
5-Formylcytosine weakens the G–C pair and imparts local conformational fluctuations to DNA duplexes†

Abstract
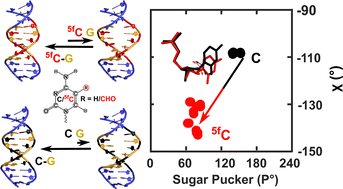
DNA epigenetic modifications such as 5-methyl (5mC), 5-hydroxymethyl (5hmC), 5-formyl (5fC) and 5-carboxyl (5caC) cytosine have unique and specific biological roles. Crystallographic studies of 5mC containing duplexes were conducted in the A-, B- or the intermediate E-DNA polymorphic forms. 5fC-modified duplexes initially observed in the disputed F-DNA architecture were subsequently crystallized in the A-form, suggesting that epigenetic modifications enable DNA sequences to adopt diverse conformational states that plausibly contribute to their function. Solution-state studies of these modifications were found in the B-DNA form, with marked differences in the conformational flexibility of 5fC containing duplexes in comparison to C/5mC containing duplexes, compromising the DNA duplex's stability. Herein, we systematically evaluate sensitive and commonly inaccessible NMR parameters to map the subtle differences between C, 5mC, and their oxidized (5hmC/5fC) counterparts. We observe that 15N/1H chemical shifts effectively report on the weakening of 5fC–G Watson–Crick base-pair H-bonding, extending the instability beyond any achievable within the sequence-specific changes in DNA. Triple 5fC containing sequences propagate the destabilization farther from the site of modifications, explaining reduced duplex stability upon multiple modifications. Additionally, scalar and residual dipolar coupling measurements unravel local sugar pucker fluctuations. One-bond 13C–1H scalar coupling measurements point towards a significant deviation away from the anticipated C2′-endo pucker for the 5fC modified nucleotide. Structural models obtained employing 13C–1H residual dipolar couplings and inter-proton distances corroborate the sugar pucker's deviation for 5fC modified DNA duplexes. The changes in the sugar pucker equilibria remain local to the 5fC modified nucleotide sans additive/long-range effects arising from multiple contiguous modifications. These observations highlight the impact of a major groove modification that alters the physical properties of DNA duplex without disturbing the Watson–Crick face. The changes observed in our studies for the 5fC containing DNA contrast with the perturbations induced by damage/lesion highlight the varied conformational preferences that modified nucleobases impart to the DNA duplex. As sequence-specific DNA transactions are rooted in the base-pair stability and pucker deviations, the observed structural perturbations for 5fC-modified DNA potentially play critical functional roles, such as protein-DNA recognition and interactions.
- This article is part of the themed collections: Emerging concepts in nucleic acids: structures, functions and applications and 2022 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

