
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorinated triphenylenes and a path to short tailed discotic liquid crystals: synthesis, structure and transport properties†
Zhe
Li‡
 a,
Mitchell
Powers
*b,
Kayla
Ivey
a,
Sonya
Adas
a,
Brett
Ellman
b,
Scott D.
Bunge
a and
Robert J.
Twieg
a
a,
Mitchell
Powers
*b,
Kayla
Ivey
a,
Sonya
Adas
a,
Brett
Ellman
b,
Scott D.
Bunge
a and
Robert J.
Twieg
a
aDepartment of Chemistry & Biochemistry, Kent State University, Kent, OH 44242, USA
bDepartment of Physics, Kent State University, Kent, OH 44242, USA. E-mail: mpower16@kent.edu
First published on 24th November 2021
Abstract
A new synthetic approach for fluorinated alkoxytriphenylene discotic liquid crystals is presented. This methodology exploits the previously described photocyclodehydrofluorination (PCDHF) reaction for the preparation of fluorinated triphenylene derivatives coupled with a variety of nucleophilic aromatic substitution (SNAr) reactions. This particular combination of reactions provides a versatile route to discotic materials with carefully controlled core fluorine and alkoxy tail content. In the course of these studies, new discotic materials with minimal tail content have been revealed. The mesogenic properties of these materials are reported, and their charge transport properties are measured using the time of flight technique.
Introduction
Columnar discotic liquid crystals have attracted attention due to their quasi-one dimensional structures that give rise to a range of novel physical properties and potential applications.1–6 The widely accepted convention is that molecules which exhibit columnar mesophases are comprised of a rigid and mostly flat aromatic core surrounded by flexible chains, as found in the prototypical discotic hexa-substituted benzene derivatives.7 Interactions between the rigid cores create a columnar structure, and the tails form a soft buffer between the columns. Within the columns, the constituent molecules can adopt orientations with comparatively large transfer integrals between conjugated aromatic cores compared to the much more limited charge transfer through the tails.8–11 These charge transport properties have motivated the study of discotic liquid crystals as a valuable class of organic semiconductors12–14 which may be employed in photovoltaic devices,15 organic light emitting diodes (OLED),16 nanowires17 and other electronic applications.Amongst the many different types of discotic liquid crystals, triphenylene derivatives are perhaps the most widely investigated due to the relative ease of their synthesis.18–23 Within this class, dipolar substitution of the core is a known technique for extending the discotic mesophase range, e.g., nitro groups,24–27 nitrile groups and halogen atoms22 have frequently been investigated. Fluorine substitution is particularly attractive due to the small size of the substituent atom which helps to maintain a flat molecular core and may allow for highly cofacial and close-packed orientations between molecules within a column.
