
Photoexcited charge manipulation in conjugated polymers bearing a Ru(II) complex catalyst for visible-light CO2 reduction†
Akinobu
Nakada
 *ab,
Ryuichi
Miyakawa
a,
Ren
Itagaki
a,
Kosaku
Kato
c,
Chinami
Takashima
d,
Akinori
Saeki
e,
Akira
Yamakata
c,
Ryu
Abe
f,
Hiromi
Nakai
dgh and
Ho-Chol
Chang
*a
*ab,
Ryuichi
Miyakawa
a,
Ren
Itagaki
a,
Kosaku
Kato
c,
Chinami
Takashima
d,
Akinori
Saeki
e,
Akira
Yamakata
c,
Ryu
Abe
f,
Hiromi
Nakai
dgh and
Ho-Chol
Chang
*a
aDepartment of Applied Chemistry, Faculty of Science and Engineering, Chuo University, 1-13-27 Kasuga, Bunkyo-ku, Tokyo 112-8551, Japan. E-mail: nakada@scl.kyoto-u.ac.jp; chang@kc.chuo-u.ac.jp
bPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
cGraduate School of Engineering, Toyota Technological Institute, 2-12-1 Hisakata, Tempaku-ku, Nagoya 468-8511, Japan
dWaseda Research Institute for Science and Engineering (WISE), Department of Chemistry and Biochemistry, School of Advanced Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku, Tokyo 169-8555, Japan
eDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
fDepartment of Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
gWaseda Research Institute for Science, Waseda University, 3-4-1 Okubo, Shinjuku, Tokyo 169-8555, Japan
hElements Strategy Initiative for Catalysts & Batteries (ESICB), Kyoto University, 1-30 Goryo-Ohara, Nishikyo-ku, Kyoto 615-8245, Japan
First published on 9th May 2022
Abstract
Conjugated polymers have emerged as promising candidates for photocatalyst materials. Design principles that maximize the synergy between the conjugated skeleton and catalyst moiety are strongly desired to be established for achieving efficient photocatalysis. Herein, the photoexcited charge manipulation was demonstrated by molecular engineering in conjugated polymers bearing a Ru(II) complex as the CO2 reduction photocatalyst. Combinational studies based on ultrafast spectroscopies and theoretical calculations revealed that the introduction of an electron-donating carbazole (Cz) skeleton in the polymer enables enhanced photoexcited charge trapping on the Ru(II)-complex catalyst moiety. The carbazole-based polymer [Cz-bpyRu]n facilitates CO2 reduction under visible light even longer than 500 nm and exhibited 7- to 15-fold greater activity than those of phenyl (Ph) and benzothiadiazole (Bt) counterparts. The findings of this study thus provide insights into molecular engineering for photoexcited charge manipulation to achieve efficient photocatalysis.
 Akinobu Nakada | Akinobu Nakada is a Senior Lecturer at Kyoto University. He received his PhD from Tokyo Institute of Technology in 2017. During 2017–2018, he joined Kyoto University as a Postdoctoral Fellow and a specially-appointed Assistant Professor. After serving as an Assistant Professor at Chuo University from 2019 to 2021, he was promoted to the present position at Kyoto University in 2022. From 2020, he concurrently serves as a PRESTO researcher of Japan Science and Technology Agency. His research interests include molecular engineering into photocatalyst materials/systems for light-to-chemical conversion. |
Introduction
Reductive conversion of CO2 into energy-added molecules has been an important subject in various fields including materials chemistry,1,2 catalysis,3,4 electrochemistry,5,6 and photochemistry,7,8 from viewpoints of both decreasing CO2 concentration and gaining energy and carbon resources.9–11 In particular, the development of photocatalyst materials, which facilitate CO2 reduction without high temperature and/or pressure, has been regarded as a promising way.7,8For the past four decades, molecular-based photocatalysts including metal complexes12,13 and semiconductor-based photocatalysts including metal oxides14–16 and mixed-anion materials16,17 have been extensively studied. High selectivity for CO2 reduction, based on well-defined and tunable active sites, is one of the advantages of molecular photocatalysts, as has been demonstrated so far.12,18 Another important priority of molecular photocatalysis is the designability of molecular orbitals in order to facilitate transfer and separation of photoexcited charges for the efficient photocatalysis even under visible-light illumination.13
On the other hand, semiconductor photocatalysts have advantageous in terms of band formation.8,15 The band-gap excitation of semiconductors generates multiple electrons and holes in the conduction and valence bands. Therefore, simultaneous multi-electron reduction and oxidation reactions, e.g., overall water splitting19–22 and CO2 reduction using water as the electron source,16 have been accomplished using a number of semiconductor photocatalysts, while such reactions have hardly been reported using a molecular photocatalyst system.23 In terms of CO2 reduction, however, semiconductor photocatalysts frequently suffer from the low selectivity of CO2 reduction by competing with efficient proton reduction.16 Recently, molecular-semiconductor hybrid photocatalysts have been developed to maximize their advantages, although it still remains a challenge for efficient photocatalytic CO2 reduction.24–26
Recently, organic conjugated polymers have emerged as new candidates for photocatalyst materials.27,28 The organic polymers not only exhibit semiconducting properties due to the extended π-conjugation but also have the molecular designability.29 To date, organic polymer-based photocatalysts have been developed for CO2 reduction under visible light.30–39 Metal-based catalytic sites31,33–35,39 are frequently required to selectively reduce CO2 although some organic polymers are reported to facilitate metal-free CO2 reduction.30,32,36–38 Hence, the design principle, that maximizes the synergy between the conjugated skeleton and catalyst moiety, is strongly desired for achieving efficient photocatalytic CO2 reduction. Several combinations of the metal-based catalytic sites and organic polymers have been developed so far,31,33–35,39 while the design principles, particularly to manipulate the excited charge carrier have not been established satisfactorily. Herein, we show strong impacts of excited charge distribution in conjugated polymer photocatalysts with a site-selectively incorporated Ru(II) complex catalyst (Fig. 1a), which are elucidated by a combination of ultrafast spectroscopy and theoretical calculation, on their photocatalytic activity for CO2 reduction.
