
5-Formylcytosine weakens the G–C pair and imparts local conformational fluctuations to DNA duplexes†
Manjula
Jaisal‡
,
Rajesh Kumar Reddy
Sannapureddi‡
 ,
Arjun
Rana
and
Bharathwaj
Sathyamoorthy
*
,
Arjun
Rana
and
Bharathwaj
Sathyamoorthy
*
Department of Chemistry, Indian Institute of Science Education and Research, Bhopal 462066, India. E-mail: bharathwaj@iiserb.ac.in
First published on 5th December 2022
Abstract
DNA epigenetic modifications such as 5-methyl (5mC), 5-hydroxymethyl (5hmC), 5-formyl (5fC) and 5-carboxyl (5caC) cytosine have unique and specific biological roles. Crystallographic studies of 5mC containing duplexes were conducted in the A-, B- or the intermediate E-DNA polymorphic forms. 5fC-modified duplexes initially observed in the disputed F-DNA architecture were subsequently crystallized in the A-form, suggesting that epigenetic modifications enable DNA sequences to adopt diverse conformational states that plausibly contribute to their function. Solution-state studies of these modifications were found in the B-DNA form, with marked differences in the conformational flexibility of 5fC containing duplexes in comparison to C/5mC containing duplexes, compromising the DNA duplex's stability. Herein, we systematically evaluate sensitive and commonly inaccessible NMR parameters to map the subtle differences between C, 5mC, and their oxidized (5hmC/5fC) counterparts. We observe that 15N/1H chemical shifts effectively report on the weakening of 5fC–G Watson–Crick base-pair H-bonding, extending the instability beyond any achievable within the sequence-specific changes in DNA. Triple 5fC containing sequences propagate the destabilization farther from the site of modifications, explaining reduced duplex stability upon multiple modifications. Additionally, scalar and residual dipolar coupling measurements unravel local sugar pucker fluctuations. One-bond 13C–1H scalar coupling measurements point towards a significant deviation away from the anticipated C2′-endo pucker for the 5fC modified nucleotide. Structural models obtained employing 13C–1H residual dipolar couplings and inter-proton distances corroborate the sugar pucker's deviation for 5fC modified DNA duplexes. The changes in the sugar pucker equilibria remain local to the 5fC modified nucleotide sans additive/long-range effects arising from multiple contiguous modifications. These observations highlight the impact of a major groove modification that alters the physical properties of DNA duplex without disturbing the Watson–Crick face. The changes observed in our studies for the 5fC containing DNA contrast with the perturbations induced by damage/lesion highlight the varied conformational preferences that modified nucleobases impart to the DNA duplex. As sequence-specific DNA transactions are rooted in the base-pair stability and pucker deviations, the observed structural perturbations for 5fC-modified DNA potentially play critical functional roles, such as protein-DNA recognition and interactions.
Introduction
DNA methyltransferases robustly incorporate and maintain the epigenetic cytosine modifications in CpG dinucleotide steps.1–3 Methylation at the 5th position of cytosine (5-methylcytosine, 5mC, Fig. 1A) is the most common epigenetic marker in DNA, with 5mC being regarded as the 5th abundant base in the genome.4–85mC modified sites are recognized to play myriad roles in cells.9–16 Ten-eleven translocation enzymes sequentially oxidize 5mC to 5-hydroxymethylcytosine17 (5hmC), 5-formylcytosine17 (5fC), and 5-carboxylcytosine18 (5caC), with thymidine DNA glycosylase and base excision repair enzymes providing a pathway towards demethylation of 5mC19,20 (Fig. 1B). Furthermore, each of these oxidized counterparts is increasingly identified to be semi-permanent, not just intermediates, and perform a wide range of unique, tissue-specific, and functional roles21–23 in, including but not limited to, genome packaging,24–26 gene expression,27,28 replication modulation,29 mutability of neighboring nucleotides,30 embryo development31 and prognosis of cancer.32 The structure–function paradigm of molecular biology thus motivates detailed biophysical characterization of these modifications.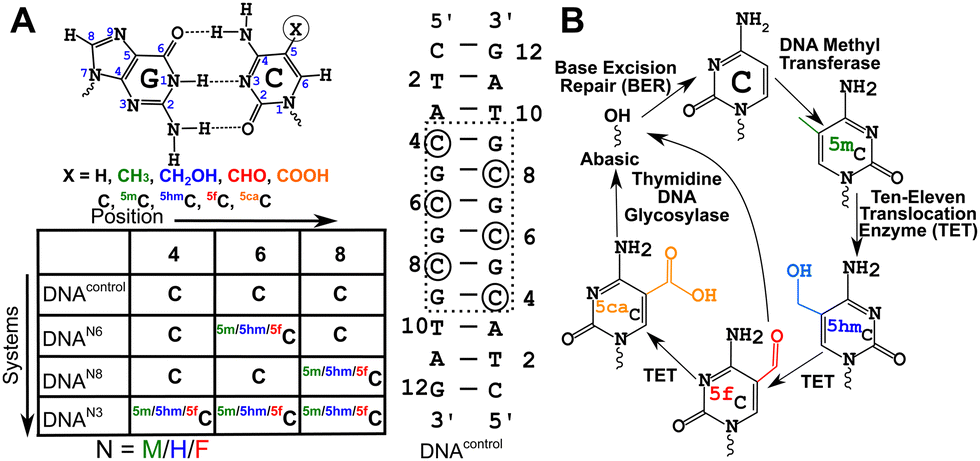 | ||
| Fig. 1 (A) Chemical structure of the cytosine-guanosine Watson–Crick pair with characteristic hydrogen (H-)bonds. Epigenetic modifications in cytosine are induced by changing the functional group in the 5th position. The palindromic dodecamer duplex DNA sequence (DNAcontrol, (5′-CTACGCGCGTAG-3′)2) studied in this work along with the suitable modifications (DNAN#, with N = M/H/F for 5mC/5hmC/5fC and # = 8/6/3 depending upon the type of modification, see Experimental methods) is introduced. Changes are introduced in the CpG repeat “core” of the duplex to avoid end-fraying conformational dynamics. (B) Methylation and subsequent demethylation are carried out by enzymes that convert C to 5mC, then to 5hmC, 5fC, and 5caC completing the cycle for the cytosine epigenetic modification. | ||
Cytosine modifications in the major groove retain the conventional Watson–Crick hydrogen (H-)bonding pattern (Fig. 1A). X-ray crystallographic studies of singly hemi-modified 5mC/5hmC/5fC in the CpG step of palindromic Drew-Dickerson dodecamer duplex DNA (5′-CGC![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) AATTNGCG-3′, referred to as DDDN, N = 5mC/5hmC/5fC modification, indicates the N-G pair) showed minimal perturbation from the B-DNA architecture.33–355mC incorporated in a G-C base-pair rich palindromic hexamer d(5′-GG
AATTNGCG-3′, referred to as DDDN, N = 5mC/5hmC/5fC modification, indicates the N-G pair) showed minimal perturbation from the B-DNA architecture.33–355mC incorporated in a G-C base-pair rich palindromic hexamer d(5′-GG![[5 with combining low line]](https://www.rsc.org/images/entities/char_0035_0332.gif)
![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) CC-3′)2 was crystallized in an intermediate E-DNA form with bases being perpendicular to the helical axis (B-form like) while the sugars sample an A-form like the C3′-endo pucker.36 The metastable E-DNA eventually equilibrates under crystallographic conditions to the A-DNA form.36 On the other hand, the triply 5fC modified palindromic dodecamer sequence (5′-CTA
CC-3′)2 was crystallized in an intermediate E-DNA form with bases being perpendicular to the helical axis (B-form like) while the sugars sample an A-form like the C3′-endo pucker.36 The metastable E-DNA eventually equilibrates under crystallographic conditions to the A-DNA form.36 On the other hand, the triply 5fC modified palindromic dodecamer sequence (5′-CTA![[f with combining low line]](https://www.rsc.org/images/entities/char_0066_0332.gif) TAG-3′, referred henceforth as DNAF3, Fig. 1A) was crystallized in a form that alters the hydration pattern stabilizing propeller twist and base-pair opening parameters, that appeared to differ significantly from A- and B-DNA forms, and hence led to a newly proposed class of architecture called the F-DNA.37 Such an observation correlated with differences in the circular dichroism (CD) signatures of DNAF3 compared to the unmodified DNA (DNAcontrol, Fig. 1A), in line with in silico modeling that predicts that the helical under-winding traps water molecules stabilizing the proposed F-DNA form.38 However, a subsequent study showed that structures of both DNAF3 and DNAcontrol sample the A-DNA form with no significant differences in the spatial arrangement of heavy atoms.39 Previously reported differences in CD signatures between DNAF3 and DNAcontrol were attributed to potential changes in the local electronic transition dipole moment rather than due to global structural perturbations of the DNA duplex.39 Hence, the next question follows whether the structure observed in the crystal form would be retained or be any different in the solution-state conditions.
TAG-3′, referred henceforth as DNAF3, Fig. 1A) was crystallized in a form that alters the hydration pattern stabilizing propeller twist and base-pair opening parameters, that appeared to differ significantly from A- and B-DNA forms, and hence led to a newly proposed class of architecture called the F-DNA.37 Such an observation correlated with differences in the circular dichroism (CD) signatures of DNAF3 compared to the unmodified DNA (DNAcontrol, Fig. 1A), in line with in silico modeling that predicts that the helical under-winding traps water molecules stabilizing the proposed F-DNA form.38 However, a subsequent study showed that structures of both DNAF3 and DNAcontrol sample the A-DNA form with no significant differences in the spatial arrangement of heavy atoms.39 Previously reported differences in CD signatures between DNAF3 and DNAcontrol were attributed to potential changes in the local electronic transition dipole moment rather than due to global structural perturbations of the DNA duplex.39 Hence, the next question follows whether the structure observed in the crystal form would be retained or be any different in the solution-state conditions.
Solution-state 1H-based nuclear magnetic resonance (NMR) studies of DNAF3 substantiated that the 5fC modification maintained the B-DNA form, as adjudged from the inter-proton distance and 1H–1H scalar coupling measurements.39 Interestingly, this study hinted at a deviation from the C2′-endo pucker only for the 5fC-modified nucleotides. Imino 1H-exchange NMR experiments performed on hemi-modified DDDN (N = 5mC/5hmC/5fC) samples showed increased base-pair opening rates for 5fC compared to the unmodified duplex suggesting subtle differences in their conformational landscape.35 Single-molecule fluorescence-based DNA cyclization assays revealed that 5fC modification imparts enhanced flexibility compared to unmodified cytosine-containing duplexes, while 5mC rigidifies the duplex.40 Steady-state and time-resolved infrared spectroscopy showed that 5fC in DNAF3 increases base-pair fluctuations reducing the cooperativity of duplex formation and thereby increasing the double-strand dissociation rate constant.41 The weakening of the duplex was attributed to the reduced pKa of the N3 nitrogen atom in 5-formyl modified cytosine that accepts the proton from the pairing guanosine nucleobase (Fig. 1A).42,43 Recently, solution-state 1H-based relaxation dispersion measurements have demonstrated an increase in the population of the single-stranded form for the 5fC containing DNA duplex44 (5′-GCGATGATCGC-3′). Additionally, it was reported that the destabilization propagates across the DNA duplex beyond the single 5fC–G fully modified base-pair. These observations suggest that 5fC modification might not alter the structure as much in comparison to cytosine or 5mC, but may interfere with the conformational fluctuations due to its unique chemical properties.
While the effect of a single site modification has been characterized, the influence of multiple contiguous modifications on DNA duplex structure is yet to be explored. Additionally, cytosine nucleotides are known to exhibit enhanced sugar puckering dynamics in comparison to other canonical nucleotides catering towards sequence-specific recognition.45,46 Therefore, a question arises whether these modifications alter such specific conformational dynamics of DNA duplexes, and whether can there be more NMR probes for measuring the same. Also, we sought to compare the destabilization/fluctuations achieved by the 5fC–G pair to what is achievable within the canonical C–G framework without modifications by only altering the primary sequence. In this study, we present NMR probes to understand the effect of single and multiple cytosine modifications (5mC, 5hmC, and 5fC) on the global structure and dynamics of DNA duplexes using solution-state NMR spectroscopy. Additionally, using these parameters we probe the presence/absence of differential sugar puckering of 5fC-containing duplexes.