In the course of a study of partly fluorinated dipolar triphenylene derivatives enabled by a novel photochemical technique,28 we have found families of triphenylene discotic liquid crystals with a continuum of alkoxy tail lengths, ranging from methoxy to the more conventional butoxy or longer chains. This is extremely unusual: the standard paradigm of a rigid core and highly flexible, entropic tails is seemingly violated by the compounds with minimal tail length. Therefore, our results provide a “bridge” between familiar triphenylene discotic liquid crystals and new, unusual, but closely related mesogens.
Synthesis
The original successful seven step synthetic route for 1,4-difluoro-2,3,6,7,10,11-hexakishexyloxytriphenylene22 is shown (Scheme 1). In this case, the ultimate fluorinated triphenylene core is formed in the very last step by a highly regioselective FeCl3 mediated oxidative ring closure of an appropriate difluorinated o-terphenyl precursor. In this synthesis the specific locations and exact identity of all of the alkoxy groups found in the final product are established in the precursors relatively early in the synthesis.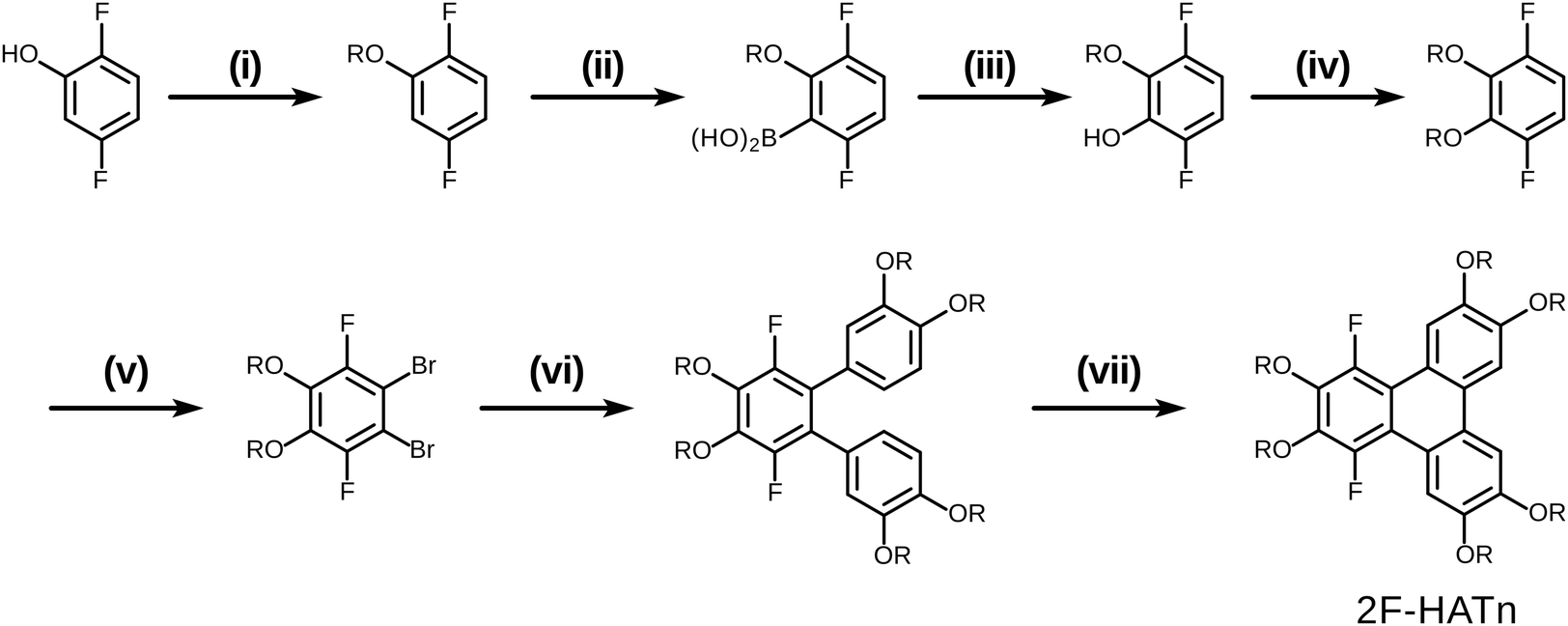 | ||
| Scheme 1 Literature fluorinated triphenylene synthesis (i) RBr/K2CO3/EtOH/reflux; (ii) (1) n-BuLi/THF/−78 °C, (2) B(OMe)3/THF/−78 °C, (3) 1 M HCl (aq); (iii) H2O2, 82%; (iv) RBr/K2CO3/EtOH/reflux, 80%; (v) Br2/DCM/0–25 °C, 98%; (vi) 0.3 mol% Pd(PPh3)4/C6H5CH3/H2O/Na2CO3/argon, 80%; (vii) (1) FeCl3/DCM, (2) MeOH, >90%. | ||
In this contribution, we describe a versatile alternative synthetic route to prepare the requisite 1,4-difluorinated-2,3,6,7,10,11-hexaalkoxy triphenylene derivatives employing a combination of the PCDHF reaction28,29 to form the fluorinated triphenylene ring coupled with SNAr methodologies to introduce some of the alkoxy tails to the fluorinated ring late in the synthetic sequence. This route offers several advantages over the existing methods. For example, tetrafluorotriphenylene compounds are easily accessed, leading to a new class of potential discotic liquid crystals. Furthermore, these same compounds are intermediates for the production of tri- and di-fluoro molecules that allow the inclusion of heterogeneous tails at a late stage of the synthesis (Schemes 5 and 7). These compounds will be designated mF-XATn, where m is the number of fluorines, n is the number of carbons in the alkoxy tails, and X is a prefix indicating the number of tails (H, hexa-; P, penta-; T, tetra-). For example, 1,2,4-trifluoro-3,6,7,10,11-pentakishexyloxytriphenylene is referred to as 3F-PAT6. Examination of this new route in retrosynthetic mode (Scheme 2) indicates that the appropriately functionalized core should be easily accessed providing that two conditions are met: first, and what was not clear at the initiation of this project, is whether one can efficiently convert an annulated 1,2,3,4-tetrafluorinated ring to the desired 1,4-difluoro-2,3-dialkoxy derivative. This question was cleared up in a satisfactory fashion by some model studies (vide infra). Second, a regiospecific PCDHF ring closure (as shown in Scheme 2) is also required. While the regiospecificity of the PCDHF reaction remains a topic under active examination, studies thus far indicate that the desired ring closure shown should at least be preferred. Very generally, steric effects tend to dictate the regiospecificity and, fortunately, the less hindered ring closure is usually favored, and thus in this case delivers the desired isomer as well.
