 CrossRef CAS.
- N. v. Korovina, N. F. Pompetti and J. C. Johnson, J. Chem. Phys., 2020, 152, 040904 CrossRef CAS PubMed.
- N. E. Geacintov, J. Burgos, M. Pope and C. Strom, Chem. Phys. Lett., 1971, 11, 504–508 CrossRef CAS.
- Y. Tomkiewicz, R. P. Groff and P. Avakian, J. Chem. Phys., 1971, 54, 4504–4507 CrossRef CAS.
- V. Gray, J. R. Allardice, Z. Zhang, S. Dowland, J. Xiao, A. J. Petty, J. E. Anthony, N. C. Greenham and A. Rao, ACS Nano, 2020, 14, 10748–10749 CrossRef CAS PubMed.
- O. Millington, S. Montanaro, A. Leventis, A. Sharma, S. A. Dowland, N. Sawhney, K. J. Fallon, W. Zeng, D. G. Congrave, A. J. Musser, A. Rao and H. Bronstein, J. Am. Chem. Soc., 2023, 145, 2499–2510 CrossRef CAS PubMed.
- G. He, K. R. Parenti, L. M. Campos and M. Y. Sfeir, Adv. Mater., 2022, 34, 2203974 CrossRef CAS PubMed.
- H. Liu, R. Wang, L. Shen, Y. Xu, M. Xiao, C. Zhang and X. Li, Org. Lett., 2017, 19, 580–583 CrossRef CAS PubMed.
- Z. Wang, H. Liu, X. Xie, C. Zhang, R. Wang, L. Chen, Y. Xu, H. Ma, W. Fang, Y. Yao, H. Sang, X. Wang, X. Li and M. Xiao, Nat. Chem., 2021, 13, 559–567 CrossRef CAS.
- N. V. Korovina, C. H. Chang and J. C. Johnson, Nat. Chem., 2020, 12, 391–398 CrossRef CAS PubMed.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. L. Steigerwald, M. Y. Sfeir and L. M. Campos, Chem, 2016, 1, 505–511 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03665k |
| This journal is © The Royal Society of Chemistry 2023 |