Heteronuclear 13C/15N chemical shifts,47–50 scalar couplings,51,52 and partial anisotropic parameters, such as residual dipolar couplings53–57 (RDCs), are sensitive in characterizing conformational properties of DNA duplexes.58 RDCs provide a relative orientation of bonds across the molecule and thus improve the global structure of DNA duplexes, that otherwise evade conventional characterization that employs inter-proton distances and 1H–1H scalar coupling measurements. The structural perturbations employing RDCs for duplexes have been well characterized for DNA comprising of A-tracts,55 nucleotides with a locked sugar pucker,56 and N1-methyladenine57 (m1A) modification. In particular, the damage modification m1A present in duplexes results in bending of the helical axis and contributes to local base-pair melting suggesting a pre-primed bent DNA for effective protein recognition toward damage repair.57
In this work, we employ an optimized sparse sampling methodology that reduces overall measurement times of two-dimensional NMR data by 75%, thus making it possible to measure heteronuclear (13C/15N) shifts and RDCs robustly at low concentration (∼100 μM) in natural isotopic abundance samples (ESI†). Application of the optimized methods reveals that 15N imino chemical shifts of the paired guanosine are sensitive to the weakening of the H-bond for 5fC modified duplexes in comparison to DNAcontrol. The triply 5fC modified sample (DNAF3, Fig. 1A) shows a weakening of H-bonds farther than the singly modified samples (DNAF6/F8, Fig. 1A) indicating propagation of base-pair destabilization. At the same time, no discernable effect is observed for the 5mC/5hmC analog. One-bond 13C–1H scalar coupling (1JCH) measurements for sugar C1′–H1′ bonds point towards deviation from the C2′-endo pucker confined to the 5fC modified nucleotides. Structural models, obtained by employing inter-proton distances and one-bond 13C–1H residual dipolar couplings (RDCs, 1DCH), indicate that 5fC modified nucleotides’ sugar moiety samples conformations away from the C2′-endo pucker, while C, 5mC, and 5hmC containing DNA duplexes do not display any appreciable excursions. Such sugar pucker perturbations are localized to 5fC modified sites, with no additive effect arising from multiple modifications next to each other. The results highlight the impact that conformational changes due to 5fC incorporation may potentially have on protein–DNA recognition.
Results
15N/1H chemical shifts indicate a weakened 5fC–G H-bond beyond all possible sequence-specific contexts
13C/15N chemical shifts are NMR parameters that provide the necessary resolution to alleviate any chemical shift degeneracy in the 1H dimension and contain critical structural information, such as the presence and strength of H-bonds, and changes to the sugar pucker and glycosyl dihedral angle.47–50 Chemical shifts are perturbed by subtle changes in atomistic/molecular interactions, such as changes in H-bonding and/or π–π stacking.49,50 To delineate chemical shift perturbations (CSP) that arise in the modified duplexes due to changes in H-bonding and ring current effects, single “fully” modified (DNAN6, Fig. 1A) and single “hemi” modified (DNAN8, Fig. 1A) samples were studied and compared with the control (DNAcontrol, 5′-CTATAG-3′, Fig. 1A). CSPs observed in the paired G5 for the hemi-modified DNAN8 samples (with C8 being modified with 5mC/5hmC/5fC, Fig. 1A) would provide the change solely due to H-bonding, while the CSPs of G7 (5′-neighbor of C8, Fig. 1A) and G9 (3′-neighbor of C8, Fig. 1A) indicate the changes due to stacking/ring current effects for 5mC/5hmC/5fC in comparison to unmodified cytosine. On the other hand, the G7 CSP from a single fully modified DNAN6 (Fig. 1A) would reflect the effect due to both H-bonding and ring current effects. Any differences in CSP observed in DNAN6versus DNAN8 (Fig. 1A) would thus aid in pointing at the effect of hemi- vs. fully modified systems. Importantly, differences in CSPs measured from DNAN3versus DNAN6 (Fig. 1A) would provide insights into potential long-range perturbations due to multiple contiguous modifications.
Firstly, the G5 imino chemical shift (associated with C8–G5 pairing) in DNAM8/H8/F8 was examined to probe the influence of modifications solely on the base pairing. G5–N1/H1 resonances shift upfield by ∼0.8/0.4 ppm and ∼0.3/0.1 ppm for 5fC and 5hmC, respectively, in comparison to unmodified C, while 5mC shows marginal downfield shifts of 0.05/0.05 ppm (Fig. 2A, B and Table S1, Fig. S3, ESI†). It is evident that amongst the C–G pairs, modification with 5fC tends to shift both G–N1/H1 resonances significantly in contrast to the control and the other epigenetic modifications. The electron donating/withdrawing characteristics of the CH3, CH2OH, and CHO functional groups present in modified cytosine are correlated to the direction of the imino 1H CSP. A chemical modification on the C alters the C[N3]–G[N1] H-bond distance which in turn causes deshielding/shielding of the G–N1/H1 spins affecting CSP relative to the unmodified cytosine.48,59 The longer (shorter) the hydrogen bond, the higher (lower) the (de)shielding of the imino group. Consequently, imino CSP is being upfield shifted for 5fC/5hmC and downfield shifted for 5mC paired G–N1/H1 in comparison to unmodified C. Prior computational studies predict a correlated change in G–N1 and G–H1 chemical shifts due to the weakening of the C–G base pair upon chemical modification of the cytosine base.48
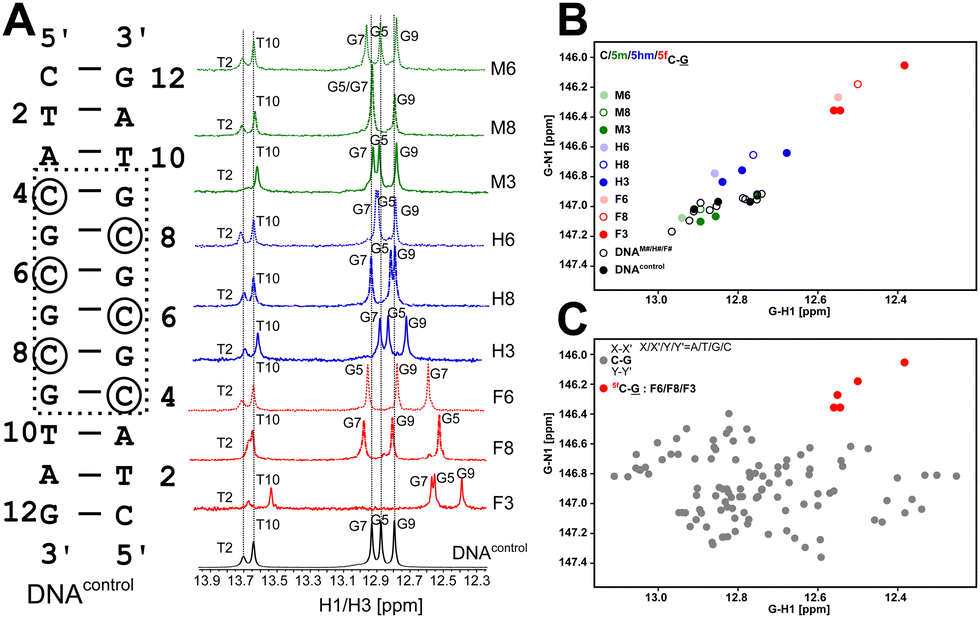 | ||
| Fig. 2 (A) 1H 1D NMR spectra acquired for DNAcontrol (bottom trace, black) and modified DNAN# (N = M/H/F for 5mC/5hmC/5fC, respectively, and # = 8/6/3 for hemi-/fully/triply modified samples) indicate stable duplex formation across samples. (B) Scatter plot of 15N–1H chemical shift correlation obtained for G–N1/H1 paired with C/5mC/5hmC/5fC chemical shifts obtained for DNAcontrol (black filled circles) and DNAN# (circles colored based on modification N = M/H/F). C–G pairs that are unmodified within modified sequences are also shown (open black squares) to indicate that only the modified cytosine experiences CSP. (C) Comparison of 5fC modified G–N1/H1 shifts (red circles) to unmodified C–G pairs across all possible trinucleotide sequence contexts (gray circle). | ||
Having assessed the effect of cytosine modification on base pairing, next is to quantify the changes that may arise due to the stacking of a chemically altered base on the 5′- and 3′-neighbors. The G7–N1/H1 resonances in DNAN8 (5′-end neighbor of C8, Fig. 2 and Fig. S3, ESI†) are downfield shifted to 0.15/0.06 ppm for 5fC, while a negligible change is observed for 5m/5hmC (Table S1 and Fig. S3, ESI†), suggesting either ring current or stacking change (or both) only for the 5fC modification. These measurements would come in handy to interpret the chemical shift perturbation for DNAN6 modifications, wherein a mere arithmetic sum of H-bonding and ring current effects would then indicate no appreciable difference between single hemi-modified (i.e., DNAN8) and single fully modified (i.e., DNAN6) cases. The magnitude and directionality of G–N1/H1 chemical shift perturbation for the C6–G7 pair in DNAN6 are in line with the observation for C8–G5 in DNAN8 sequences across all modifications (N = M/H/F). Such an observation suggests that base pairing affects the chemical shifts more significantly than the effect of modified ring current effects. Importantly, the G7–N1/H1 shifts in DNAN6 (for all modifications) show a simple arithmetic sum of chemical shift perturbation due to H-bonding and 3′ neighbor effect, indicating no significant structural changes from single hemi-modified to single fully modified systems (Table S2, ESI†).
Next, the question arises whether single versus multiple modifications cause any differential effects on the DNA duplex. Like the observation in DNAN6 systems, G5–N1/H1 and G7–N1/H1 chemical shift changes in DNAN3 (for all modifications) are simple arithmetic sums of a single fully (6th position) and hemi-modified (8th position) chemical shift. The only exception is observed with the magnitude of the G9–N1/H1 chemical shift change that arises due to inherent differences in the dinucleotide step (A vs. G). Noticeably, in DNAF3, the T10–N3/H3 and T2–N3/H3 nuclei experience a significant upfield shift to 0.25/0.13 ppm and 0.11/0.03 ppm suggesting a weakening of pairing that is two base pairs away from the sight of 5fC modification (Fig. 2A and Table S1, Fig. S3, ESI†) for the triply 5fC modified system. This observation is in agreement with complementary infra-red41 and NMR44 experiments, where the rate of duplex association is markedly reduced while that of dissociation is increased upon 5fC incorporation.
It is intriguing to comprehend the implications of the upfield shift of imino resonances of 5fC–G pairs in the context of the DNA duplex structure. Comparison of the measured shifts for the imino resonances of C–G pairs across primary sequence contexts would yield insights into how the 5fC–G pair differs from the canonical unmodified C–G pair. This was carried out by generating DNA samples consisting of trinucleotide steps in the non-terminal regions of dodecamer duplexes with C–G being the middle base pair (i.e., 5′-XY-3′ paired “•” with 5′-Y′X′-3′) flanked by canonical Watson–Crick pairs (X–X′ and Y–Y′). The first nearest neighbors to the C–G pair on both 5′- and 3′-ends were sampled across all possible trinucleotides (X/X′/Y/Y′ = A/T/G/C) resulting in 16 combinations, with a minimum of four replicates for each combination (unpublished data). The average G–N1/H1 chemical shift for all C–G pairs is observed to be 146.9/12.75 ppm (110 data points, Fig. 2C), agreeing well with the data obtained for DNAcontrol. The 5mC and 5hmC modified G–N1/H1 resonate at 147.0/12.87 ppm and 146.7/12.79 ppm, respectively, with 5 data points each across DNAM#/H# (Fig. 2B). Interestingly, for the 5fC modified base-pair, G–N1/H1 are well resolved from the entire cluster of C–G canonical pairs and resonate at 146.2/12.51 ppm (5 data points across DNAF#) – upfield shifted in both 15N and 1H dimensions (Fig. 2C). The significant average upfield shift for G–N1/H1 paired to 5fC in comparison to 5mC/5hmC and the entire C–G cluster indicates that the destabilization achieved for C–G upon formylation is beyond the scope that is achievable for any given trinucleotide primary sequence of DNA. This is an important observation given the fact that C–G pairs tend to impart stability to the DNA duplex in comparison to A–T pairs. The 5fC modification, in contrast, relaxes this property and contributes to the necessary level of destabilization beyond the scope achievable from the primary sequence, yet suitably retaining the Watson–Crick pairing that is essential for biomolecular processes.
Amino 1H spins present in the cytosine nucleobase (C–H41/H42) also corroborate the above observations. 1H chemical shifts of C–H41, which is also involved in the formation of Watson–Crick H-bonding, are relatively downfield shifted at the 5m/5hm/5fC nucleotide position. On the other hand, the chemical shift of C–H42 experiences an upfield shift for 5mC (0.30–0.40 ppm) and 5hmC (0.10–0.14 ppm), while 5fC modification results in a significant downfield shift (∼1.5 ppm). This observation supports the formation of a intranucleobase H-bond between the formyl group's carbonyl oxygen (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and the amino proton (H42) of 5-formyl cytosine.60 This intramolecular H-bonding of 5fC restricts formyl substituent conformation and hence forces it to be in plane with the cytosine aromatic ring, consistent with the previous reports.35 The small magnitude of chemical shift perturbation for 5m/5hmC indicates these bases do not make such type of H-bonding (CHO H-bond for instance), with prior crystallographic studies involving 5hmC containing DNA providing evidence that the orientation of CH2OH precludes such intramolecular H-bond formation with C(H42).34 Such an intramolecular H-bond excludes the interaction of water molecules at this site, which is otherwise available with the CH3 and CH2OH modifications.61
O) and the amino proton (H42) of 5-formyl cytosine.60 This intramolecular H-bonding of 5fC restricts formyl substituent conformation and hence forces it to be in plane with the cytosine aromatic ring, consistent with the previous reports.35 The small magnitude of chemical shift perturbation for 5m/5hmC indicates these bases do not make such type of H-bonding (CHO H-bond for instance), with prior crystallographic studies involving 5hmC containing DNA providing evidence that the orientation of CH2OH precludes such intramolecular H-bond formation with C(H42).34 Such an intramolecular H-bond excludes the interaction of water molecules at this site, which is otherwise available with the CH3 and CH2OH modifications.61
Following the characterization of 15N/1H imino/amino shifts, changes in 13C/1H were pursued for the aromatic base [C–C6/H6 and G–C8/H8]. As anticipated, C–C6/H6 was highest for the modified base due to the change in the functional group present in the 5th position, with upfield shift (3.3/0.2 ppm) for 5mC and downfield shifts for 5fC (13.3/0.9 ppm) and 5hmC (1.7/0.04 ppm) (Fig. S3, ESI†). Importantly, G5–C8/H8 nucleotide DNAF8 (5fC pair) experiences a downfield shift of 0.3/0.04 ppm (Fig. S3, ESI†), sensing the weakening of 5fC–G H-bond strength propagated by the aromaticity of the nucleobase. Next, 13C–C8 CSP of G7 in DNAN6 samples was analyzed to probe for any effects that may arise due to single contiguous modifications in the DNA duplex, versus a hemi-modified case (DNAN8). We observe a simple arithmetic sum of the H-bonding and ring-current changes manifested by the 5′/3′-neighbor (as adjudged from DNAN8) for all the cytosine modifications (Table S2, ESI†), without any exceptions. This suggests that the modifications do not confer any additive effect in terms of structural perturbations beyond the site of change. A similar observation is made when comparing 13C-C8 CSP of G5, G7, and G9 for DNAN3 samples, potentially indicating minimal changes along the major groove of the DNA duplex due to multiple contiguous modifications present in the system. Like aromatic 13C–H chemical shift perturbations, the furanose ring was most affected for the modified bases, with 5fC–C1′/H1′ nuclei experiencing the highest magnitude of 0.7–0.9/∼0.02 ppm (Fig. S3, ESI†). Although C1′ shifts report on sugar pucker equilibria,49,62 their interpretation, in this case, is affected due to the strong influence of ring current effects. Thus, furanose 13C/1H shifts are not further interpreted.