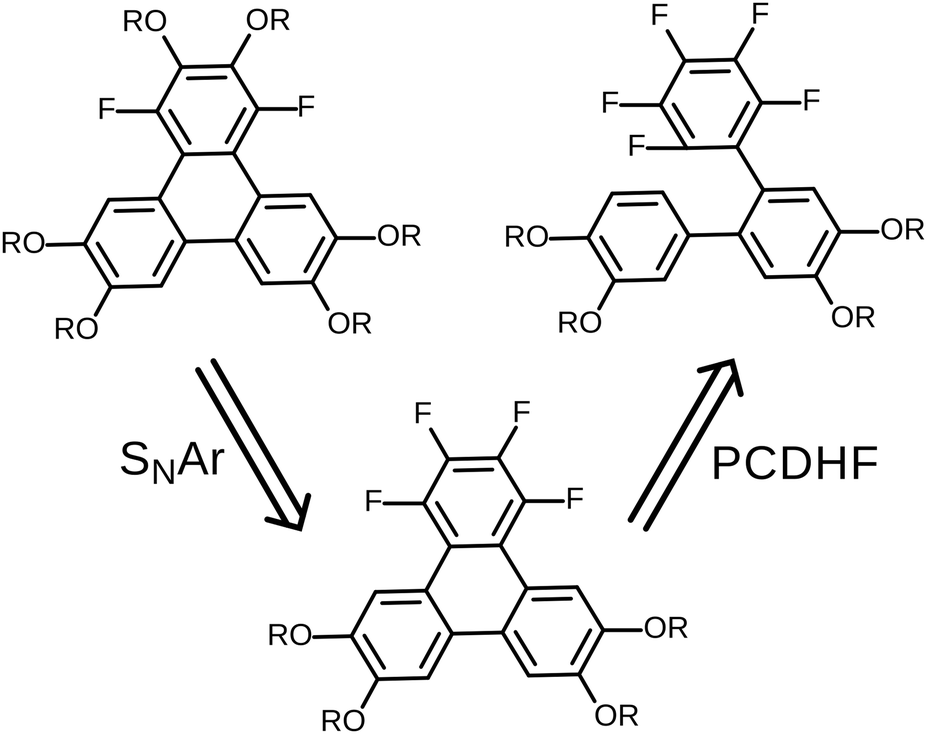 | ||
| Scheme 2 Retrosynthetic analysis for fluorinated alkoxytriphenylenes. The viability of this synthesis approach requires regiospecificity in both of the SNAr and PCDHF steps. | ||
It is well known that most pentafluorinated benzene derivatives undergo preferential nucleophilic aromatic substitution reaction (SNAr) at the most activated para-fluorine atom site and often even exclusively at this site.30,31 Such reactivity of pentafluorinated rings has been widely exploited and has even found prior use for the synthesis of calamitic liquid crystals.32–34 However, the location(s) and multiplicity of nucleophilic aromatic substitution in the case of 1,2,3,4-tetrafluorobenzene derivatives appears to be much less clearly documented in general and appears to not have been applied to liquid crystal synthesis. In order to clarify this situation prior to the initiation of a full synthesis of discotic triphenylenes based on the route in Scheme 2, some simple model substrates and test reactions were examined (Scheme 3). Here, 1,2,3,4-tetrafluorotriphenylene was selected as the test model precursor as it had already been efficiently prepared from the precursor 2,3,4,5,6-pentafluoro-1,1′:2′,1′′-terphenyl via the photochemical PCDHF approach as shown in Scheme 3.28
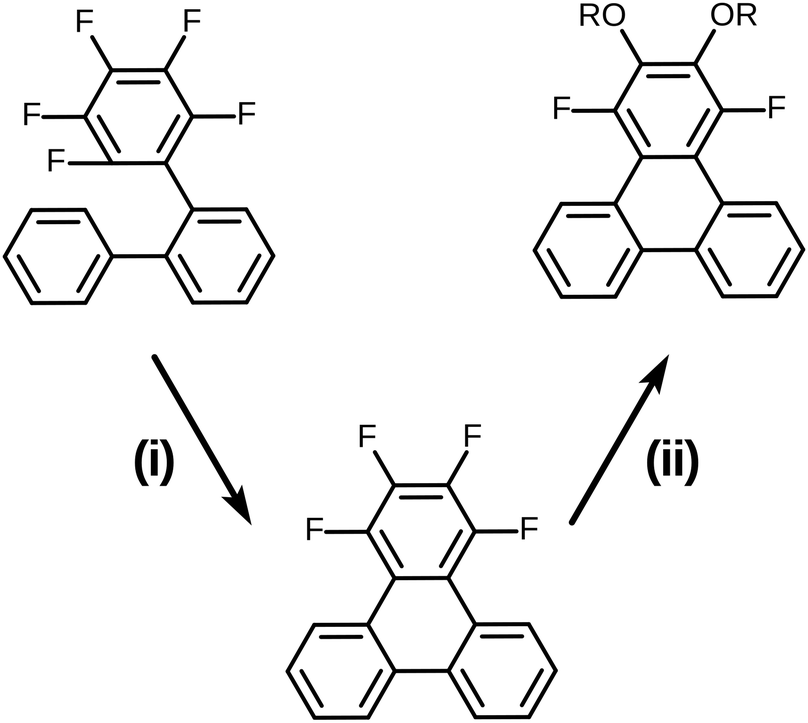 | ||
| Scheme 3 Synthesis of the model SNAr substrate and its use in the double SNAr reaction. (i) PCDHF/acetonitrile 94%; (ii) t-BuOK (8 eq.) in excess R-OH/toluene. | ||
From the onset, the initial SNAr reaction was expected to predominate at either of the structurally degenerate 2 and 3 positions of 1,2,3,4-tetrafluorotriphenylene. However, the possibility of a second reaction and the site of a second reaction were both much less certain. In order to evaluate multiple SNAr alkoxylation, different simple primary alcohols were deprotonated in the presence of potassium t-butoxide (typically 8.0 equivalents potassium t-butoxide per equivalent of 1,2,3,4-tetrafluorotriphenylene in an excess amount of alcohol). Use of potassium t-butoxide is convenient as it has special properties that make it useful (an enhanced basicity relative to most primary and secondary alcohols promoting exchange and a very low intrinsic nucleophilic activity). When a low boiling point alcohol such as methanol or ethanol was used some toluene was added as co-solvent to increase the solubility of 1,2,3,4-tetrafluorotriphenylene. In the case of methanol, after three hours of refluxing, the disubstituted product 1,4-difluoro-2,3-dimethoxytriphenylene was easily obtained in 60% yield. Ethanol proved to be significantly less reactive and after five hours of refluxing, only the monosubstituted product 2-ethoxy-1,3,4-trifluorotriphenylene was obtained in 75% yield. A Dean–Stark trap was utilized in order to push the second SNAr reaction to occur. After the removal of excess ethanol, the reaction temperature increased and a mixture of the mono-substituted product 2-ethoxy-1,3,4-trifluorotriphenylene (33% yield) and the disubstituted product 2,3-diethoxy-1,4-difluorotriphenylene (55% yield) were obtained. Higher boiling point primary alcohols with potassium t-butoxide were also able to react with 1,2,3,4-tetrafluorotriphenylene to achieve dialkoxylation. For example, in the case of 1-hexanol the product 1,4-difluoro-2,3-bis(hexyloxy)triphenylene