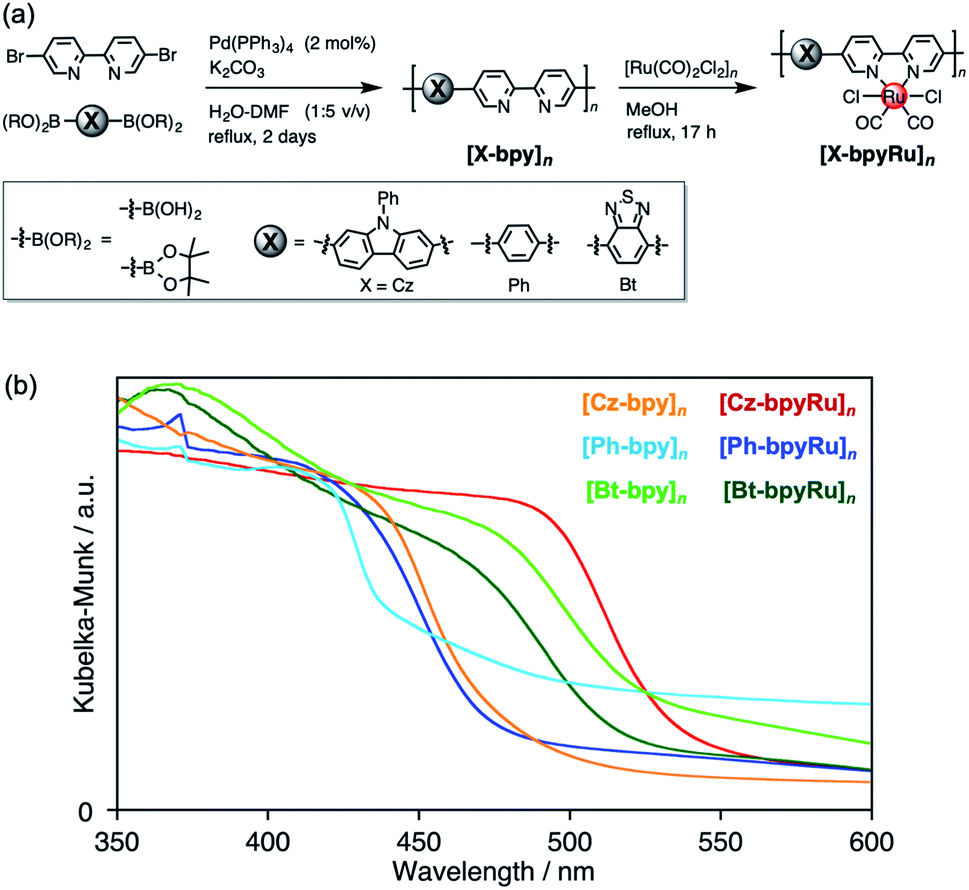 | ||
| Fig. 1 (a) Synthetic procedures and (b) UV-vis diffuse reflectance spectra of the polymer photocatalysts developed in this study. | ||
Results and discussion
[X-bpy]n (X = Ph, Bt, and Cz) were synthesized by the Suzuki–Miyaura cross-coupling reaction between 5,5′-dibromo-2,2′-bipyridine (Br2bpy) and diboronic acids or esters corresponding to the X moieties (X-(B(OR)2)2) with the aid of the Pd(PPh3)4 catalyst (Fig. 1a). MALDI-TOF-MS spectra of the obtained materials gave repeated peaks with interpeak m/z differences corresponding to the X-bpy unit (Fig. S1†), indicative of the formation of –[X-bpy]– conjugated networks. The maximum numbers of connected units detected in the MALDI-TOF-MS spectra were 16, 32, and 19 for [Cz-bpy]n, [Ph-bpy]n, and [Bt-bpy]n, respectively. ATR-FT-IR spectra of [X-bpy]n, which exhibited specific peaks similar to the model monomers corresponding to the X and bpy units, supported that the X and bpy skeletons were maintained (Fig. S2–S4†).A Ru(II)-complex catalyst moiety was introduced by refluxing [Ru(CO)2Cl2]n and the polymers [X-bpy]n in methanol. The obtained powder exhibited characteristic CO vibrations at 2054 and 1990 cm−1 assignable to the Ru(bpy)(CO)2Cl2-type coordination environment regardless of the type of X, indicative of successful post-complexation of the Ru(CO)2Cl2 unit to the bipyridine moiety in [X-bpy]n as the ligand (Fig. S2–S4†). Based on elemental analyses, the introduced Ru complex moieties were estimated to be at least ca. 2/3 of the bpy skeleton for all [X-bpyRu]n. The Ru(II) complex-incorporated polymers are denoted as [X-bpyRu]n. Scanning electron microscope (SEM) images and XRD patterns suggested that the obtained materials had indefinite low-crystalline structures (Fig. S5 and S6†). We also tried to directly synthesize [Cz-bpyRu]n by Suzuki–Miyaura cross-coupling between Cz-(B(OR)2)2 and the as-synthesized Ru(Br2bpy)(CO)2Cl2. However, the CO vibration peaks in IR spectra were shifted to 2019 and 1923 cm−1 after the reaction (Fig. S7†), suggesting that the Ru(bpy)(CO)2Cl2-type coordination environment was not maintained, but there was a formation of a [Ru(bpy)(CO)2]n-type network with Ru–Ru bonds.40,41 Therefore, we concluded that the post complexation method is more suitable for the precise synthesis of the conjugated polymer with structurally well-defined Ru(II) complex catalyst, and will describe the post-complexation polymer unless otherwise noted.
In contrast to the colorless building-block monomers (Fig. S8†), the obtained polymers exhibited largely red-shifted absorption covering the visible region due to the formation of the extended π-conjugation (Fig. 1b), as reported for conventional conjugated polymers.27,28 The band positions for each material, which were estimated by the bandgaps (Eg) and the ionization energies determined using photoelectron yield spectroscopy (PYS),42 are shown in Fig. 2. Focusing on the Ru non-incorporated materials, electron-deficient Bt (Eg = 2.25 eV) and electron-rich Cz (Eg = 2.56 eV) made impacts the narrowing bandgaps compared with [Ph-bpy]n (Eg = 2.69 eV).