The magnitude of 13C–1H scalar coupling indicates a local deviation from the C2'-endo pucker at 5fC modified sites
Prior NMR studies involving DNAF3 hinted at the deviation of the 5fC sugars away from the C2′-endo pucker, adjudged from the cross-peak intensities observed in the NOESY data across furanose ring protons.39 Scalar couplings between protons connected via three covalent bonds (3JHH) are immensely useful in characterizing ring puckers, especially for nucleic acids.63,64 These are measured conventionally using the double-quantum filtered 1H–1H COSY experiment, where deoxyribose sugars populated heavily close to the C2′-endo pucker show substantial Σ3JHH between H1′–H2′/H2′′ (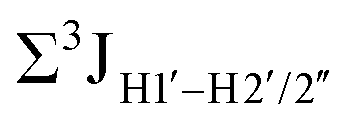 , 10–15 Hz).51,65 On the other hand, deoxyribose sugars averaging in their C3′-endo pucker are expected to display a reduction of such a measurement such that
, 10–15 Hz).51,65 On the other hand, deoxyribose sugars averaging in their C3′-endo pucker are expected to display a reduction of such a measurement such that 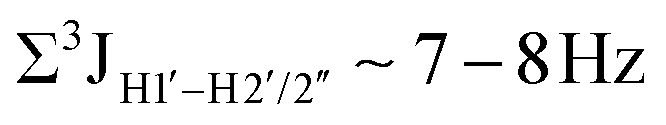 .65 Previous report on 5fC modified duplexes documented small reductions (0.5–1 Hz) in
.65 Previous report on 5fC modified duplexes documented small reductions (0.5–1 Hz) in 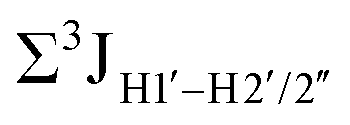 , with the NOESY data indicating an excursion away from the C2′-endo pucker for the formyl-modified cytosines adjudged from the inter-proton distances obtained from the NOESY experiment.39 However,
, with the NOESY data indicating an excursion away from the C2′-endo pucker for the formyl-modified cytosines adjudged from the inter-proton distances obtained from the NOESY experiment.39 However, 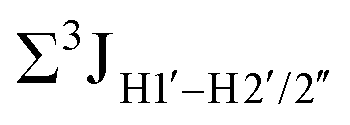 (ΣH1′) measurements are relatively insensitive, requiring a significant population change (∼30%) away from the C2′-endo pucker to effect a substantial reduction of the coupling (∼1 Hz) given the precision of the measurements (∼0.5 Hz).65 Thus, other probes would be convenient for mapping subtle pucker changes. One-bond heteronuclear scalar couplings (e.g.,
(ΣH1′) measurements are relatively insensitive, requiring a significant population change (∼30%) away from the C2′-endo pucker to effect a substantial reduction of the coupling (∼1 Hz) given the precision of the measurements (∼0.5 Hz).65 Thus, other probes would be convenient for mapping subtle pucker changes. One-bond heteronuclear scalar couplings (e.g., 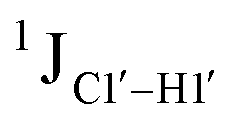 ) are influenced by torsion angles (including pucker and glycosyl angle) and C–H bond lengths making them attractive probes to highlight sugar pucker changes.52,66,67
) are influenced by torsion angles (including pucker and glycosyl angle) and C–H bond lengths making them attractive probes to highlight sugar pucker changes.52,66,67
Beginning with the DNAcontrol system, we observe that the position of the cytosine in the sequence influences the magnitude of the 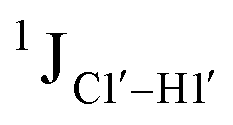 coupling magnitude. For instance,
coupling magnitude. For instance, 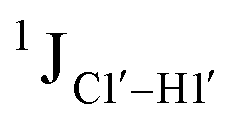 for the cytosine nucleotide in the RG (R = purine, A or G) trinucleotide step is found to be ∼166 Hz, while 5′-T (cytosine positioned at the 5′-end of the DNA strand) averages ∼172 Hz. This is expected as conformational degrees of freedom allow 5′-terminal cytosine to sample a broader range of puckers and glycosyl torsion angles. No significant difference in
for the cytosine nucleotide in the RG (R = purine, A or G) trinucleotide step is found to be ∼166 Hz, while 5′-T (cytosine positioned at the 5′-end of the DNA strand) averages ∼172 Hz. This is expected as conformational degrees of freedom allow 5′-terminal cytosine to sample a broader range of puckers and glycosyl torsion angles. No significant difference in 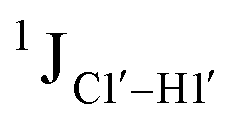 (
(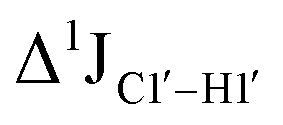 , relative to DNAcontrol) is observed for all nucleotides present in DNAM# and DNAH# within the measurement uncertainty (±2 Hz) (Fig. 3A). On the other hand,
, relative to DNAcontrol) is observed for all nucleotides present in DNAM# and DNAH# within the measurement uncertainty (±2 Hz) (Fig. 3A). On the other hand, 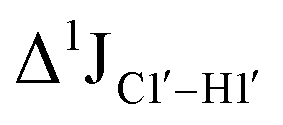 for singly modified 5fC6 (in DNAF6) and 5fC8 (DNAF8) results in an increase of 5–6 Hz, while the unmodified cytosine nucleotides within these samples show no change (Fig. 3A). All 5fC-modified nucleotides in DNAF3 also exhibit an increase of 3–6 Hz (Fig. 3A). No significant changes were observed for aromatic 13C–1H 1JCH (adenine C2–H2, pyrimidine C6–H6, purine C8–H8), indicating the reliability of the scalar coupling measurements (Fig. S4, ESI†). An increase in
for singly modified 5fC6 (in DNAF6) and 5fC8 (DNAF8) results in an increase of 5–6 Hz, while the unmodified cytosine nucleotides within these samples show no change (Fig. 3A). All 5fC-modified nucleotides in DNAF3 also exhibit an increase of 3–6 Hz (Fig. 3A). No significant changes were observed for aromatic 13C–1H 1JCH (adenine C2–H2, pyrimidine C6–H6, purine C8–H8), indicating the reliability of the scalar coupling measurements (Fig. S4, ESI†). An increase in 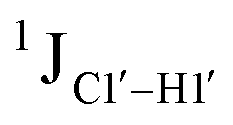 indicates a deviation from the C2′-endo sugar pucker as predicted from a computational study involving ribose sugars for a given anti glycosyl dihedral angle, with C3′-endo being predicted to have a coupling of 178 Hz, 10 Hz increase over the C2′-endo conditions.52 NMR data analysis across 2D spectra (NOESY, HMQC, and HSQC) of 5fC modified DNA (DNAF#) rules out any evidence of 5fC/G syn orientation. Hence, the increased
indicates a deviation from the C2′-endo sugar pucker as predicted from a computational study involving ribose sugars for a given anti glycosyl dihedral angle, with C3′-endo being predicted to have a coupling of 178 Hz, 10 Hz increase over the C2′-endo conditions.52 NMR data analysis across 2D spectra (NOESY, HMQC, and HSQC) of 5fC modified DNA (DNAF#) rules out any evidence of 5fC/G syn orientation. Hence, the increased 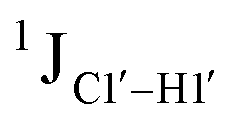 of 5fC potentially arises due to the shift in sugar pucker equilibrium from C2′-endo and plausibly subtle changes in the glycosidic dihedral angle.52,66,67
of 5fC potentially arises due to the shift in sugar pucker equilibrium from C2′-endo and plausibly subtle changes in the glycosidic dihedral angle.52,66,67
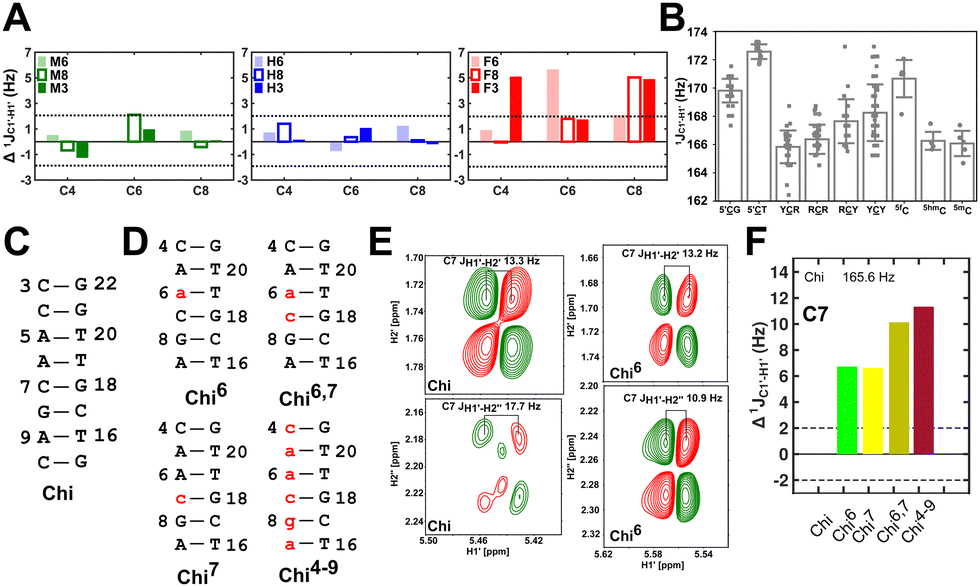 | ||
Fig. 3 (A) Changes to one-bond 13C–1H sugar C1′–H1′ heteronuclear scalar coupling magnitudes (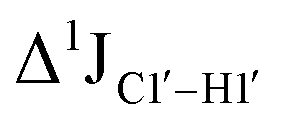 , in Hz) for nucleotide positions 4, 6, and 8 upon cytosine modification across DNAN# samples. Measurement uncertainty (2 Hz) is marked with dotted lines, with 5fC modification (in red) showing significant changes relative to unmodified cytosine. (B) , in Hz) for nucleotide positions 4, 6, and 8 upon cytosine modification across DNAN# samples. Measurement uncertainty (2 Hz) is marked with dotted lines, with 5fC modification (in red) showing significant changes relative to unmodified cytosine. (B) 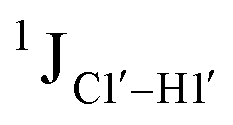 scalar coupling magnitude for cytosine nucleotides juxtaposed between being purine (R)/pyrimidine (Y) neighbors within a trinucleotide step. 5′-Terminal cytosine (5′G and 5′T, ∼170–172 Hz) displays a higher magnitude relative to cytosine present in the core of the helix (166–168 Hz). 5mC and 5hmC show no significant difference (∼166 Hz), while 5fC modification introduces a ∼6 Hz difference (RR versus5fC). (C) Non-palindromic model system (Chi) was studied to chart the deviation of the sugar pucker from C2′-endo conformation by introducing ribose sugars. (D) Secondary structures of ribose containing the “Chi” system, with ribose sugars marked in red and with small alphabets. (E) Subset of DQF-COSY spectra highlighting the reduction in scalar coupling magnitude for cytosine nucleotides juxtaposed between being purine (R)/pyrimidine (Y) neighbors within a trinucleotide step. 5′-Terminal cytosine (5′G and 5′T, ∼170–172 Hz) displays a higher magnitude relative to cytosine present in the core of the helix (166–168 Hz). 5mC and 5hmC show no significant difference (∼166 Hz), while 5fC modification introduces a ∼6 Hz difference (RR versus5fC). (C) Non-palindromic model system (Chi) was studied to chart the deviation of the sugar pucker from C2′-endo conformation by introducing ribose sugars. (D) Secondary structures of ribose containing the “Chi” system, with ribose sugars marked in red and with small alphabets. (E) Subset of DQF-COSY spectra highlighting the reduction in 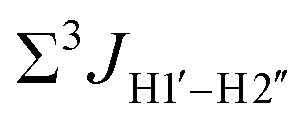 for Chi6 (6th position adenine changed to ribose) in comparison to Chi. (F) Change in one-bond 13C–1H C1′–H1′ scalar coupling by 6–11 Hz upon single (Chi6, Chi7), double (Chi6,7), and multiple (Chi4\–9) ribose incorporations (relative to Chi). for Chi6 (6th position adenine changed to ribose) in comparison to Chi. (F) Change in one-bond 13C–1H C1′–H1′ scalar coupling by 6–11 Hz upon single (Chi6, Chi7), double (Chi6,7), and multiple (Chi4\–9) ribose incorporations (relative to Chi). | ||
The 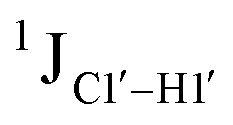 magnitude is also influenced by the 13C–1H bond distance.66 Formyl being an electron-withdrawing group might affect the bond lengths of base C6–H6 and furanose C1′–H1′ due to the resonance effect in aromatic rings. Although C6–H6 chemical shifts are most affected by C5 modifications of cytosine, Δ1JC6–H6 for all nucleobases (including modified cytosine) remains within ±2 Hz across all systems (DNAN#, Fig. S4, ESI†). And, if it was the bond distance that caused a change in
magnitude is also influenced by the 13C–1H bond distance.66 Formyl being an electron-withdrawing group might affect the bond lengths of base C6–H6 and furanose C1′–H1′ due to the resonance effect in aromatic rings. Although C6–H6 chemical shifts are most affected by C5 modifications of cytosine, Δ1JC6–H6 for all nucleobases (including modified cytosine) remains within ±2 Hz across all systems (DNAN#, Fig. S4, ESI†). And, if it was the bond distance that caused a change in 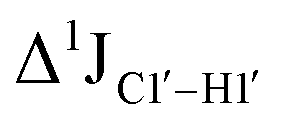 , then irrespective of the position and across samples (i.e., DNAF6/F8 and DNAF3) the magnitude of change would have remained constant. The mere fact that 5fC modified in the sixth position in DNAF6 (∼6 Hz) and DNAF3 (∼3 Hz) are different suggests that the change in scalar coupling is not due to bond-distance changes. Additionally, a comparison of high-resolution (∼1 Å) crystal structures of the cytosine nucleotide (BOXGIE, CCDC 114593) and 5fC (RAKLOG, CCDC 843055) showed no substantial increase in the C1′–H1′ bond length, supporting the fact that the
, then irrespective of the position and across samples (i.e., DNAF6/F8 and DNAF3) the magnitude of change would have remained constant. The mere fact that 5fC modified in the sixth position in DNAF6 (∼6 Hz) and DNAF3 (∼3 Hz) are different suggests that the change in scalar coupling is not due to bond-distance changes. Additionally, a comparison of high-resolution (∼1 Å) crystal structures of the cytosine nucleotide (BOXGIE, CCDC 114593) and 5fC (RAKLOG, CCDC 843055) showed no substantial increase in the C1′–H1′ bond length, supporting the fact that the 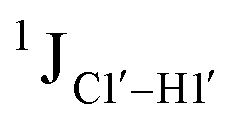 change is not due to change in bond length but due to other structural factors (pucker and glycosyl dihedral angle).