could be obtained in 25% isolated yield. The reactions of the low boiling alcohols under conditions other than atmospheric pressure have not been investigated and the precise reaction conditions to cleanly obtain monoalkoxy or dialkoxy adducts remain to be identified. However, the preliminary studies undertaken here clearly indicate that the desired products needed for discotic liquid crystal synthesis are accessible by this process. The model study indicates that the initial SNAr single reaction occurs at the 2-site and the subsequent SNAr reaction occurs as the 3-site (reaction at sites 1 and 4 were not observed). The initial SNAr reaction is significantly faster than the second SNAr reaction (due to complementary electronic and steric factors) and the rates of both reactions can be influenced by steric factors. It is clear that the fluorine atoms on the 2,3-positions in 1,2,3,4-tetrafluoro compounds are significantly more reactive in the SNAr reaction than those at the 1,4-positions. This outcome exactly fits our needs which require the conversion of a 1,2,3,4-tetrafluoro-6,7,10,11-tetraalkoxytriphenylene intermediate into the desired 2,3,6,7,10,11-hexaalkoxytriphenylene product (Scheme 1). Structure analysis by X-ray diffraction demonstrates this outcome.
The assignment of the extent and location of alkoxide substitution in these SNAr reactions was supported by proton and fluorine NMR spectroscopy. In addition, X-ray crystallography analysis (Fig. 1; see ESI† for details) was performed on 1,4-difluoro-2,3-dimethoxytriphenylene (the diadduct obtained by reaction of potassium methoxide with 1,2,3,4-tetrafluorotriphenylene). This single crystal analysis unequivocally demonstrates that the methoxy groups substitute the fluorine atoms at the 2,3-positions. The crystal structure itself is of some interest with 1,4-difluoro-2,3-dimethoxytriphenylene crystallizing in the monoclinic space group P21/c with 4 molecules in the unit cell. The two methoxy groups in each molecule are located on the same side of the molecular plane with opposing torsion angles of about 60° as shown in Fig. 1. The steric disposition of these adjacent methoxy groups in the crystal of 1,4-difluoro-2,3-dimethoxytriphenylene is unusual and different from that in nonfluorinated systems. This pair of methoxy groups in each molecule makes it impossible for full coplanar overlap of the adjacent triphenylene molecules. Instead, the molecules have a larger stacking distance of about 4 Å (this may be compared to a distance of ∼3.5 Å for HATn compounds) and there is a highly coplanar but shifted stack of triphenylene molecules. Two close C–O–H–C contacts exist between methoxy groups on adjacent molecules within a stack. Each molecule also has a C–F–H–C contact between a fluorine atom and the hydrogen on a methoxy group situated on neighboring molecules on adjacent stacks. Presumably the intra-stack interactions help stabilize the columns while the C–F–H–C contacts affect the relative offset of each stack along the column direction relative to its neighbors. While it is unclear to what extent this offset stacking will manifest itself in compounds with other alkoxy groups and/or alkoxy tails on all of the rings, it is promising that the molecular core is not significantly distorted by the steric interactions between the fluorines and the adjacent hydrogens in the non-fluorinated rings.