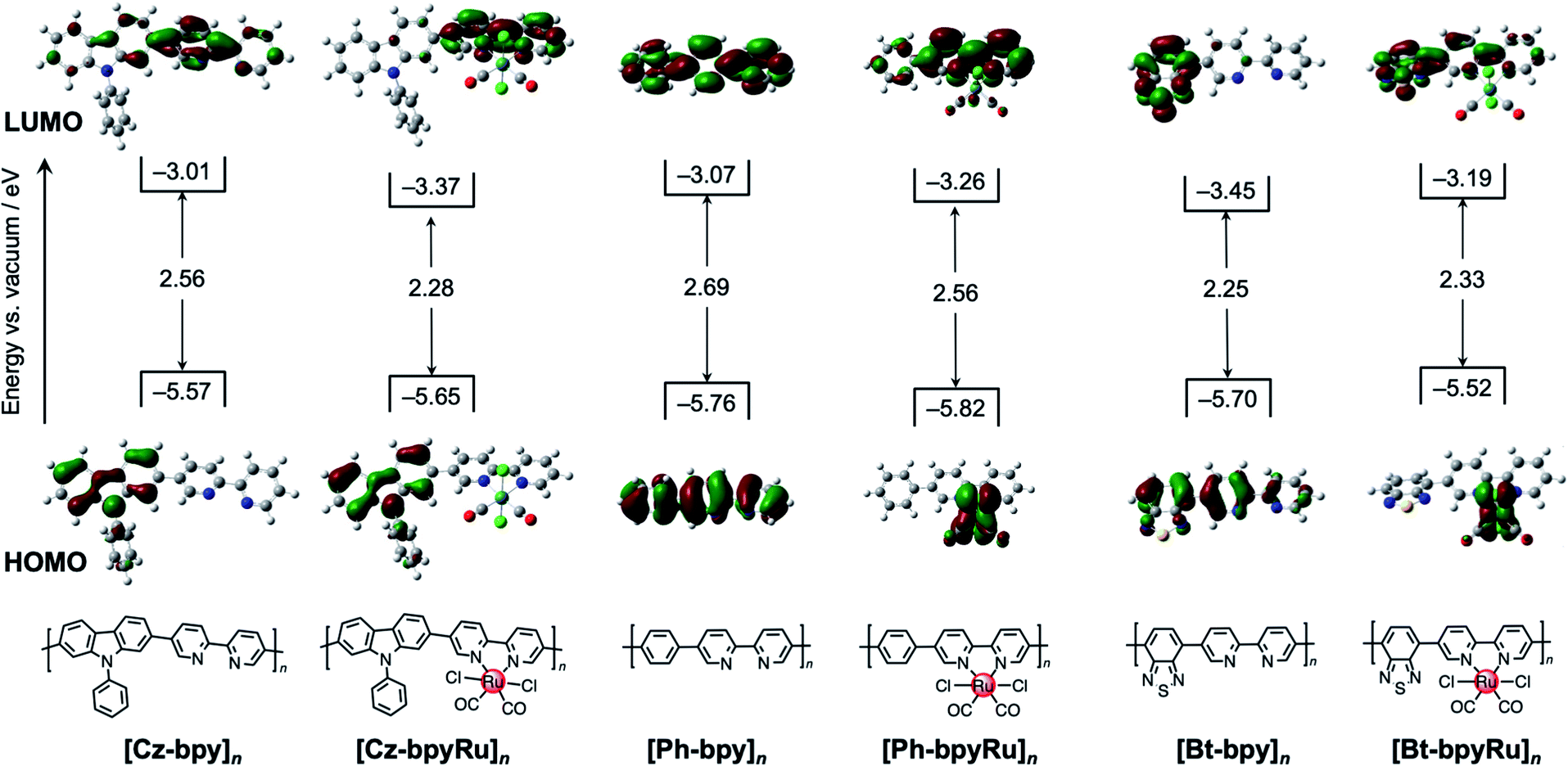 | ||
| Fig. 2 Experimentally-estimated band positions of [X-bpyM]n (X = Cz, Ph, and Bt; M = none and Ru) along with calculated HOMO (lower) and LUMO (upper) distributions of X-bpyM. | ||
To gain further insights into the roles of X moieties in the electron transition, we conducted a theoretical calculation of molecular orbitals for the model monomers X-bpy (Fig. 2). Regardless of the X moiety, the frontier orbitals were located around the π-conjugated skeletons. The π–π*-based HOMO–LUMO distribution shows, however, different nature depending on the X moiety. In the cases of Ph-bpy, both HOMO and LUMO are delocalized on the whole conjugated skeleton. In contrast, charge-transfer characters can be found in the HOMO–LUMO electron transition in the cases of X = Cz and Bt, but the roles of Cz and Bt are different. The electron-rich Cz in Cz-bpy appeared to be HOMO for donating an electron to the bpy unit, whereas electron-deficient Bt serves as an acceptor (i.e., LUMO in Bt-bpy). The character of donor–acceptor charge transfer should decrease the Eg values of [Cz-bpy]n and [Bt-bpy]n, in which the electron-donating Cz increases the HOMO energy while the electron-withdrawing Bt decreases the LUMO energy, respectively.
Notably, the introduction of the Ru(II) complex unit further modified the HOMO–LUMO potentials of the polymers. In the case of X = Ph, LUMO energies decreased with a slightly decreased bandgap (ca. 0.1 eV) after introducing the Ru(II) complex. Since LUMO are delocalized over the whole π-conjugated system in Ph-bpy and Ph-bpyRu (Fig. 2), these potential shifts can be understood by stabilization of the π-conjugated system through coordination of the bpy moiety to the Ru(II) center.43 At a glance, the X = Cz system exhibits a similar trend to that of X = Ph, whereas a larger degree of bandgap narrowing from [Cz-bpy]n (Eg = 2.6 eV) to [Cz-bpyRu]n (Eg = 2.3 eV) was obviously observed as a specific feature of the X = Cz system (Fig. 2). The HOMO and LUMO of Cz-bpyRu separately distribute at the Cz and bpy moieties, respectively. Therefore, LUMO localized on bpy was selectively stabilized by the coordination to the Ru(II) center, leading to a decreased bandgap in the case of [Cz-bpyRu]n. Interestingly, introducing the Ru(II) complex to [Bt-bpy]n increased both HOMO–LUMO energies with a slightly expanded bandgap (Fig. 2). The Ru d orbitals participated in the formation of HOMO of Bt-bpyRu, in contrast to the X = Cz system in which both HOMO–LUMO are constructed by the organic conjugated chain without the direct contribution of Ru d orbitals. The different origins of HOMO between Bt-bpy (π orbitals) and Bt-bpyRu (Ru d orbitals) might elevate the HOMO level by introducing the Ru(II) complex moiety, although the reason for the destabilization of LUMO by introducing the Ru(II) complex moiety is unclear at this stage. From these results, we can conclude that the opposite trends in the HOMO–LUMO shifts, when introducing the Ru(II) complex moiety, are caused by the electron-donating or withdrawing nature of unit X. The photoexcited charge distribution for X-bpyRu was evaluated by the differential Mulliken population between the ground and excited states. The differential charges on the bpyRu moiety were −0.057, 0.001, and 0.021 for Cz-bpyRu, Ph-bpyRu, and Bt-bpyRu, respectively (Table S1†). These results suggest that Cz-bpyRu generates photoexcited electrons relatively located on the bpyRu moiety, while Bt-bpyRu generates it on the opposite side. As mentioned in the Introduction, not only the HOMO–LUMO gap but also its potential and spatial distribution are important to manipulate photoexcited charge carriers to be effectively utilized at the catalytic moiety. Thus, the direct or indirect cooperation of π-the conjugated system and metal d-orbitals will expand the tunability of HOMO–LUMO level, gap, and distribution for the development of suitable photocatalyst materials.
Prior to photocatalysis studies, behaviors of photoexcited carriers in the polymer materials were evaluated by means of transient spectroscopy. Fig. 3 shows transient photoconductivity signals of [X-bpyM]n, which were obtained using time-resolved microwave conductivity (TRMC) measurements upon laser excitation (λ = 355 nm). The photoconductivity values ϕΣμ, where ϕ, the quantum efficiency of charge carrier generation and Σμ, the sum of photogenerated carrier mobilities, are increased within the instrumental time resolution (∼10−7 s) and then gradually decreased due to charge recombination and/or trapping. The maximum photoconductivity values ϕΣμmax44 were similar among three [X-bpy]n compounds (compare grey bars in Fig. 3d). The incorporation of the Ru(II) complex moiety into the bpy ligand led to the decreased ϕΣμmax in the case of X = Cz (Fig. 3a and d). Since the main structures of the polymers are maintained by post complexation of the Ru(CO)2Cl2 unit (Fig. S2–S4†), the decreased photoconductivity of [Cz-bpyRu]n compared with [Cz-bpy]n is likely due to the charge trapping at the Ru(II) complex moiety. A similar phenomenon was observed in the case of X = Ph although the degree was smaller than that of X = Cz (Fig. 3b and d). In contrast, the [Bt-bpyM]n system showed negligible change trapping by incorporating the Ru complex moiety (Fig. 3c and d).