change is not due to change in bond length but due to other structural factors (pucker and glycosyl dihedral angle).
To put things in perspective regarding 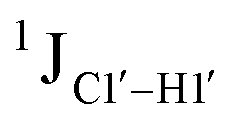 scalar coupling measurements, similar data were measured for cytosine present across trinucleotide repeats and in the 5′/3′ termini of duplex DNA (unpublished data). The presence of cytosine in the 5′-terminus observed for 5′-G and 5′-T results in 169.8 ± 0.8 and 172.6 ± 0.5 Hz, respectively, while the 3′-terminal G-3′ displays an average of 167.3 ± 1.3 Hz (Fig. 3B). Penultimate to 5′/3′-termini results in reduction (166.7 ± 1.1 for 5′-GC and 166.0 ± 1.1 Hz for TG-3′) in the magnitude with respect to the termini by 1–3 Hz. Similar measurements across the RR, RY, YR, and YY (where R = purine and Y = pyrimidine) trinucleotide steps within the “core” of the duplex resulted in 166.4 ± 1.0, 167.6 ± 1.5, 165.8 ± 1.2, and 168.2 ± 2.0 Hz, respectively, with the highest magnitude and spread of measured scalar couplings for the YY (Fig. 3B) step. The observations are thus consistent with the fact that the cytosine nucleotide tends to sample a larger conformational pool68 depending on the available degrees of freedom, with
scalar coupling measurements, similar data were measured for cytosine present across trinucleotide repeats and in the 5′/3′ termini of duplex DNA (unpublished data). The presence of cytosine in the 5′-terminus observed for 5′-G and 5′-T results in 169.8 ± 0.8 and 172.6 ± 0.5 Hz, respectively, while the 3′-terminal G-3′ displays an average of 167.3 ± 1.3 Hz (Fig. 3B). Penultimate to 5′/3′-termini results in reduction (166.7 ± 1.1 for 5′-GC and 166.0 ± 1.1 Hz for TG-3′) in the magnitude with respect to the termini by 1–3 Hz. Similar measurements across the RR, RY, YR, and YY (where R = purine and Y = pyrimidine) trinucleotide steps within the “core” of the duplex resulted in 166.4 ± 1.0, 167.6 ± 1.5, 165.8 ± 1.2, and 168.2 ± 2.0 Hz, respectively, with the highest magnitude and spread of measured scalar couplings for the YY (Fig. 3B) step. The observations are thus consistent with the fact that the cytosine nucleotide tends to sample a larger conformational pool68 depending on the available degrees of freedom, with 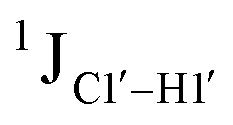 measurements reflecting the same. The increase in
measurements reflecting the same. The increase in 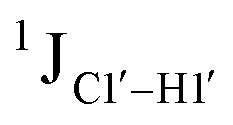 by 3–6 Hz suggests that 5fC modification to the RG step makes it behave like the YY step, the most conformationally flexible trinucleotide present.
by 3–6 Hz suggests that 5fC modification to the RG step makes it behave like the YY step, the most conformationally flexible trinucleotide present.
To further validate the results obtained from 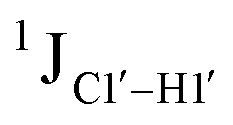 , control experiments were performed with ribose sugars in a non-palindromic DNA duplex (Fig. 3C, reference “Chi” system) anticipated to force pucker equilibria away from C2′-endo.69,70 In this sequence, ribose sugars were strategically positioned to increase the population of the C3′-endo pucker on the cytosine nucleotide (C7). Positioning the ribose sugar in A6 (Fig. 3D, Chi6) results in an increase of
, control experiments were performed with ribose sugars in a non-palindromic DNA duplex (Fig. 3C, reference “Chi” system) anticipated to force pucker equilibria away from C2′-endo.69,70 In this sequence, ribose sugars were strategically positioned to increase the population of the C3′-endo pucker on the cytosine nucleotide (C7). Positioning the ribose sugar in A6 (Fig. 3D, Chi6) results in an increase of 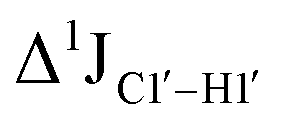 of ∼7 Hz, accompanied by a decrease in ΣH1′ (H1′–H2′′) of ∼7 Hz (Fig. 3E) indicating the pucker equilibria shifting towards C3′-endo. This is validated by ribose sugar modification for Chi at positions C7 (Chi7), A6 and C7 (Chi6,7), and C4–A9 (Chi4–9), where C7
of ∼7 Hz, accompanied by a decrease in ΣH1′ (H1′–H2′′) of ∼7 Hz (Fig. 3E) indicating the pucker equilibria shifting towards C3′-endo. This is validated by ribose sugar modification for Chi at positions C7 (Chi7), A6 and C7 (Chi6,7), and C4–A9 (Chi4–9), where C7 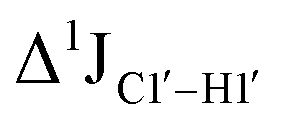 increased by 7–12 Hz (Fig. 3F), and by the disappearance of the H1′–H2′ cross peak in the DQF-COSY spectrum. Hence a change in
increased by 7–12 Hz (Fig. 3F), and by the disappearance of the H1′–H2′ cross peak in the DQF-COSY spectrum. Hence a change in 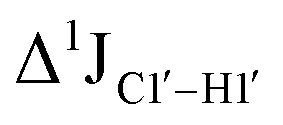 for 5fC modified nucleotides indicates puckering away from C2′-endo by a small yet significant degree.
for 5fC modified nucleotides indicates puckering away from C2′-endo by a small yet significant degree.
Residual dipolar coupling measurements reiterate that 5fC modified sites deviate in pucker/glycosyl angle
RDC measurements have the capability of mapping global structural changes, in addition to local perturbations.45,53,54,56 Comparison of RDCs for an A-tract DNA duplex versus a randomized sequence clearly indicates the helical bending observed in the former.55,57,71–73 RDCs would further complement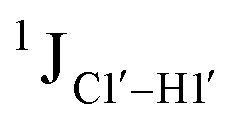 measurements in probing sugar pucker changes for 5fC modified DNA duplexes. In particular, C1′–H1′, C2′–H2′/2′′ and C3′–H3′ RDCs are sensitive to the changes in the pseudorotation angle.45 Since sugar moieties display fast exchange across the different puckers, RDC measurements have been interpreted as a population-weighted average across C2′-endo and C3′-endo puckers. Such studies on DDD have shown that cytosine sugar present in the core ends up sampling 20–30% C3′-endo pucker, followed by thymidine (2–20%) and purines45 (0–4%). RDCs measured for DNAcontrol also reiterate their ability to discriminate pucker differences as C4 present in an AG shows a lowered
measurements in probing sugar pucker changes for 5fC modified DNA duplexes. In particular, C1′–H1′, C2′–H2′/2′′ and C3′–H3′ RDCs are sensitive to the changes in the pseudorotation angle.45 Since sugar moieties display fast exchange across the different puckers, RDC measurements have been interpreted as a population-weighted average across C2′-endo and C3′-endo puckers. Such studies on DDD have shown that cytosine sugar present in the core ends up sampling 20–30% C3′-endo pucker, followed by thymidine (2–20%) and purines45 (0–4%). RDCs measured for DNAcontrol also reiterate their ability to discriminate pucker differences as C4 present in an AG shows a lowered 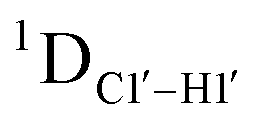 (3.5 Hz) in comparison to C6/C8 (11–13 Hz) that is present in the GG step (1DC6-H6 for C4/C6/C8 19–21 Hz). Structure refinement of DNAcontrol with NOE-derived distances and RDCs indicates that the C4 sugar pucker averages around the O4′-endo while C6/C8 sample the C1′-exo to C2′-endo pucker (see the next section).
(3.5 Hz) in comparison to C6/C8 (11–13 Hz) that is present in the GG step (1DC6-H6 for C4/C6/C8 19–21 Hz). Structure refinement of DNAcontrol with NOE-derived distances and RDCs indicates that the C4 sugar pucker averages around the O4′-endo while C6/C8 sample the C1′-exo to C2′-endo pucker (see the next section).
Measuring RDCs and correlating the measured values across DNAcontrol and modified systems (DNAN#) would aid in characterizing any global bending that may be present upon cytosine modification. To start with, a good RDC agreement (Pearson's coefficient of R2 ∼ 0.95 and RDC RMSD ∼ 1.2 Hz, Fig. 4A) was observed for concentrated (2.7 mM, uniform Nyquist NMR data sampling and conventional Fourier transform processing) and diluted (500 μM, 25% sparse sampling and compressed sensing processing) DNAcontrol samples indicating that the sparse methodology for limited concentration samples works as efficiently (within the experimental uncertainty of ∼2 Hz) as the routinely employed conventional methods.