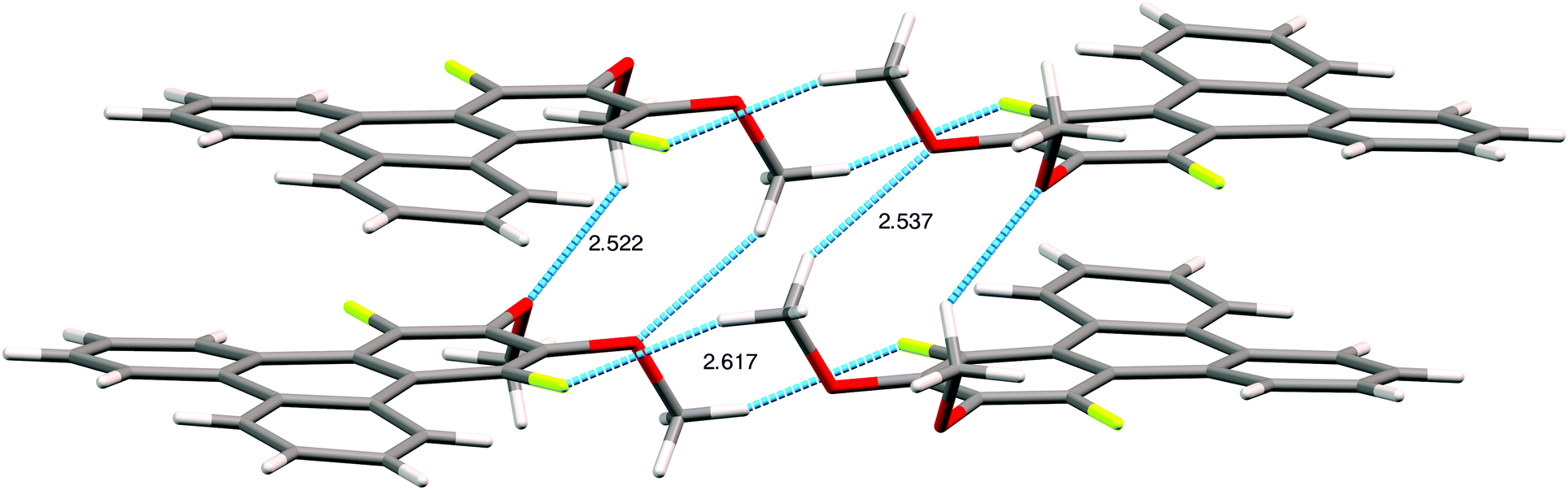 | ||
| Fig. 1 A stack of three 1,4-difluoro-2,3-dimethoxytriphenylenes and an adjacent stack of two 1,4-difluoro-2,3-dimethoxytriphenylenes showing the pairs of methoxy groups and their intermolecular H-bonding interactions contributing to formation of shifted stacks and also to the H–F intermolecular short contacts. | ||
Having examined and qualified the two most uncertain components of the planned synthesis, the preparation of a known fluorinated and alkoxy substituted triphenylene target 2F-HATn was undertaken.22 A crucial intermediate compound required in the overall synthesis is the o-terphenyl 6 (Scheme 4). Veratrole 1 is a readily available starting material which can be treated with iodine and periodic acid in methanol to provide 4,5-diiodoveratrol 2 in high yield.35 By use of an excellent technique involving the copper-mediated decarboxylation of potassium pentafluorobenzoate in diglyme,36 one of the two iodine atoms in compound 2 could be selectively substituted to provide 2,3,4,5,6-pentafluoro-2′-iodo-4′,5′-dimethoxy-1,1′-biphenyl 3. This fluorinated biphenyl was then coupled with phenylboronic acid 5 which itself can also be synthesized from veratrole. The monobromination of veratrol occurs under mild conditions at room temperature37 to provide 4-bromoveratrol 4. The 4-bromoveratrol was then converted to the corresponding boronic acid 5 and coupled with biphenyl 3via a Suzuki reaction, yielding the desired 1,2,3,4-tetrafluoro-6,7,10,11-tetramethoxy-o-terphenyl 6. The final elaboration of o-terphenyl 6 to the difluorinated hexaalkoxytriphenylenes 10 is shown in Scheme 5. Here the o-terphenyl 6 underwent the PCDHF reaction (irradiation at 254 nm in acetonitrile) to yield 1,2,3,4-tetrafluoro-6,7,10,11-tetramethoxytriphenylene 7, which conveniently precipitated out of the solution directly in 89% yield without the need for any further purification. After the deprotection of the four methoxy groups using BBr3 in dichloromethane, the crude 9,10,11,12-tetrafluorotriphenylene-2,3,6,7-tetraol 8 was obtained in almost quantitative yield and subsequently treated directly with a primary alkyl halide and potassium carbonate in DMF to yield the respective 1,2,3,4-tetrafluoro-6,7,10,11-tetraalkoxytriphenylene (4F-TATn) 9. Finally, the previously discussed SNAr technique was applied to 9 and the pair of fluorine atoms at the 2,3-positions were selectively substituted with alkoxy groups in sequential SNAr reactions to give the desired 2F-HATn derivatives 10 as final products (2F-HAT5 10a has six pentyloxy groups, while 2F-HAT6 10b has six hexyloxy groups). Note that this approach also provides the opportunity to readily introduce different alkoxy groups between the 6,7,10,11-positions and the 2,3-positions. For example, 2F-HAT(6/12) 10c-i was prepared with four hexyloxy groups at the 6,7,10,11-positions and two dodecyloxy groups at the 2,3-positions while, vice versa, 2F-HAT(12/6) 10c-ii, was prepared with four dodecyloxy groups at the 6,7,10,11-positions and two hexyloxy groups at the 2,3-positions.