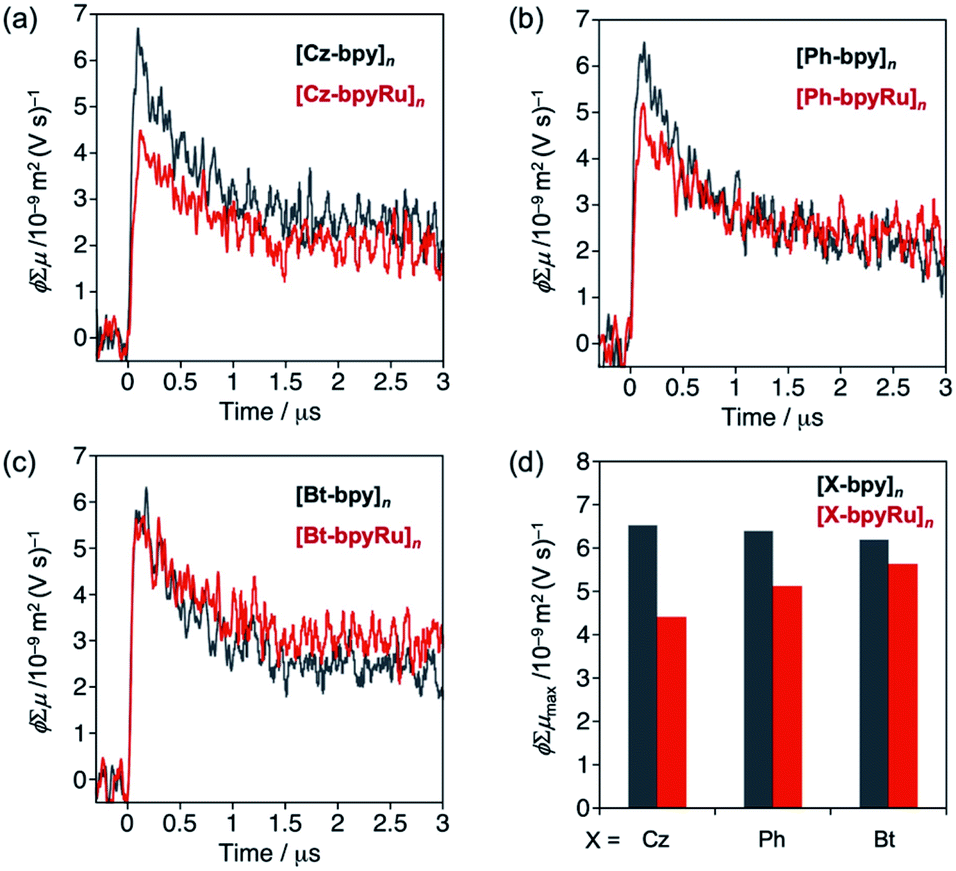 | ||
| Fig. 3 Transient conductivities of (a) [Cz-bpyM]n, (b) [Ph-bpyM]n, and (c) [Bt-bpyM]n (M = none (grey) or Ru (red)), along with (d) their maximum values (ϕΣμmax) obtained by photoexcitation at λ = 355 nm. | ||
For an in-depth understanding of the origin of photoexcited charge trapping, time-resolved infrared (TR-IR) absorption measurements were performed. We focused on the characteristic absorption corresponding to the CO vibration in the Ru(bpy)(CO)2Cl2 moiety, which reflects the changes in electronic densities at the Ru center.45 Upon laser excitation (λ = 420 nm) to [Cz-bpyRu]n, absorption bands at around 2069 and 2007 cm−1 decreased, whereas lower-energy bands at around 2045 and 1977 cm−1 increased within 1 ps (Fig. 4a). Such derivative-type differential spectra approximately centered at absorption peaks of the ground state (2060 and 1994 cm−1) are characteristic of the simultaneous decay and rise of the ground state and a new state, respectively.46 Kubiak et al. reported that one-electron reduction of Ru(bpyR2)(CO)2Cl2 (bpyR2 = 6,6′-dimethythyl-2,2′-bipyridine) induces lower-energy shifts of CO vibration with 25–30 cm−1 owing to the enhanced back donation from the electron-rich Ru center.45 Hence, it is strongly suggested that the photoexcitation of [Cz-bpyRu]n generates a reduced Ru complex moiety through the excited charge trapping. Although [Ph-bpyRu]n and [Bt-bpyRu]n showed similar spectroscopic changes (Fig. 4b and c), [Cz-bpyRu]n exhibited the largest generation of the reduced catalyst moiety (Fig. 4d). The trends are in good agreement with those observed in TRMC (vide supra, Fig. 3). From the aforementioned transient spectroscopy, we can conclude that photoexcited electrons are captured at the Ru(II) complex moiety more effectively in [Cz-bpyRu]n compared with the X = Ph and Bt counterparts, in association with their LUMO distribution at the Ru(II) complex moiety (see Fig. 2).