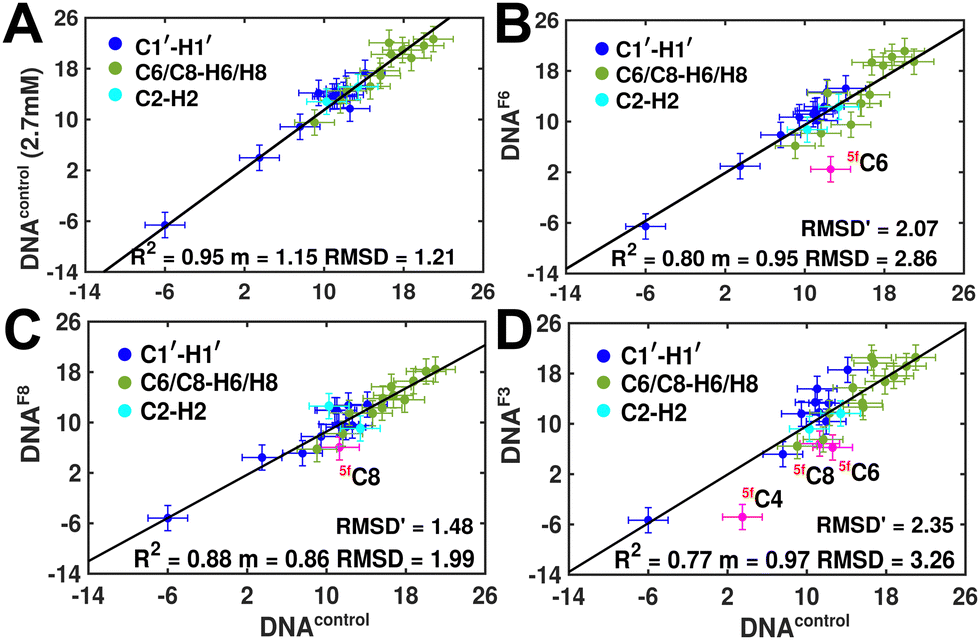 | ||
| Fig. 4 Experimentally measured RDC correlation scatter plots to highlight the differences that arise between DNAcontrol and DNAF#, with sugar (C1′–H1′, blue) and nucleobase (C6/C8–H6/H8 and C2–H2, in green and cyan, respectively) RDCs displayed. (A) Comparison of RDCs measured between DNAcontrol (2.7 mM, y-axis) using conventional NMR data acquisition and DNAcontrol (500 mM, x-axis) with 25% sparse sampling NMR methods. Data were best fit with a linear function (solid black line) without an intercept, with the slope varying depending upon subtle changes in Pf1 alignment media concentrations known to arise during sample preparation. RDC RMSD is calculated between the x- and y-axis to highlight that low-concentration sparse sampling methods work within experimental uncertainties (2 Hz, represented by error bars). Scatter of RDCs measured for DNAF6 (B), DNAF8 (C) and DNAF3 (D) plotted against DNAcontrol, with C1′–H1′ RDC of the 5fC modified RDC marked in pink. RMSD′ reported in panels (B)–(D) indicates measurement difference with DNAcontrol when 5fC modified nucleotide measurement is removed. | ||
RDCs measured for 5mC and 5hmC modified samples (DNAM# and DNAH#) correlate well with DNAcontrol (R2 in the range of 0.86–0.91 and RMSD < 2 Hz, Fig. S9, ESI†), indicating similarity in their overall structure. Strikingly, significant RDC differences are observed for DNAF6 and DNAF3 (R2 0.75–0.80, RMSD 3.0–3.5 Hz, Fig. 4B and D) but within the experimental uncertainty for DNAF8 (R2 0.88, RMSD 2.0 Hz, Fig. 4C) pointing at differences between single hemi-modified (DNAF8) and single fully modified (DNAF6) systems. Noticeably, 5fC–C1′–H1′ RDC is the only data point (indicated in pink color in Fig. 4B and D) that deviates by 6–10 Hz reduction in the correlation plot. Removal of these 5fC C1′–H1′ RDC outliers improves the correlation (R2 ∼ 0.90, RMSD′ < 2 Hz, Fig. S9, ESI†), implying only a change in the local structure for DNAF6/F3 with no apparent helical bending that is any different from DNAcontrol.
The RDC measurement also helps rule out the possibility of C–H bond length changes for the C1′–H1′ bond vector. A back-of-the-envelope calculation suggests that a ∼6 Hz decrease in RDC (given an alignment and B-DNA structure for DNA and DNAN#) requires an increase of ∼0.25 Å in the C1′–H1′ bond length, which is rather unlikely. The 5fC selective deviations corroborate with the ∼6 Hz increase in 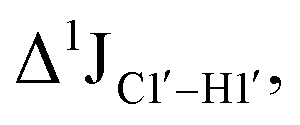 suggesting a local structural perturbation induced by 5fC plausibly due to changes in sugar pucker equilibria away from canonical C2′-endo conformation for B-DNA.
suggesting a local structural perturbation induced by 5fC plausibly due to changes in sugar pucker equilibria away from canonical C2′-endo conformation for B-DNA.
It is pertinent to note here that the magnitude of terminal 5′-T C1′–H1′ RDCs is in the range of −5 to −8 Hz across DNAcontrol and DNAN# samples (Table S1, ESI†). This scenario yet again highlights that 5fC alters the local structure in terms of pucker and glycosyl dihedral angle for the RG step; however, it does not make it as flexible as the terminal cytosine nucleotides.
Structure refinement supports the change in the pucker at 5fC modified sites
Following the detailed analysis of NMR parameters, the next step was to refine the structure using the NOESY and RDC data acquired for all the samples. Firstly, NOESY cross peak connectivity across the base (H6/H8) and sugar protons (H1′/H2′/H2′′) qualitatively confirms that all DNA duplexes are in the right-handed helix in solution and close to B-form conformation.58,74–76 The weak NOE cross-peak of inter and intranucleotide H6/H8–H1′ and intranucleotide H6/H8–H2′′ and the strong intensity of intranucleotide H6/H8–H2′ qualitatively describe a high anti glycosyl torsional angle and a C2′-endo sugar conformation for 5m/5hm/5fC DNA.Next, the characterization of the structures sampled by DNAcontrol and DNAN# was pursued using inter-proton distances and RDCs as constraints. As the number of measurements/constraints are significantly small given the total number of degrees of freedom available for nucleic acids,45 the aim here was to avoid overfitting the NMR data yet obtain a (low-resolution) conformational model for DNAcontrol and DNAN# that may highlight any differences in the DNA duplex upon modification. Also, as the modifications are in the major groove with no effect on Watson–Crick pairing, the unmodified cytosine nucleobase was refined against the measured NMR parameters for each of the DNAN# modified sequences. Thus, the measured data (inter-proton distances and RDCs, Table S3, ESI†) were supplied to refine initialized from “idealized” B-DNA geometry using the XPLOR-NIH structure refinement program77 (see Experimental methods).
Upon refinement, DNA systems studied (DNAcontrol and DNAN#) continue to sample an overall B-DNA as anticipated and predicted in previous studies (Fig. S5, ESI†).39 Notably, RDCs refine the B-DNA structure where back-prediction of RDCs measured for DNAN# with the DNAcontrol structure (and vice versa) yields experimentally derived correlations (Table S5, ESI†). It indicates that refined structures mimic conformations sampled across these modifications. Structural analysis of refined conformers was performed to determine base pairs, base-pair step parameters, sugar pucker using 3DNA, and Curves+ to determine DNA helical curvature (methods, Table S4, ESI†). Parameters that are used to define intra-basepair78 (shear, stretch, stagger, buckle, propeller, and opening) and inter-basepairs78 (shift, slide, rise, roll, tilt, and twist) and dihedral angles (backbone: α, β, γ, δ, ε, ζ; glycosidic dihedral angle χ; and sugar: ν0–ν4) follow the anticipated distribution about the canonical B-DNA geometry without any exceptions. No differences between average helical bending (within the measurement uncertainty and structural noise) and major groove widths were observed between DNAcontrol and DNAN#.
Sugar pucker analysis of the refined structures agrees with the inferences derived from one-bond scalar 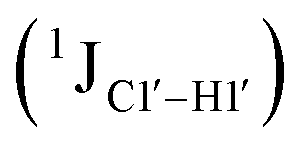 and residual
and residual 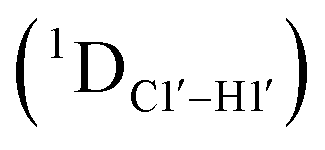 dipolar coupling measurements. Sugar puckers in B-DNA are known to sample conformations about the C2′-endo puckers, with drifts commonly observed towards O4′-endo. This expectation is preserved for DNAcontrol and DNAM#/H# systems (Fig. 5A). Mainly, the AG (
dipolar coupling measurements. Sugar puckers in B-DNA are known to sample conformations about the C2′-endo puckers, with drifts commonly observed towards O4′-endo. This expectation is preserved for DNAcontrol and DNAM#/H# systems (Fig. 5A). Mainly, the AG (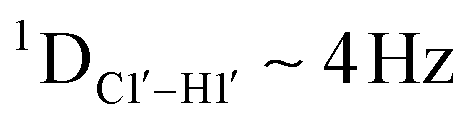 for C4) versus GG (
for C4) versus GG (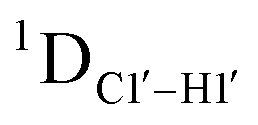 11–14 Hz for C6 and C8) trinucleotide step indicates a discernable difference in the pucker equilibria corroborating the RDC measurements for these steps in DNAcontrol (Fig. 5A).
11–14 Hz for C6 and C8) trinucleotide step indicates a discernable difference in the pucker equilibria corroborating the RDC measurements for these steps in DNAcontrol (Fig. 5A).
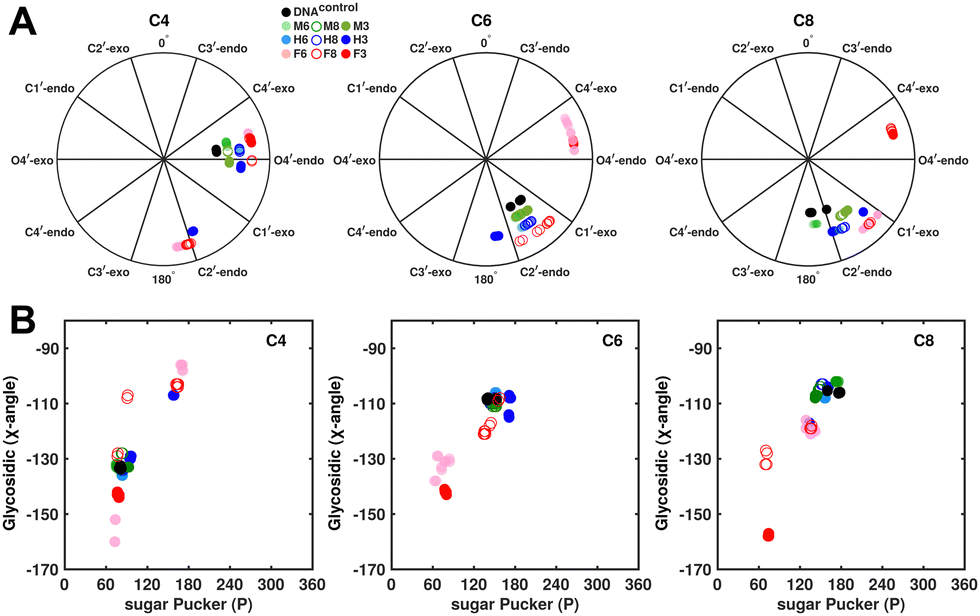 | ||
| Fig. 5 (A) Pseudorotation phase angle plots at cytosine nucleotides C4, C6, and C8 of refined DNA structures to compare the sugar conformation of unmodified and modified DNA. (B) Variation in the glycosidic dihedral angle as a function of the sugar pucker for the refined DNA structures. Black, green, blue, and red colored data points correspond to DNAcontrol, DNAM#, DNAH#, and DNAF#, respectively. | ||
In the single 5fC-modified systems, it is observed that the C6 nucleotide in DNAF6 shows more extensive excursions towards O4′-endo compared to DNAcontrol. In contrast, DNAF8 shows to a lesser extent, in agreement with the coupling measurements and highlights the difference between single hemi-modified and single fully modified 5fC systems. DNAF3 alters the pucker clearly for C6 and C8 away from C2′-endo, while C4, which is already at O4′-endo, is altered to a smaller extent. Additionally, pucker changes tend to affect the glycosidic torsional (χ-)angle, as observed for A- (C3′-endo, χ = −150°) and B-DNA (C2′-endo, χ = −110°). A correlation was plotted between sugar pucker and χ (Fig. 5B) for the refined DNA structures to see whether a similar effect persists upon 5fC modification. Indeed, for nucleotides C6 (DNAF6) and C8 (DNAF3), C4 is affected in DNAcontrol and DNAN# due to its presence in the AG step (Fig. 5B). In contrast, all complementary base-paired guanosine nucleotides (i.e., G5, G7, and G9) exist in C2′-endo with χ near −100°, pointing to the relative orientation between base and sugar changing locally at the 5fC site.
Further, to assess whether any correlated change occurs in the phosphate backbone due to alteration in the pucker, the phosphate backbone dihedral angles ε and ζ were measured from the refined structures to see whether BI (ε − ζ < 0) and BII (ε − ζ > 0) equilibria get affected. The correlation of the sugar pucker to ε − ζ indicates that all cytosine nucleotides in DNAcontrol and DNAN# are in BI backbone conformation (Fig. S6, ESI†), without exceptions. Indeed, the results are analyzed conservatively, as without 31P chemical shifts and scalar coupling (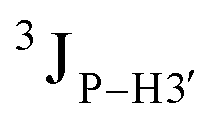 and
and 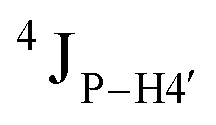 ) measurements the observations cannot be further refined/validated. Thus, 5fC modification in duplex DNA alters sugar pucker equilibria without significant changes to other conformational and structural properties.
) measurements the observations cannot be further refined/validated. Thus, 5fC modification in duplex DNA alters sugar pucker equilibria without significant changes to other conformational and structural properties.