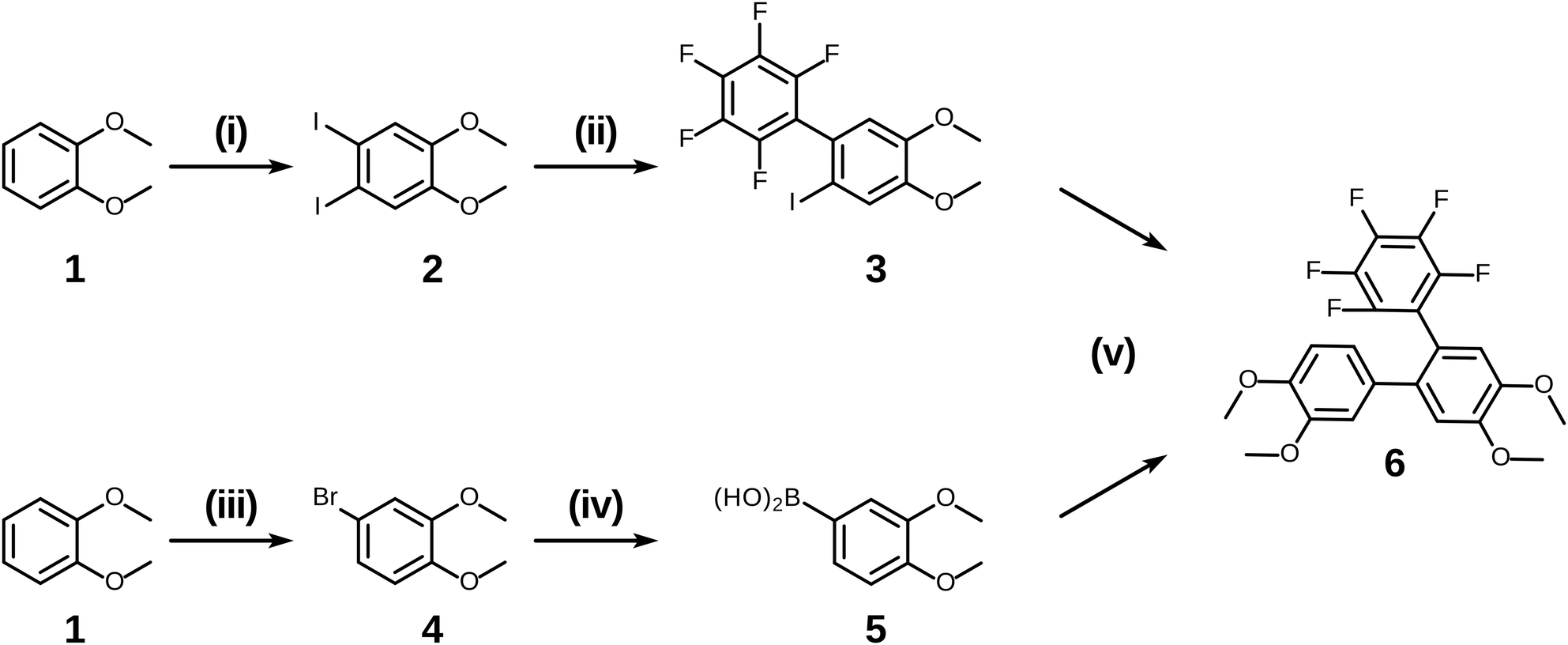 | ||
| Scheme 4 The synthesis of o-terphenyl intermediate 6. (i) I2/periodic acid/methanol, 89%; (ii) PhF5COOK/CuI (0.1 eq.)/diglyme/150 °C, 55%; (iii) NBS/silica Gel/CHCl3/rt, 92%; (iv) (1) n-BuLi/−78 °C, (2) triisopropyl borate/−78 °C, (3) HCl/rt, 51%; (v) Pd(PPh3)4/1,4-dioxane/water, 73%. | ||
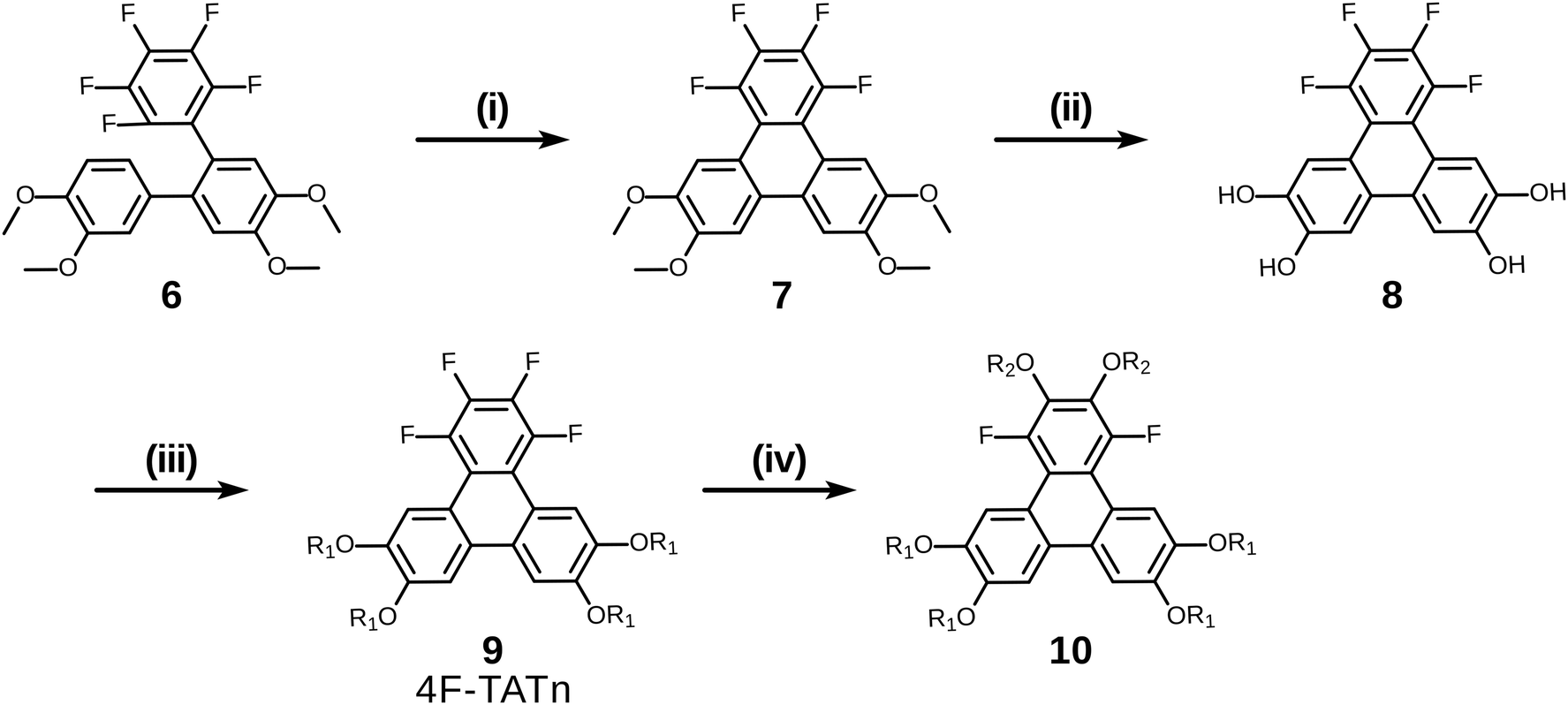 | ||
| Scheme 5 The synthesis of hexakisalkoxytriphenylene discotic liquid crystals. (i) PCDHF, 89%; (ii) BBr3/DCM, 98%; (iii) R1-I/K2CO3/DMF, 87% for R1 = C6H13; (iv) R2-OH/t-BuOK, 58% for R2 = C6H13, 81% for R2 = C12H25. | ||
Of course, for the synthesis of hexakisalkoxytriphenylene discotic liquid crystals with six identical alkoxy tails, it was possible to treat 1,2,3,4-tetrafluoro-6,7,10,11-tetramethoxytriphenylene 7 with potassium methoxide (t-BuOK in MeOH) to give 1,4-difluoro-2,3,6,7,10,11-hexamethoxytriphenylene 11. All the methoxy groups can be cleaved in molten Py·HCl salt (or BBr3 in dichloromethane) to give 1,4-difluorotriphenylene-2,3,6,7,10,11-hexaol 12. This hexaol was used as crude material in the next alkylation step, giving the desired product. As long as this kind of product is a subset of previously discussed 10, we label it as 10′ for convenience (Scheme 6).