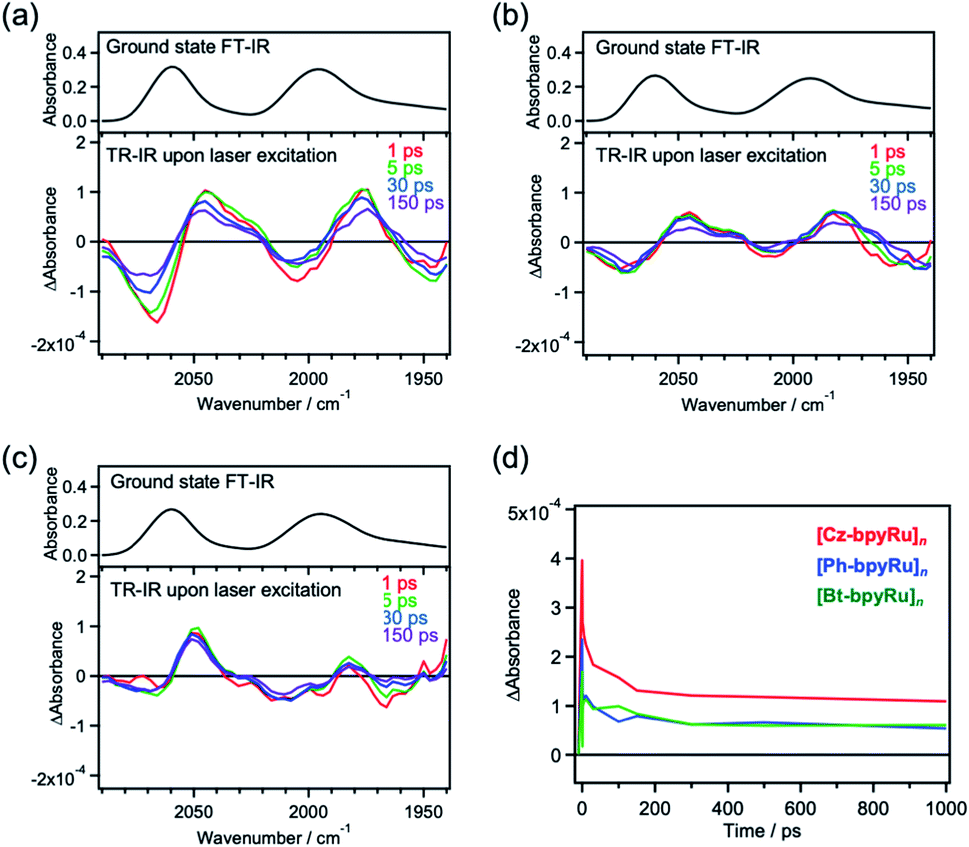 | ||
| Fig. 4 Ground state FT-IR (upper) and TR-IR (lower) spectra of (a) [Cz-bpyRu]n, (b) [Ph-bpyRu]n, and (c) [Bt-bpyRu]n, along with (d) time courses of the differential absorbance at 2045 cm−1 and 2069 cm−1 (red: [Cz-bpyRu]n, blue: [Ph-bpyRu]n, and green: [Bt-bpyRu]n). | ||
Photocatalytic CO2 reduction activities of the polymers were evaluated in a MeCN-TEOA (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) dispersion upon visible-light (λ > 400 nm) irradiation (Table 1). [Cz-bpyRu]n generated formate as the main product together with H2 and a small amount of CO (Fig. 5a and entry 1 in Table 1). In the absence of photoirradiation, CO2, or TEOA, almost no activity was found (entries 2–4, Table 1), indicating that this reaction represents a photochemical reduction of CO2 using TEOA as an electron donor. By using [Cz-bpy]n instead of [Cz-bpyRu]n, CO2 reduction products became negligible and the main product switched to H2 (entry 5 in Table 1). Therefore, it is strongly suggested that the Ru(II) complex moiety acts as the catalyst for CO2 reduction. After 12 h photoirradiation, the photocatalysis deactivated (Fig. 5a). In ATR-FT-IR spectra, characteristic CO vibrations corresponding to Ru(bpy)(CO)2Cl2-type coordination environment were shifted to 2019 and 1923 cm−1 after photolysis, suggesting the formation of a [Ru(bpy)(CO)2]n-type network with Ru–Ru bonds (Fig. S9†). On the other hand, IR spectra corresponding to the organic skeleton were almost unchanged. Hence, we conclude that the changes in the Ru catalyst moiety have likely caused the deactivation. Although the stability leaves much room for improvement, the wavelength dependency of the formate formation clearly indicates that [Cz-bpyRu]n effectively utilizes its HOMO–LUMO photoexcitation even more than 500 nm for photocatalytic reaction (Fig. 5b). Maeda and co-workers reported that copolymerized organic polymers C3N4, which were modified with Ru(2,2′-bipyridine-4,4′-diphosphonic acid)(CO)2Cl2, facilitate the photocatalytic formate formation under λ > 500 nm visible light, while the quantum efficiency at λ > 500 nm was not reported.47,48 The external quantum efficiencies for the formate formation (EQEformate) by the present [Cz-bpyRu]n polymer were estimated to be 0.44% and 0.38% at 505 and 525 nm, respectively. These values are comparable to or better than that of a triazine-based conjugated polymer for CO formation,30 which is one of the few examples of obtaining reliable quantum efficiency at λ > 500 nm visible light by using organic polymer-based photocatalysts.
:1 v/v) dispersion upon visible-light (λ > 400 nm) irradiation (Table 1). [Cz-bpyRu]n generated formate as the main product together with H2 and a small amount of CO (Fig. 5a and entry 1 in Table 1). In the absence of photoirradiation, CO2, or TEOA, almost no activity was found (entries 2–4, Table 1), indicating that this reaction represents a photochemical reduction of CO2 using TEOA as an electron donor. By using [Cz-bpy]n instead of [Cz-bpyRu]n, CO2 reduction products became negligible and the main product switched to H2 (entry 5 in Table 1). Therefore, it is strongly suggested that the Ru(II) complex moiety acts as the catalyst for CO2 reduction. After 12 h photoirradiation, the photocatalysis deactivated (Fig. 5a). In ATR-FT-IR spectra, characteristic CO vibrations corresponding to Ru(bpy)(CO)2Cl2-type coordination environment were shifted to 2019 and 1923 cm−1 after photolysis, suggesting the formation of a [Ru(bpy)(CO)2]n-type network with Ru–Ru bonds (Fig. S9†). On the other hand, IR spectra corresponding to the organic skeleton were almost unchanged. Hence, we conclude that the changes in the Ru catalyst moiety have likely caused the deactivation. Although the stability leaves much room for improvement, the wavelength dependency of the formate formation clearly indicates that [Cz-bpyRu]n effectively utilizes its HOMO–LUMO photoexcitation even more than 500 nm for photocatalytic reaction (Fig. 5b). Maeda and co-workers reported that copolymerized organic polymers C3N4, which were modified with Ru(2,2′-bipyridine-4,4′-diphosphonic acid)(CO)2Cl2, facilitate the photocatalytic formate formation under λ > 500 nm visible light, while the quantum efficiency at λ > 500 nm was not reported.47,48 The external quantum efficiencies for the formate formation (EQEformate) by the present [Cz-bpyRu]n polymer were estimated to be 0.44% and 0.38% at 505 and 525 nm, respectively. These values are comparable to or better than that of a triazine-based conjugated polymer for CO formation,30 which is one of the few examples of obtaining reliable quantum efficiency at λ > 500 nm visible light by using organic polymer-based photocatalysts.
| Entry | Photocatalyst | Control | Product/μmol | ||
|---|---|---|---|---|---|
| Formate | CO | H2 | |||
|
a 2 mg of photocatalyst powder in an MeCN-TEOA (2 mL; 4:1 v/v) dispersion was irradiated at λ > 400 nm for 12 h under CO2 atmosphere.
b Under N2 atmosphere.