Discussion
The effect of 5m/5hm/5fC on the stability and structural properties of the DNA duplexes has been studied employing various spectroscopic techniques. Thermal melting studies show that 5mC increases the duplex stability by ∼5 °C, and 5hm/5fC tends to reverse the impact of stability afforded by 5mC.37,41,42,795hmC has a melting temperature similar to that of unmodified DNA, whereas 5fC destabilizes the DNA duplex by ∼3 °C.41,42,44,57 Contrastingly, the presence of 5fC in duplex RNA results in increased stability with a ∼5 °C increase in the melting temperature, due to increased stacking interactions with neighboring base pairs.80 In addition to DNA and RNA duplexes, formation of i-motifs in cytosine-rich DNA sequences is also altered by the presence of these epigenetic modifications where C–C+ pairs are formed.81 The fact that additional protonation is required to stably form C–C+ pairs, the addition of CH2OH and CHO groups stabilizes i-motifs at a lower pH (∼0.1 units relative to unmodified cytosine), while 5mC increases the same by 0.1–0.2 units.81Prior studies have pointed out that CHO (5fC) and COOH (5caC) modifications in cytosine change the pKa of the H-bond accepting N3 nitrogen atom that was predicted to cause a weakening of the H-bond for DNA duplexes.42,43,82 Computational studies performed on such modified cytosine duplex systems report that the calculated isotropic chemical shift of both the imino proton (1H) and nitrogen (15N) shows a correlated change with the increasing or decreasing H-bond distance in the C–G base pair.48 Geometry optimized and energy minimized structures of C–G pairs predict an increase in the G:N1–H1⋯N3:C distance upon varying C from 5mC to 5hmC, 5fC, and 5caC, the longest being for the 5fC–G base pair.59 Such a weakening of the H-bond is attributed to enhanced base-pair opening rates35 and increased population of single-stranded DNA.41,44 However, direct measurement of structural changes in duplex DNA upon 5fC modification would be convenient and aid in characterizing other pertinent modifications in nucleic acids.
Our results of 15N/1H chemical shifts of the guanosine base paired with the modified cytosine provide an unbiased way of assessing local structural changes. Notably, the measurements are made without the need for 15N-isotopically enriched samples, demonstrating 13C/15N chemical shift measurements to be a viable approach to studying modified nucleotides – an unexplored treasure trove in terms of epigenetics, damage/lesion, and epitranscriptomics. 15N/1H chemical shifts measured from the complementary G paired to 5mC, and 5fC modified nucleotides show significant downfield and upfield shifts, respectively, indicating the strengthening and weakening of the H-bond. In addition, the weakening of the 5fC–G base-pair propagates beyond the modification site, as reported for DNAF3, substantiating the previous findings that 5fC destabilizes the whole DNA duplex.44,82 Thus, measurement of 15N chemical shifts could proxy as an indicator of strengthening/weakening akin to the chemical exchange saturation transfer type experiments. This also explains that 5fC containing DNA templates display reduced substrate specificity of dGTP incorporation as observed experimentally.30 The insertion of dGMP opposite to 5fC is less efficient in comparison with the insertion of dGMP opposite to unmodified C, with dAMP/dTMP being more frequently misincorporated.83
DNA duplexes are known to exhibit exchange across lowly populated conformational states (such as Hoogsteen and tautomeric forms) that have been implicated in various functional roles.84–88 As G–C pair Hoogsteen pair formation requires C–N3 protonation, we speculate that lowered pKa for cytosine (4.5 units) upon 5-formyl incorporation (2.1 units) would reduce the Hoogsteen population. Also, prior studies have indicated that 5-formyl substitution could potentially drive cytosine to a lesser-known imino tautomer rather than the conventional amino form.89 To keep the three H-bonds between the G–C pair, then such a change would force the paired guanosine to sample the enol (Genol) form away from the keto form. Interestingly, the formation of Genol has been documented to shift the G–N1 chemical shift (in the context of the dG·dT wobble pair) downfield by 30–50 ppm.90,91 However, we observe for the 5fC–G pair a moderate 0.8 ppm upfield shift of the 15N–N1 paired guanosine indicating that such a tautomeric base pair formation appears less likely.
Crystal structures of the DNA duplex containing 5mC36 and 5fC37 have reported significant deviations from B-DNA. However, prior solution NMR studies refuted such claims based on NOE-based distances, indicating only subtle differences in the 5fC-modified nucleotides.39 In our studies, complementing NOEs, heteronuclear 13C/15N chemical shifts, and coupling-based measurements aid in confirming that the overall structure of 5m/5hm/5fC DNA does not deviate from that of canonical B-DNA. RDCs are effective probes for global structural perturbations and our results provide no evidence favoring the presence of E- or F-DNA forms under solution conditions. Heteronuclear scalar and residual dipolar couplings aid in capturing subtle variations in the local structure upon 5fC incorporation. Combined analysis across various NMR parameters shows that 5fC influences the local nucleotide structure in the sugar pucker and the glycosyl dihedral angle.
Contrary to common misconception, the DNA duplex embeds subtle differences on top of the uniform double-helix structure based on the primary sequence. For instance, sequence-specific variation in structure is essential for indirect DNA readout carried out by regulatory proteins.92 Conformational flexibility of DNA allows for the torsion angles to sample sparsely populated states and is often functionally relevant. Hoogsteen base pair formation for A–T and C–G pairs is a good example and is known to induce helical bending and increase the propensity of DNA damage in the Watson–Crick phase.57,88,93 Similarly, in B-DNA, 2′-deoxyriboses sugar moieties primarily pucker proximal to the C2′-endo region, transgressing to the C3′-endo conformation at 5–20% population based on the nucleobase type.94 This is not surprising given that the C2′-endo form in B-form DNA is only marginally more stable than the C3′-endo form by ∼1 kcal mol−1, with transitions occurring in the pico-nanoseconds timescale (energy barrier 2–5 kcal mol−1).68,95–97 Molecular dynamics simulation shows that C2′-endo to C3′-endo transitions occur stochastically and are uncooperative.94 Hence, individual sugar puckering is rapid and such effects cannot be directly studied by spectroscopy as they do not dramatically impact the average duplex structure. Importantly, C3′-endo conformations are more commonly observed in pyrimidine (especially for C) nucleotides than in purine.45,46 The lifetime and population of C3′-endo conformation increase to 20% for C located in the CG, CA, and TG steps compared to other dinucleotide steps, with CA, TG, TA, and CG being the most flexible steps in the DNA duplex.46,985fC exploits this unique property of C, enhances the flexibility of DNA and establishes itself as a distinct cytosine modification over the other 5mC and 5hmC. Such a facet of 5fC, in addition to weakened H-bonds, enables duplex DNA containing the modification to transiently sample locally melted and flexible states that results in faster duplex cyclization rates for 5fC in comparison to C/5mC/5hmC. The rate increases with multiple 5fC modifications in the sequence.40
It is well documented now that the chemical structure of the modifications in the 5th position of the cytosine base serves as a mode of recognition and binding of proteins.25,99–102 For instance, 5fC modification strongly interacts with transcriptional regulators, DNA repair factors and chromatin regulators.25 The CHO group present in 5fC is known to form covalent interactions with the amine groups present in proteins such as methyltransferases103 and histones.104 The motivation in our study was to interrogate the plausible effects that transcend the chemical structure and potentially drive conformational changes that modulate the properties of the double helical DNA structure. Our results unequivocally indicate that 5fC introduction into the DNA duplex results in the sampling of C–G conformations that are not accessible within any sequence context. Hence, the weakening of H-bond strength achieved due to the formyl modification in the 5fC–G pair enhances the base opening rate,35 local fluctuations,41 and double-strand DNA dissociation constant resulting in reduced DNA duplex stability44 in comparison to any possible canonical primary sequence containing Watson–Crick base pairs. This is important as transcription factors are known to exploit the weakened base pair towards recognition.105 Hence, because of base-pair wobbling around the 5fC–G base-pair, the duplex achieves an enhanced degree of flexibility. Weakening of the 5fC–G H-bond increases the probability of 5fC base flipping and un-base stacking over the other 5mC and 5hmC, which may assist TDG in recognizing. Therefore, the base flipping into the catalytic pocket of the thymidine DNA glycosylase/base-excision repair106 enzymes is plausibly facilitated.
Another factor to highlight here is the difference between epigenetic and damage modifications in duplex DNA. For instance, 1-methyadenine (m1A) is a known form of DNA damage with a methyl group inhibiting Watson–Crick pairing and facilitating Hoogsteen pairing.57 Such a modification is found to enhance local fluctuations in the millisecond time scale. In contrast, 5fC epigenetic modification enhances conformational flexibility in the faster pico-nanosecond time scale motion (as no appreciable resonance broadening is observed in the NMR spectra of DNAF#) contrasting the effect of epigenetic versus a damage (m1A) modification in the conformational landscape of DNA duplexes. This potentially underlines the fact that damage modifications that severely affect the function of DNA duplexes cause more alarming conformational changes in comparison to epigenetic modifications that play more than one given role in the biological context. A thorough structural mapping of damage and natural modifications would aid in testing/refining this hypothesis.
Conclusions
Cytosine epigenetic modifications are reported to sample a wide range of polymorphic structures. Our study shows that all the cytosine modifications do not deviate from the B-DNA duplex structure, although prior crystallographic reports have suggested the same. We present heteronuclear chemical shifts and scalar couplings as effective probes to map subtle variations arising from chemical modifications in DNA. These NMR probes reveal the weakening of the G–C H-bonding upon formyl modification. The subtle differences between single and multiple 5fC modifications are evidently observed with these measurements. Notably, the change in the pucker/glycosyl angle for the 5fC modified duplexes highlights the fact that cytosine uniquely manages to change the local flexibility of the duplex thereby enhancing its functionality within the context of duplex DNA. Such a feature is brought about with no change in the canonical base pair, hence not affecting the integral function of DNA. Also, the fundamental paradigm of structure–function within molecular biology is expanded to include conformational flexibility that provides distinctive avenues for encoding information within the limited chemical space of nucleotides. Their alterations to the physical properties of duplex DNA upon 5fC modification throw light on the role of epigenetic modifications in their biological function.Experimental methods
Choice of the primary sequence
DNA oligonucleotides were prepared with the palindromic sequence 5′-CTAGGGTAG-3′; 4th/6th/8th positions were modified with 5mC/5hmC/5fC in various samples (Fig. 1A). The choice of the sequence was motivated by the (CpG)n repeat sequence that also has abundant data available across crystallography,37,39 solution-state NMR,39 infrared spectroscopy,41 and computational studies.38 Additionally, the system enables careful dissection of chemical shift perturbations that arise solely due to base-pairing (8th position, single hemi-modified) and a combination of base-pairing and stacking (6th position, single fully modified). The sequence also sports a CpG repeat sequence that allows one to understand the effect of single versus multiple contiguous modifications. 5mC/5hmC/5fC modified duplexes are labeled as DNAM#/DNAH#/DNAF# (# = 6, 8, or 3 for single fully, single hemi-modified, or triple modification, respectively). The sample without any modifications serving as the control is denoted as DNAcontrol.
Sample preparation
DNAcontrol was purchased from Integrated DNA Technologies (IDT USA) and modified DNAN# (N = M/H/F) from Keck Oligonucleotide Synthesis Resource (W. M. Keck Foundation) synthesized using phosphoramidite chemistry107 and purified with RP-HPLC (purity >99% from mass spectrometry). DNA oligonucleotides were used as is, without any further purification. Duplexes were annealed by heating single-strands (∼200 μM concentration) in pure water to 95 °C for 10 min and cooling the sample at room temperature. The duplexes were then subjected to centrifugal concentration using 3 kDa cut-off filters (EMD Millipore) with the NMR buffer (15 mM sodium phosphate pH 7.4, 25 mM sodium chloride, 0.1 mM ethylene diamine tetraacetate (EDTA), 10% D2O for field-frequency locking, 50 μM trimethylsilyl propanoic acid (TSP) as an internal standard for chemical shift referencing). The final duplex DNA concentrations for DNAcontrol and modified DNAN# were between 90 and 250 μM. Partial anisotropic alignment was achieved by adding 20–25 mg mL−1 filamentous Pf1 phage108 to the sample, keeping the DNA duplex concentration as similar as possible to the isotropic condition.NMR spectroscopy
NMR experiments were performed employing a 700 MHz 1H Larmor precession frequency Bruker Avance-III spectrometer equipped with a cryogenically cooled triple {1H, 13C}, 15N channel resonance probe at 298 K. Chemical shifts were referenced using TSP to 0 ppm on the indirect 13C dimension (following appropriate spectral aliasing) and direct 1H dimension. The 1H imino 1D NMR spectra of the DNAcontrol and DNAN# samples show characteristic resonances between 12 and 14 ppm (Fig. 2A), indicating stable duplex formation facilitated by Watson–Crick pairing. The 1H chemical shifts of DNAcontrol and DNAF3 were observed to be in excellent agreement (±0.02 ppm) with previously published values.3913C–C7 shifts of modifiedH3, H2OH, and HO groups fall in the expected ∼15, ∼60, and ∼191 ppm indicating their proper incorporation in the sites of interest in all the DNA systems (DNAN#) studied. 1H shifts of the aldehyde C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) O proton resonating at 9.2 ppm in DNAF# samples against 9.5 ppm for the free base indicate stacking accompanied by duplex formation. In addition, observation of the CO resonance at 9.2 ppm indicates the insignificant population of the geminal diol C(O)2 form, which resonates at ∼5 ppm.109
O proton resonating at 9.2 ppm in DNAF# samples against 9.5 ppm for the free base indicate stacking accompanied by duplex formation. In addition, observation of the CO resonance at 9.2 ppm indicates the insignificant population of the geminal diol C(O)2 form, which resonates at ∼5 ppm.109
Data were acquired using TopSpin 3.6pl5, with sparse Poisson-Gap110 sampling scheduling done using the macro ‘nusPGSv3’ (PGS_TS3.2 distribution) obtained from the Wagner's lab (gwagner.med.harvard.edu). Two-dimensional (2D) heteronuclear correlations 13C–1H and 15N-1H were obtained using the sensitivity-enhanced adiabatic heteronuclear single quantum coherence (HSQC with 13C adiabatic pulses with water flip-back)111 and band-Selective Optimized Flip Angle Short Transient (SOFAST-) heteronuclear multiple quantum coherence (HMQC)112,113 spectroscopy, respectively, from the Bruker pulse program library. The 13C and 15N spectral widths (with carrier position) were optimized to obtain maximal resolution (64 ms t1,max) to 8 (83) and 16 (153) ppm, respectively, by spectral aliasing with minimal signal overlap/loss. The scheduling lists were generated with 5–30% (5% increments), 50%, 75%, and 95% sampling to obtain the optimum level of sampling, providing a robust measurement of chemical shifts and scalar couplings. Data were then processed using multi-dimensional decomposition114 (qMDD 2.5 v3b) followed by NMRPipe115 and analyzed using NMRFAM-SPARKY.116 The details of the performance of sparse sampling methodology to measure chemical shifts and couplings robustly and reliably are provided in the ESI.†
The 2D nuclear Overhauser effect (NOESY, 100, 150, and 200 ms mixing time) and double-quantum filtered correlation (DQF-COSY) spectra were acquired with the 3-9-19 WATERGATE water suppression scheme and uniform sampling with an inter-scan delay of 2.5 and 1.5 s, respectively.1111H–1H correlation 2D data were acquired using conventional Nyquist sampling. 1JCH and 1DCH couplings were measured for samples under isotropic and anisotropic conditions, respectively, from the frequency difference between the doublets obtained from 13C–1H 2D HSQC without decoupling in the direct detect 1H dimension.