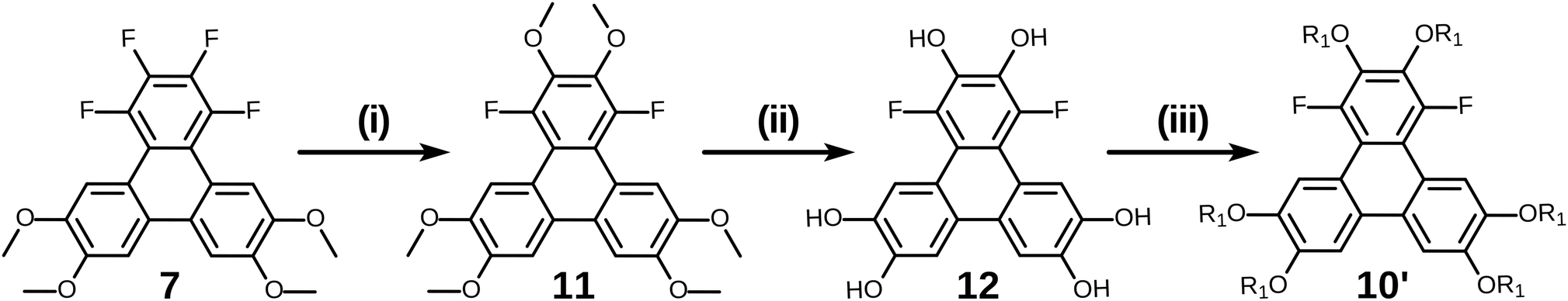 | ||
| Scheme 6 The synthesis of hexakisalkoxytriphenylene discotic liquid crystals with identical alkoxy tails. (i) t-BuOK in MeOH, 90%; (ii) Py·HCl, heating, product was used as crude; (iii) R1-I/K2CO3/DMF, 39% for R1 = C5H11. | ||
With a simple modification of this synthetic route involving only changing the order of some steps, pentaalkoxytriphenylenes could also be easily accessed (Scheme 7). Rather than initial PCDHF reaction, the terphenyl 6 was instead treated with BBr3 in dichloromethane to cleave the four methyl ethers producing crude 2′′,3′′,4′′,5′′,6′′-pentafluoro-[1,1′:2′,1′′-terphenyl]-3,4,4′,5′-tetraol 13. Next, 13 was reacted with an alkyl halide R1X to yield the pentafluorotetraalkoxyterphenyl 14. In this case the single para-fluorine atom in the pentafluoroterphenyl 14 is uniquely most activated and the SNAr substitution with the alkoxide derived from R2OH occurred cleanly at only that position and gave the alkoxy derivative 15. Finally, triphenylene synthesis was accomplished by PCDHF under UV irradiation to give 1,2,4-trifluoro-3,6,7,10,11-pentaalkoxytriphenylene (3F-PATn) 16 as the final product.