|
|||||
| 1 | [Cz-bpyRu]n | —a | 24.7 | 0.8 | 7.8 |
| 2 | [Cz-bpyRu]n | Without light | N.D. | N.D. | N.D. |
| 3 | [Cz-bpyRu]n | Without CO2b | N.D. | Trace | N.D. |
| 4 | [Cz-bpyRu]n | Without TEOA | N.D. | 0.5 | Trace |
| 5 | [Cz-bpy]n | —a | 0.5 | N.D. | 26.6 |
| 6 | [Ph-bpyRu]n | —a | 3.6 | 0.4 | 0.5 |
| 7 | [Bt-bpyRu]n | —a | 1.7 | 0.3 | 0.3 |
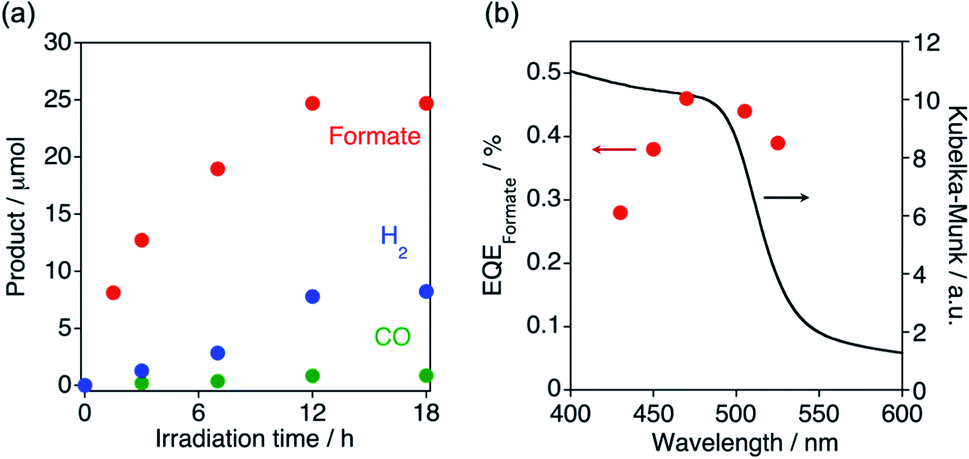 | ||
| Fig. 5 (a) Time courses of photocatalytic reaction and (b) action spectra of external quantum efficiency of formate formation (EQEformate) by [Cz-bpyRu]n (2 mg) in an MeCN-TEOA (2 mL; 4:1 v/v) dispersion under CO2 atmosphere. | ||
Notably, [Cz-bpyRu]n exhibited 7- to 15-folds higher photocatalytic activity for CO2 reduction than [Ph-bpyRu]n and [Bt-bpyRu]n (compare entries 1, 6, and 7 in Table 1). Although the bulk structures possibly affect photocatalytic performance, all the [X-bpyRu]n polymers have similar indefinite low-crystalline bulk structures (vide supra, Fig. S5 and S6†). The different visible-light absorbability (Fig. 1a) might be a reason for their distinct photocatalytic activities. However, the trend of the formate-formation activity did not change even if a light centered at λ = 430 nm, where all [X-bpyRu]n can enough absorb, was irradiated (Fig. S10†). In addition, [Cz-bpyRu]n has a smaller driving force for both redox reactions than [Ph-bpyRu]n (Fig. 2). Therefore, the much better photocatalytic activity of [Cz-bpyRu]n most probably originated from its effective capturing of photogenerated electrons at the catalytic Ru(II)-complex moiety, as demonstrated by means of transient spectroscopies (Fig. 3 and 4). From these results, we can conclude that precise HOMO–LUMO tuning via molecular engineering with an appropriate choice of the building block X in [X-bpyRu]n (Cz, Ph, or Bt) enables effective charge trapping at the structurally well-defined Ru(II) complex catalyst unit, leading to the much efficient CO2 photoreduction.
Conclusions
In this work, we constructed conjugated polymers bearing a Ru(II) complex catalyst in the bipyridine-ligand moiety and unveiled the relationship between their molecular structures, HOMO–LUMO distributions, excited charge trapping, and photocatalytic activity for CO2 reduction. The introduction of an electron-donating carbazole skeleton enables LUMO localization and effective trapping of the photoexcited electrons at the Ru(II) catalyst moiety, engaging in 7- to 15-folds efficient CO2 reduction compared with phenyl and benzothiadiazole counterparts. Hence, in this study, we demonstrate the effectiveness of the manipulation of local photoexcited charge distributions by the molecular engineering of organic polymers with a site-selectively incorporated molecular catalyst. The carbazole-based polymer [Cz-bpyRu]n utilizes visible light at more than 500 nm with a potential photocatalytic efficiency of CO2 reduction, which is comparable to or better than that of the organic-polymer photocatalyst reported previously.30 On the other hand, we should note that the activity of [Cz-bpyRu]n has still much room for improvement, possibly due to their indefinite low-crystalline bulk structure. Recently, research on crystal- and pore-structure engineering of organic-polymer photocatalysts including covalent organic frameworks have been accelerated so far.28,33 Therefore, we believe that this strategy of manipulating photoexcited charge carriers expands the opportunity to develop efficient photocatalysts by coupling with well-established bulk engineering.Experimental
General procedure
1H NMR (400 MHz) spectra were measured on a JEOL ECZ-500R spectrometer. Elemental analyses were carried out on a PerkinElmer 2400 II CHN analyzer. UV-vis absorption and diffuse reflectance spectra were recorded at room temperature on Shimadzu U1900i and U2600i spectrophotometers, respectively. Solid-state FT-IR spectra were measured on a Thermo-Fisher Nicolet 6700 spectrometer equipped with a Smart-Orbit (Diamond) attenuated total reflection (ATR) accessory. Solution FT-IR spectra were measured on a JASCO FT/IR-4100 spectrometer. MALDI-TOF-MS spectra were measured on a Shimadzu MALDI-8020 mass spectrometer. Scanning electron microscope (SEM) images were obtained using a Hitachi MC1000 at 10 kV after coating Pt on the samples. Powder X-ray diffraction patterns were recorded on a Rigaku Ultima IV diffractometer with Cu Kα radiation. Photoelectron yield spectra (PYS) were measured on a Bunko Keiki BIP-KV202GD apparatus. The PYS measurements provide the lowest ionization energies corresponding to the valence-band maximum potentials.42,49Acetonitorile (MeCN; dehydrated, >99.5%), methanol (MeOH; dehydrated, >99.6%), CHCl3 (>99.0%), CH2Cl2 (dehydrated, >99.5%), and K2CO3 (>99.5%) were purchased from Kanto Chemical Co. Inc. 5,5-dibromo-2,2′-bipyridine (>98.0%), 9-phenyl-2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9H-carbazole (>96.0%), 1,4-phenylenediboronic acid, 9-phenylcarbazole (>98.0%), 2,1,3-benzothiadiazole (>99.0%) were purchased from Tokyo Chemical Industry Co., Ltd. Tetrakis(triphenylphosphine)palladium (Pd(PPh3)4; >90.0%), triethanolamine (TEOA; >98.0%), p-toluenesulfonic acid (>99.0%), ethylenediamine tetraacetic acid (>99.0%), and Bis-Tris (>99.0%) were purchased from Wako Pure Chemical Industries. These materials were used without any further purification. [Ru(CO)2Cl2]n was prepared according to a literature procedure.50
Synthesis
:1, v/v; 12 mL) solution containing 5,5′-dibromo-2,2′-bipyridine (314.8 mg, 1.0 mmol), 9-phenyl-2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9H-carbazole (497.9 mg, 1.0 mmol), K2CO3 (1040 mg, 7.5 mmol), and Pd(PPh3)4 (23.5 mg, 0.02 mmol) was stirred under reflux for 2 days under N2 atmosphere. The obtained precipitate was filtered and washed with water (20 mL), MeOH (20 mL), and CH2Cl2 (20 mL). Yield 256 mg.