Analysis of the NOESY spectra, structure refinement and analysis
2D NOESY data were analyzed for all samples to obtain inter-proton distances required for structure refinement protocols.57,58 Briefly, the H5–H6 distance in cytosine was referenced to 2.45 Å, the methyl cross-peaks were calibrated with the H6–H7# distance in thymine to 3.00 Å, and the H2′–H2′′ distances to 1.76 Å.117 The distances obtained were then relaxed by 50% to obtain the lower and upper limit constraints for the structure refinement, as described earlier.57XPLOR-NIH77 version 2.41 was used for structure refinement following a simulated annealing protocol. As DNAcontrol and DNAN# are palindromic in nature, the C2-axis of symmetry was input as a constraint. While data for the modified systems were used, the unmodified cytosine base was employed for the structure refinement protocols as a proxy for 5mC, 5hmC, and 5fC modifications, as only the trends of structural perturbations were sought from such refinements. Alignment tensor parameters (Da and Dr – the axial and rhombic components of the tensor) were optimized for the DNA duplexes based on the measured RDC datasets.54 As imino 1H shifts were observed in the characteristic 12–14 ppm region indicative of Watson–Crick base pairs, H-bond constraints were incorporated in the structure refinement protocol. Dihedral angles (except for ε and ζ angles) were constrained as described earlier. Phosphate backbone dihedral angles were not constrained to assess changes in the BI/BII populations upon modified cytosine incorporation. Fifty structures were annealed starting from the idealized B-DNA geometry, and the five structures having no restraint violations were used for further structural analysis. The number of restraints and the summary of structure refinement for each system are listed in Table S3 (ESI†).
Structural analysis of the refined conformers was performed to determine inter- and intra-base pair parameters using 3DNA,89 while helical bending was assessed using CURVES+.19 RDC comparisons (Table S5, ESI†) were generated by fitting experimental RDCs to refined DNA structures with the module calcTensor (single value decomposition for best-fitting experimental measurements to back-predicted values) present in XPLOR-NIH.77
Author contributions
B. S. conceptualized, acquired funding, supervised the investigation, methodology and formal analysis, and wrote the manuscript. M. J. carried out the methods, data curation and analysis, with R. K. R. S. sharing the load and validating the datasets across the entire project. M. J. and R. K. R. S worked in editing the manuscript. A. R. performed analysis of a sub-section of the dataset in this project, with supervision from M. J. and R. K. R. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Indian Institute of Science Education and Research (IISER) Bhopal for providing the necessary research infrastructure. We would like to thank IISER Bhopal for allowing access to the 700 MHz NMR facility at IISER Bhopal and Mr Rajbeer Singh for timely support in the maintenance of the spectrometer. This work was supported by the Science and Engineering Research Board via the Early Career Research grant (ECR/2016/001196) and the start-up research grant (INST/CHM/2016047) from IISER Bhopal to B. S. M. J. thanks CSIR for the fellowship and research support. R. K. R. S. thanks IISER Bhopal for the research fellowship.Notes and references
- T. H. Bester, Gene, 1988, 74, 9–12 CrossRef PubMed.
- M. Okano, S. Xie and E. Li, Nat. Genet., 1998, 19, 219–220 CrossRef CAS PubMed.
- S. Xie, Z. Wang, M. Okano, M. Nogami, Y. Li, W.-W. He, K. Okumura and E. Li, Gene, 1999, 236, 87–95 CrossRef CAS PubMed.
- G. R. Wyatt, Nature, 1950, 166, 237–238 CrossRef CAS PubMed.
- M. Ehrlich and R. Y. Wang, Science, 1981, 212, 1350–1357 CrossRef CAS PubMed.
- M. Ehrlich, M. A. Gama-Sosa, L. H. Huang, R. M. Midgett, K. C. Kuo, R. A. McCune and C. Gehrke, Nucleic Acids Res., 1982, 10, 2709–2721 CrossRef CAS PubMed.
- A. P. Bird, Nature, 1986, 321, 209–213 CrossRef CAS PubMed.
- R. Lister and J. R. Ecker, Genome Res., 2009, 19, 959–966 CrossRef CAS PubMed.
- T. Mohandas, R. S. Sparkes and L. J. Shapiro, Science, 1981, 211, 393–396 CrossRef CAS PubMed.
- J. L. Swain, T. A. Stewart and P. Leder, Cell, 1987, 50, 719–727 CrossRef CAS PubMed.
- W. Reik, A. Collick, M. L. Norris, S. C. Barton and M. A. Surani, Nature, 1987, 328, 248–251 CrossRef CAS PubMed.
- E. Li, C. Beard and R. Jaenisch, Nature, 1993, 366, 362–365 CrossRef CAS PubMed.
- A. P. Wolffe and M. A. Matzke, Science, 1999, 286, 481–486 CrossRef CAS PubMed.
- P. A. Jones and D. Takai, Science, 2001, 293, 1068–1070 CrossRef CAS PubMed.
- P. A. Jones, Nat. Rev. Genet., 2012, 13, 484–492 CrossRef CAS PubMed.
- D. P. Barlow and M. S. Bartolomei, Cold Spring Harb Perspect Biol, 2014, 6, a018382 CrossRef PubMed.
- M. Tahiliani, K. P. Koh, Y. Shen, W. A. Pastor, H. Bandukwala, Y. Brudno, S. Agarwal, L. M. Iyer, D. R. Liu, L. Aravind and A. Rao, Science, 2009, 324, 930–935 CrossRef CAS PubMed.
- S. Ito, L. Shen, Q. Dai, S. C. Wu, L. B. Collins, J. A. Swenberg, C. He and Y. Zhang, Science, 2011, 333, 1300–1303 CrossRef CAS PubMed.
- C. Blanchet, M. Pasi, K. Zakrzewska and R. Lavery, Nucleic Acids Res., 2011, 39, W68–73 CrossRef CAS PubMed.
- R. M. Kohli and Y. Zhang, Nature, 2013, 502, 472–479 CrossRef CAS PubMed.
- M. Bachman, S. Uribe-Lewis, X. Yang, M. Williams, A. Murrell and S. Balasubramanian, Nat. Chem., 2014, 6, 1049–1055 CrossRef CAS PubMed.
- M. Bachman, S. Uribe-Lewis, X. Yang, H. E. Burgess, M. Iurlaro, W. Reik, A. Murrell and S. Balasubramanian, Nat. Chem. Biol., 2015, 11, 555–557 CrossRef CAS PubMed.
- T. Carell, M. Q. Kurz, M. Muller, M. Rossa and F. Spada, Angew. Chem., Int. Ed., 2018, 57, 4296–4312 CrossRef CAS PubMed.
- J. S. Choy, S. Wei, J. Y. Lee, S. Tan, S. Chu and T. H. Lee, J. Am. Chem. Soc., 2010, 132, 1782–1783 CrossRef CAS PubMed.
- M. Iurlaro, G. Ficz, D. Oxley, E. A. Raiber, M. Bachman, M. J. Booth, S. Andrews, S. Balasubramanian and W. Reik, Genome Biol., 2013, 14, R119 CrossRef PubMed.
- C. X. Song, K. E. Szulwach, Q. Dai, Y. Fu, S. Q. Mao, L. Lin, C. Street, Y. Li, M. Poidevin, H. Wu, J. Gao, P. Liu, L. Li, G. L. Xu, P. Jin and C. He, Cell, 2013, 153, 678–691 CrossRef CAS PubMed.
- E. A. Raiber, D. Beraldi, G. Ficz, H. E. Burgess, M. R. Branco, P. Murat, D. Oxley, M. J. Booth, W. Reik and S. Balasubramanian, Genome Biol., 2012, 13, R69 CrossRef PubMed.
- F. Neri, D. Incarnato, A. Krepelova, S. Rapelli, F. Anselmi, C. Parlato, C. Medana, F. Dal Bello and S. Oliviero, Cell Rep., 2015, 10, 674–683 CrossRef CAS PubMed.
- D. Ji, C. You, P. Wang and Y. Wang, Chem. Res. Toxicol., 2014, 27, 1304–1309 Search PubMed.
- M. W. Kellinger, C. X. Song, J. Chong, X. Y. Lu, C. He and D. Wang, Nat. Struct. Mol. Biol., 2012, 19, 831–833 CrossRef CAS PubMed.
- C. O'Neill, Animal Front., 2015, 5, 42–49 CrossRef.
- T. M. Storebjerg, S. H. Strand, S. Hoyer, A. S. Lynnerup, M. Borre, T. F. Orntoft and K. D. Sorensen, Clin Epigenetics, 2018, 10, 105 CrossRef PubMed.
- D. Renciuk, O. Blacque, M. Vorlickova and B. Spingler, Nucleic Acids Res., 2013, 41, 9891–9900 CrossRef CAS PubMed.
- L. Lercher, M. A. McDonough, A. H. El-Sagheer, A. Thalhammer, S. Kriaucionis, T. Brown and C. J. Schofield, Chem. Commun., 2014, 50, 1794–1796 RSC.
- M. W. Szulik, P. S. Pallan, B. Nocek, M. Voehler, S. Banerjee, S. Brooks, A. Joachimiak, M. Egli, B. F. Eichman and M. P. Stone, Biochemistry, 2015, 54, 1294–1305 CrossRef CAS PubMed.
- J. M. Vargason, B. F. Eichman and P. S. Ho, Nat. Struct. Biol., 2000, 7, 758–761 CrossRef CAS PubMed.
- E. A. Raiber, P. Murat, D. Y. Chirgadze, D. Beraldi, B. F. Luisi and S. Balasubramanian, Nat. Struct. Mol. Biol., 2015, 22, 44–49 CrossRef CAS PubMed.
- K. Krawczyk, S. Demharter, B. Knapp, C. M. Deane and P. Minary, Bioinformatics, 2018, 34, 41–48 CrossRef CAS PubMed.
- J. S. Hardwick, D. Ptchelkine, A. H. El-Sagheer, I. Tear, D. Singleton, S. E. V. Phillips, A. N. Lane and T. Brown, Nat. Struct. Mol. Biol., 2017, 24, 544–552 CrossRef CAS PubMed.
- T. T. Ngo, J. Yoo, Q. Dai, Q. Zhang, C. He, A. Aksimentiev and T. Ha, Nat. Commun., 2016, 7, 10813 CrossRef CAS PubMed.
- P. J. Sanstead, B. Ashwood, Q. Dai, C. He and A. Tokmakoff, J. Phys. Chem. B, 2020, 124, 1160–1174 CrossRef CAS PubMed.
- Q. Dai, P. J. Sanstead, C. S. Peng, D. Han, C. He and A. Tokmakoff, ACS Chem. Biol., 2016, 11, 470–477 CrossRef CAS PubMed.
- D. Herschlag and M. M. Pinney, Biochemistry, 2018, 57, 3338–3352 CrossRef CAS PubMed.
- R. C. A. Dubini, A. Schon, M. Muller, T. Carell and P. Rovo, Nucleic Acids Res., 2020, 48, 8796–8807 CrossRef CAS PubMed.