The same protocol was applied for the syntheses of [Ph-bpy]n and [Bt-bpy]n by using 1,4-phenylenediboronic acid and 4,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,1,3-benzothiadiazole, respectively, as the starting material instead of 9-phenyl-2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9H-carbazole. Their characterization data are shown in Fig. S1–S4.†
Route A. A MeOH dispersion (5 mL) containing [Ru(CO)2Cl2]n (22.8 mg, 0.10 mmol) and [Cz-bpy]n (42.9 mg, 0.10 mmol) was refluxed with stirring for 17 h under the N2 atmosphere. After refluxing, the obtained precipitate was filtered and washed with MeOH (20 mL). Yield 62.9 mg. FT-IR (ATR): vCO = 2054, 1990 cm−1. Elemental analysis calcd. for C30H19Cl2N3O2Ru (Cz-bpyRu): C, 57.61; H, 3.06; N, 6.72. Found: C, 57.50; H, 3.20; N, 6.81.
Route B. A DMF–H2O (5
:1, v/v; 12 mL) solution containing Ru(5,5-dibromo-2,2′-bipyridine)(CO)2Cl2 (27.0 mg, 0.05 mmol), 9-phenyl-2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9H-carbazole (24.8 mg, 0.05 mmol), K2CO3 (55.2 mg, 0.40 mmol), and Pd(PPh3)4 (1.0 mg, 1 μmol) was stirred at reflux for 2 days under a N2 atmosphere. The obtained precipitate was filtered and washed with water (20 mL) and MeOH (20 mL). Yield 14.4 mg. FT-IR (ATR): vCO = 2019, 1923 cm−1.
The protocol same as used in Route A was applied for the syntheses of [Ph-bpyRu]n and [Bt-bpyRu]n by using [Ph-bpy]n and [Bt-bpy]n, respectively, as starting materials instead of [Cz-bpy]n. Elemental analysis calcd. for C52H36Cl4N6O4Ru3 ([Ph-bpyRu]2[Ph-bpy]1): C, 54.18; H, 3.15; N, 7.29. Found: C, 55.16; H, 3.15; N, 7.47. Elemental analysis calcd. for C88H50Cl8N10O8Ru4S5 ([Bt-bpyRu]4[Bt-bpy]1): C, 44.72; H, 2.13; N, 11.85. Found: C, 44.52; H, 2.31; N, 11.82. Further characterization data are shown in Fig. S1–S7.†
Time-resolved microwave conductivity measurement
Time-resolved microwave conductivity experiments were performed using a third harmonic generator (THG; 355 nm) of an Nd:YAG laser (Continuum Inc., Surelite II, 5–8 ns pulse duration, 10 Hz) as the excitation source (9.1 × 1015 photons per cm2 per pulse) and X-band microwave (∼9.1 GHz) as the probe. The photoconductivity Δσ was obtained using eqn (1)| Δσ = ΔPr/(APr) | (1) |
| Σμ = μ+ + μ− | (2) |
| ϕΣμ = Δσ/(eI0Flight) | (3) |
Time-resolved infrared absorption spectroscopy
The time-resolved infrared absorption measurements were performed using the spectrometer described previously.51 The powder sample was mixed with KCl powder, pelletized, and set in an IR cell filled with 20 torr N2. The measurements were performed using the pump-probe method with a Ti: sapphire regenerative amplifier (Spectra Physics, Solstice. 90 fs duration, 1 kHz repetition rate) and optical parametric amplifiers (OPAs; Spectra Physics, TOPAS Prime). The sample was photoexcited by a 420 nm pulse (0.5 μJ per pulse, 500 Hz) from the OPA. The probe MIR pulse was generated by the difference frequency generation between the signal and idler from the OPA in an AgGaS2 crystal and was detected with a 128-channel linear MCT array detector (Infrared Systems Development, FPAS-0144). The MIR signal was analysed after subtraction of smooth backgrounds from the spectra.Photocatalytic reactions
A MeCN-TEOA dispersion (2 mL) of [X-bpyM]n (X = Cz, Ph, or Bt; M = none or Ru) was placed in a Pyrex test tube (inner diameter: 10 mm; volume: 8.4 mL), degassed with CO2 or N2 bubbling (20 min), and sealed using a rubber septum prior to the photoirradiation. The sample tube was placed in a LED merry-go-round apparatus (Iris-MG-S, Cell System Inc.) and irradiated with visible light (λ > 400 nm) under stirring. The gaseous products, i.e., CO and H2 were analyzed using a Shimadzu GC-2010 gas chromatograph (MS-5A column, Ar carrier) combined with a TCD detector. The formate produced in the liquid phase was analyzed using a Shimadzu LC-20AT HPLC system equipped with two Shimadzu Shim-pack FAST-OA columns (100 × 7.8 mm) and a Shimadzu CDD-10A conductivity detector. An aqueous solution containing p-toluenesulfonic acid (0.95 g L−1) was used as the eluent at a flow rate of 0.8 mL min−1 (column temperature: 313 K). After column separation, the eluent was mixed with an aqueous solution containing p-toluenesulfonic acid (0.95 g L−1), and ethylenediaminetetraacetic acid (0.03 g L−1), and Bis-Tris (4.18 g L−1). For wavelength dependency measurements, a LED lamp with an independent wavelength centered at 430, 450, 470, 505, or 525 nm (CL-H1 series, Asahi Spectra Co.) was used at a fixed photon flux of 2.1 × 10−7 einstein per s, instead of the aforementioned LED merry-go-round system. The external quantum efficiencies of photocatalytic formate formation (EQEformate) were determined on the basis of eqn (4):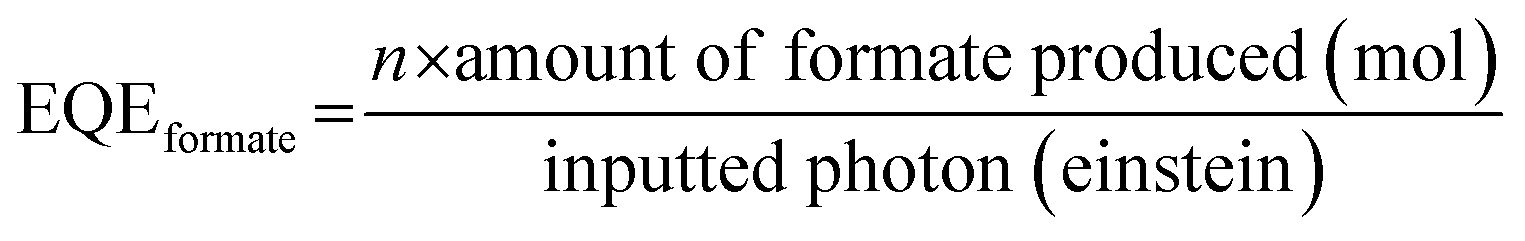 | (4) |
Computational details
Geometry optimizations were performed using the density functional theory (DFT) with ωB97X-D exchange-correlation functional.52 For core electrons of Ru ([Ar]3d10), Stuttgart/Dresden pseudopotentials53 at the multi Dirac–Fock level were used. Corresponding double-ζ basis sets were adopted to valence orbitals of Ru. For other elements, namely, C, H, Cl, N, O and S, correlation-consistent basis sets at double-ζ level termed cc-pVDZ54,55 were employed. Harmonic vibrational frequencies were also calculated analytically to confirm whether the optimized structure converged to an equilibrium geometry point. For the optimized structure, single-point calculations were performed using time-dependent DFT with the ωB97X-D functional to obtain the electronic structure in the T1 state. Mulliken atomic charges in the S0 and T1 states were evaluated at the S0 optimized structure. All calculations were performed by Gaussian 09 program.56Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST PRESTO grant JPMJPR20T5 (Controlled Reaction), by JSPS KAKENHI grants JP19K23652 and JP19H02736 as well as by MEXT KAKENHI grants JP20H05113 (I4LEC) and JP18H05517 (Hydrogenomics), and by the Research Promotion Fund from the Promotion and Mutual Aid Corporation for Private Schools of Japan.Notes and references
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- Y. Zheng, W. Zhang, Y. Li, J. Chen, B. Yu, J. Wang, L. Zhang and J. Zhang, Nano Energy, 2017, 40, 512–539 CrossRef CAS.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. Kenis, C. A. Kerfeld, R. H. Morris, C. H. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS.