- Z. Wu, F. Delaglio, N. Tjandra, V. B. Zhurkin and A. Bax, J. Biomol. NMR, 2003, 26, 297–315 CrossRef CAS PubMed.
- E. N. Nikolova, G. D. Bascom, I. Andricioaei and H. M. Al-Hashimi, Biochemistry, 2012, 51, 8654–8664 CrossRef CAS PubMed.
- Y. F. He, B. Z. Li, Z. Li, P. Liu, Y. Wang, Q. Tang, J. Ding, Y. Jia, Z. Chen, L. Li, Y. Sun, X. Li, Q. Dai, C. X. Song, K. Zhang, C. He and G. L. Xu, Science, 2011, 333, 1303–1307 CrossRef CAS PubMed.
- J. Czernek, R. Fiala and V. Sklenar, J. Magn. Reson., 2000, 145, 142–146 CrossRef CAS PubMed.
- S. L. Lam and L. M. Chi, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 56, 289–310 CrossRef CAS PubMed.
- J. M. Fonville, M. Swart, Z. Vokacova, V. Sychrovsky, J. E. Sponer, J. Sponer, C. W. Hilbers, F. M. Bickelhaupt and S. S. Wijmenga, Chemistry, 2012, 18, 12372–12387 CrossRef CAS PubMed.
- S. S. Wijmenga and B. N. M. van Buuren, Prog. Nucl. Magn. Reson. Spectrosc., 1998, 32, 287–387 CrossRef CAS.
- S. Nozinovic, P. Gupta, B. Furtig, C. Richter, S. Tullmann, E. Duchardt-Ferner, M. C. Holthausen and H. Schwalbe, Angew. Chem., Int. Ed., 2011, 50, 5397–5400 CrossRef CAS PubMed.
- M. R. Hansen, L. Mueller and A. Pardi, Nat. Struct. Biol., 1998, 5, 1065–1074 CrossRef CAS PubMed.
- A. Vermeulen, H. Zhou and A. Pardi, J. Am. Chem. Soc., 2000, 122, 9638–9647 CrossRef CAS.
- D. MacDonald, K. Herbert, X. Zhang, T. Pologruto and P. Lu, J. Mol. Biol., 2001, 306, 1081–1098 CrossRef CAS PubMed.
- Z. Wu, M. Maderia, J. J. Barchi, Jr., V. E. Marquez and A. Bax, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 24–28 CrossRef CAS PubMed.
- B. Sathyamoorthy, H. Shi, H. Zhou, Y. Xue, A. Rangadurai, D. K. Merriman and H. M. Al-Hashimi, Nucleic Acids Res., 2017, 45, 5586–5601 CrossRef CAS PubMed.
- B. Sathyamoorthy, R. K. R. Sannapureddi, D. Negi and P. Singh, J Magn Reson Open, 2022, 10–11, 100035 CrossRef.
- J. Jerbi and M. Springborg, J. Comput. Chem., 2017, 38, 1049–1056 CrossRef CAS PubMed.
- M. Munzel, U. Lischke, D. Stathis, T. Pfaffeneder, F. A. Gnerlich, C. A. Deiml, S. C. Koch, K. Karaghiosoff and T. Carell, Chemistry, 2011, 17, 13782–13788 CrossRef PubMed.
- H. Hashimoto, Y. O. Olanrewaju, Y. Zheng, G. G. Wilson, X. Zhang and X. Cheng, Genes Dev., 2014, 28, 2304–2313 CrossRef PubMed.
- K. L. Greene, Y. Wang and D. Live, J. Biomol. NMR, 1995, 5, 333–338 CrossRef CAS PubMed.
- F. J. Van de Ven and C. W. Hilbers, Eur. J. Biochem., 1988, 178, 1–38 CrossRef CAS PubMed.
- R. V. Hosur, G. Govil and H. T. Miles, Magn. Reson. Chem., 1988, 26, 927–944 CrossRef CAS.
- L. J. Rinkel, M. R. Sanderson, G. A. van der Marel, J. H. van Boom and C. Altona, Eur. J. Biochem., 1986, 159, 85–93 CrossRef CAS PubMed.
- A. S. Serianni, J. Wu and I. Carmichael, J. Am. Chem. Soc., 2002, 117, 8645–8650 CrossRef.
- J. T. Fischer and U. M. Reinscheid, Eur. J. Org. Chem., 2006, 2074–2080 CrossRef CAS.
- N. Foloppe and A. D. MacKerell, Biophys. J., 1999, 76, 3206–3218 CrossRef CAS PubMed.
- B. Schneider, Z. Moravek and H. M. Berman, Nucleic Acids Res., 2004, 32, 1666–1677 CrossRef CAS PubMed.
- J. S. Richardson, B. Schneider, L. W. Murray, G. J. Kapral, R. M. Immormino, J. J. Headd, D. C. Richardson, D. Ham, E. Hershkovits, L. D. Williams, K. S. Keating, A. M. Pyle, D. Micallef, J. Westbrook, H. M. Berman and R. N. A. O. Consortium, RNA, 2008, 14, 465–481 CrossRef CAS PubMed.
- A. Barbic, D. P. Zimmer and D. M. Crothers, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2369–2373 CrossRef CAS PubMed.
- K. McAteer, A. Aceves-Gaona, R. Michalczyk, G. W. Buchko, N. G. Isern, L. A. Silks, J. H. Miller and M. A. Kennedy, Biopolymers, 2004, 75, 497–511 CrossRef CAS PubMed.
- R. Stefl, H. Wu, S. Ravindranathan, V. Sklenar and J. Feigon, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1177–1182 CrossRef CAS PubMed.
- J. Feigon, J. M. Wright, W. Leupin, W. A. Denny and D. R. Kearns, J. Am. Chem. Soc., 1982, 104, 5540–5541 CrossRef CAS.
- D. R. Hare, D. E. Wemmer, S.-H. Chou, G. Drobny and B. R. Reid, J. Mol. Biol., 1983, 171, 319–336 CrossRef CAS PubMed.
- M. A. Weiss, D. J. Patel, R. T. Sauer and M. Karplus, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 130–134 CrossRef CAS PubMed.
- C. Schwieters, J. Kuszewski and G. Mariusclore, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 48, 47–62 CrossRef CAS.
- X. J. Lu and W. K. Olson, Nat. Protoc., 2008, 3, 1213–1227 CrossRef CAS PubMed.
- A. Thalhammer, A. S. Hansen, A. H. El-Sagheer, T. Brown and C. J. Schofield, Chem. Commun., 2011, 47, 5325–5327 RSC.
- R. Wang, Z. Luo, K. He, M. O. Delaney, D. Chen and J. Sheng, Nucleic Acids Res., 2016, 44, 4968–4977 CrossRef CAS PubMed.
- E. P. Wright, M. A. S. Abdelhamid, M. O. Ehiabor, M. C. Grigg, K. Irving, N. M. Smith and Z. A. E. Waller, Nucleic Acids Res., 2020, 48, 55–62 CrossRef CAS PubMed.
- R. C. A. Dubini, E. Korytiakova, T. Schinkel, P. Heinrichs, T. Carell and P. Rovo, ACS Phys Chem Au, 2022, 2, 237–246 CrossRef CAS PubMed.
- N. Karino, Y. Ueno and A. Matsuda, Nucleic Acids Res., 2001, 29, 2456–2463 CrossRef CAS PubMed.
- E. N. Nikolova, E. Kim, A. A. Wise, P. J. O'Brien, I. Andricioaei and H. M. Al-Hashimi, Nature, 2011, 470, 498–502 CrossRef CAS PubMed.
- H. S. Alvey, F. L. Gottardo, E. N. Nikolova and H. M. Al-Hashimi, Nat. Commun., 2014, 5, 4786 CrossRef CAS PubMed.
- A. L. Stelling, A. Y. Liu, W. Zeng, R. Salinas, M. A. Schumacher and H. M. Al-Hashimi, Angew. Chem., Int. Ed., 2019, 58, 12010–12013 CrossRef CAS PubMed.
- H. Zhou, B. Sathyamoorthy, A. Stelling, Y. Xu, Y. Xue, Y. Z. Pigli, D. A. Case, P. A. Rice and H. M. Al-Hashimi, Biochemistry, 2019, 58, 1963–1974 CrossRef CAS PubMed.
- Y. Xu, A. Manghrani, B. Liu, H. Shi, U. Pham, A. Liu and H. M. Al-Hashimi, J. Biol. Chem., 2020, 295, 15933–15947 CrossRef CAS PubMed.
- M. Banyay, M. Sarkar and A. Gräslund, Biophys. Chem., 2003, 104, 477–488 CrossRef CAS PubMed.
- I. J. Kimsey, K. Petzold, B. Sathyamoorthy, Z. W. Stein and H. M. Al-Hashimi, Nature, 2015, 519, 315–320 CrossRef CAS PubMed.
- E. S. Szymanski, I. J. Kimsey and H. M. Al-Hashimi, J. Am. Chem. Soc., 2017, 139, 4326–4329 CrossRef CAS PubMed.
- R. Rohs, S. M. West, A. Sosinsky, P. Liu, R. S. Mann and B. Honig, Nature, 2009, 461, 1248–1253 CrossRef CAS PubMed.
- E. N. Nikolova, H. Zhou, F. L. Gottardo, H. S. Alvey, I. J. Kimsey and H. M. Al-Hashimi, Biopolymers, 2013, 99, 955–968 CAS.
- A. Perez, F. J. Luque and M. Orozco, J. Am. Chem. Soc., 2007, 129, 14739–14745 CrossRef CAS PubMed.
- A. Saran, D. Perahia and B. Pullman, Theor. Chim. Acta, 1973, 30, 31–44 CrossRef CAS.
- N. Foloppe and A. D. MacKerell, J. Phys. Chem. B, 1998, 102, 6669–6678 CrossRef CAS.
- W. K. Olson, J. Am. Chem. Soc., 2002, 104, 278–286 CrossRef.
- M. A. el Hassan and C. R. Calladine, J. Mol. Biol., 1996, 259, 95–103 CrossRef CAS PubMed.
- A. M. Deaton and A. Bird, Genes Dev., 2011, 25, 1010–1022 CrossRef CAS PubMed.
- O. Yildirim, R. Li, J. H. Hung, P. B. Chen, X. Dong, L. S. Ee, Z. Weng, O. J. Rando and T. G. Fazzio, Cell, 2011, 147, 1498–1510 CrossRef CAS PubMed.
- M. Mellen, P. Ayata, S. Dewell, S. Kriaucionis and N. Heintz, Cell, 2012, 151, 1417–1430 CrossRef CAS PubMed.
- C. Rausch, F. D. Hastert and M. C. Cardoso, J. Mol. Biol., 2019, 432(6), 1731–1746 CrossRef PubMed.
- K. Sato, K. Kawamoto, S. Shimamura, S. Ichikawa and A. Matsuda, Bioorg. Med. Chem. Lett., 2016, 26, 5395–5398 CrossRef CAS PubMed.
- F. Li, Y. Zhang, J. Bai, M. M. Greenberg, Z. Xi and C. Zhou, J. Am. Chem. Soc., 2017, 139, 10617–10620 CrossRef CAS PubMed.
- A. Afek, H. Shi, A. Rangadurai, H. Sahay, A. Senitzki, S. Xhani, M. Fang, R. Salinas, Z. Mielko, M. A. Pufall, G. M. K. Poon, T. E. Haran, M. A. Schumacher, H. M. Al-Hashimi and R. Gordan, Nature, 2020, 587, 291–296 CrossRef CAS PubMed.
- W. Yang, Cell Res., 2008, 18, 184–197 CrossRef CAS PubMed.
- A. A. Tanpure and S. Balasubramanian, ChemBioChem, 2017, 18, 2236–2241 CrossRef CAS PubMed.
- G. M. Clore, M. R. Starich and A. M. Gronenborn, J. Am. Chem. Soc., 1998, 120, 10571–10572 CrossRef CAS.
- F. L. Zott, V. Korotenko and H. Zipse, ChemBioChem, 2022, 23, e202100651 CrossRef CAS PubMed.
- S. G. Hyberts, K. Takeuchi and G. Wagner, J. Am. Chem. Soc., 2010, 132, 2145–2147 CrossRef CAS PubMed.
- J. Cavanagh, N. Skelton, W. Fairbrother, M. Rance and I. Palmer, Arthur, Protein NMR Spectroscopy, Academic Press, 2006 Search PubMed.
- J. Farjon, J. Boisbouvier, P. Schanda, A. Pardi, J. P. Simorre and B. Brutscher, J. Am. Chem. Soc., 2009, 131, 8571–8577 CrossRef CAS PubMed.
- B. Sathyamoorthy, J. Lee, I. Kimsey, L. R. Ganser and H. Al-Hashimi, J. Biomol. NMR, 2014, 60, 77–83 CrossRef CAS PubMed.
- K. Kazimierczuk and V. Y. Orekhov, Angew. Chem., Int. Ed., 2011, 50, 5556–5559 CrossRef CAS PubMed.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS PubMed.
- W. Lee, M. Tonelli and J. L. Markley, Bioinformatics, 2015, 31, 1325–1327 CrossRef PubMed.
- J. D. Baleja, M. W. Germann, J. H. van de Sande and B. D. Sykes, J. Mol. Biol., 1990, 215, 411–428 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04837j |
| ‡ Contributed equally to this work. |
| This journal is © the Owner Societies 2023 |