- C. Costentin, M. Robert and J. M. Saveant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- S. Yoshino, T. Takayama, Y. Yamaguchi, A. Iwase and A. Kudo, Acc. Chem. Res., 2022, 55, 966–977 CrossRef CAS PubMed.
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Nat. Commun., 2018, 9, 772 CrossRef PubMed.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2016, 7, 70–88 CrossRef.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- K. Maeda, Phys. Chem. Chem. Phys., 2013, 15, 10537–10548 RSC.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- Y. H. Hong, Y. M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2022, 144, 695–700 CrossRef CAS PubMed.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, S. Isabell and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- T. Morikawa, S. Sato, K. Sekizawa, T. M. Suzuki and T. Arai, Acc. Chem. Res., 2022, 55, 933–943 CrossRef CAS PubMed.
- A. Nakada, H. Kumagai, M. Robert, O. Ishitani and K. Maeda, Acc. Mater. Res., 2021, 2, 458–470 CrossRef CAS.
- C. Dai and B. Liu, Energy Environ. Sci., 2020, 13, 24–52 RSC.
- J. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- C. Yang, W. Huang, L. C. da Silva, K. A. I. Zhang and X. Wang, Chem.–Eur. J., 2018, 24, 17454–17458 CrossRef CAS PubMed.
- H.-P. Liang, A. Acharjya, D. A. Anito, S. Vogl, T.-X. Wang, A. Thomas and B.-H. Han, ACS Catal., 2019, 9, 3959–3968 CrossRef CAS.
- X. Yu, Z. Yang, B. Qiu, S. Guo, P. Yang, B. Yu, H. Zhang, Y. Zhao, X. Yang, B. Han and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 632–636 CrossRef CAS PubMed.
- Z. Fu, X. Wang, A. M. Gardner, X. Wang, S. Y. Chong, G. Neri, A. J. Cowan, L. Liu, X. Li, A. Vogel, R. Clowes, M. Bilton, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 543–550 RSC.
- X. Y. Dong, Y. N. Si, Q. Y. Wang, S. Wang and S. Q. Zang, Adv. Mater., 2021, 33, 2101568 CrossRef CAS PubMed.
- J. Li, K. Ma, Y. He, S. Ren, C. Li, X.-B. Chen, Z. Shi and S. Feng, Catal.: Sci. Technol., 2021, 11, 7300–7306 RSC.
- G. E. M. Schukraft, R. T. Woodward, S. Kumar, M. Sachs, S. Eslava and C. Petit, ChemSusChem, 2021, 14, 1720–1727 CrossRef CAS PubMed.
- L. Hou, G. Tang, H. Huang, S. Yin, B. Long, A. Ali, G.-J. Deng and T. Song, J. Mater. Chem. A, 2022, 10, 5287–5294 RSC.
- S. Barman, A. Singh, F. A. Rahimi and T. K. Maji, J. Am. Chem. Soc., 2021, 143, 16284–16292 CrossRef CAS PubMed.
- R. Kuriki and K. Maeda, Phys. Chem. Chem. Phys., 2017, 19, 4938–4950 RSC.
- G. Gerbaud, J. M. Mouesca, S. Hediger, S. Chardon-Noblat, F. Lafolet, A. Deronzier and M. Bardet, Phys. Chem. Chem. Phys., 2010, 12, 15428–15435 RSC.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- Y. Nakayama, S. Machida, D. Tsunami, Y. Kimura, M. Niwano, Y. Noguchi and H. Ishii, Appl. Phys. Lett., 2008, 92, 153306 CrossRef.
- A. A. Vl
![[c with combining breve]](https://www.rsc.org/images/entities/char_0063_0306.gif) ek, Coord. Chem. Rev., 1982, 43, 39–62 CrossRef.
ek, Coord. Chem. Rev., 1982, 43, 39–62 CrossRef. - A. Saeki, S. Yoshikawa, M. Tsuji, Y. Koizumi, M. Ide, C. Vijayakumar and S. Seki, J. Am. Chem. Soc., 2012, 134, 19035–19042 CrossRef CAS PubMed.
- C. W. Machan, M. D. Sampson and C. P. Kubiak, J. Am. Chem. Soc., 2015, 137, 8564–8571 CrossRef CAS PubMed.
- A. Yamakata, T.-a. Ishibashi and H. Onishi, Bull. Chem. Soc. Jpn., 2002, 75, 1019–1022 CrossRef CAS.
- K. Shibata, K. Kato, C. Tsounis, T. Kanazawa, D. Lu, S. Nozawa, A. Yamakata, O. Ishitani and K. Maeda, Sol. RRL, 2019, 4, 1900461 CrossRef.
- C. Tsounis, R. Kuriki, K. Shibata, J. J. M. Vequizo, D. Lu, A. Yamakata, O. Ishitani, R. Amal and K. Maeda, ACS Sustainable Chem. Eng., 2018, 6, 15333–15340 CrossRef CAS.
- A. Nakada, M. Higashi, T. Kimura, H. Suzuki, D. Kato, H. Okajima, T. Yamamoto, A. Saeki, H. Kageyama and R. Abe, Chem. Mater., 2019, 31, 3419–3429 CrossRef CAS.
- P. A. Anderson, G. B. Deacon, K. H. Haarmann, F. R. Keene, T. J. Meyer, D. A. Reitsma, B. W. Skelton, G. F. Strouse, N. C. Thomas, J. A. Treadway and A. H. White, Inorg. Chem., 1995, 34, 6145–6157 CrossRef CAS.
- A. Yamakata, M. Kawaguchi, N. Nishimura, T. Minegishi, J. Kubota and K. Domen, J. Phys. Chem. C, 2014, 118, 23897–23906 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, J. Chem. Phys., 2007, 126, 124101–124112 CrossRef PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev. C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta02183h |
| This journal is © The Royal Society of Chemistry 2022 |