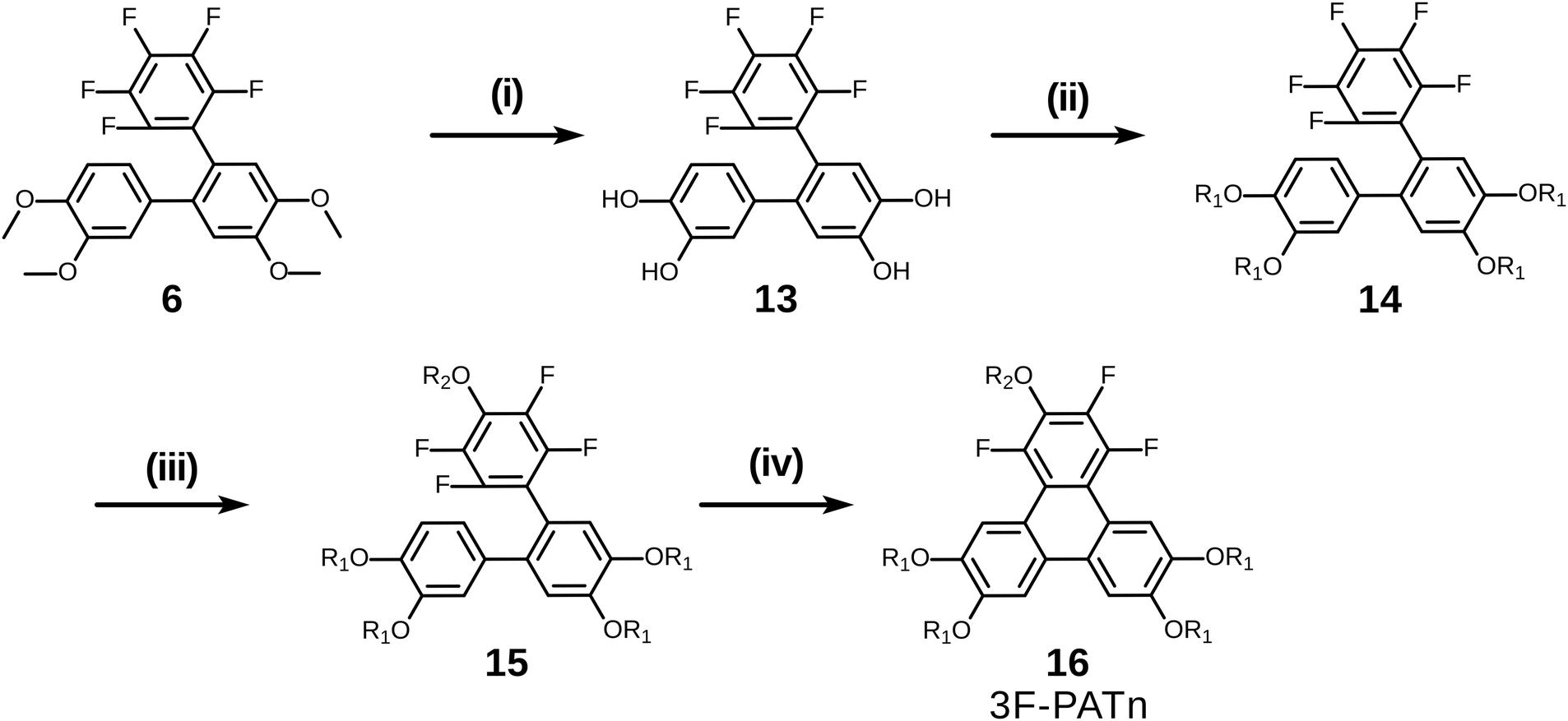 | ||
| Scheme 7 The synthesis of pentakisalkoxytriphenylene discotic liquid crystals. (i) BBr3/DCM, ∼100%; (ii) R1-I/K2CO3/DMF, 68% for R1 = C6H13; (iii) R2-OH/t-BuOK, 70% for R2 = C6H13; (iv) PCDHF, 55% for R1 = R2 = C6H13. | ||
Compared with 2F-HATn, 3F-PATn differs only in having an alkoxy tail replaced with an additional fluorine atom. While the option has not been exercised here, note that this approach also provides the opportunity to introduce different alkoxy groups between the 6,7,10,11-positions and the 3-position.
Results and discussion
Phase behavior
Using the synthetic methodologies just described, several fluorinated alkoxytriphenylenes with different numbers, locations and identities of alkoxy groups as summarized in Table 1 have been prepared. In these triphenylenes, there are either two fluorines and six alkoxy groups (10a, 10b, 10c-i, 10c-ii), three fluorines and five alkoxy groups (16a, 16b) or four fluorines and four alkoxy groups (7, 9a, 9b, 9c). The phase behavior of these substances has been investigated by differential scanning calorimetry (DSC), polarized optical microscopy (POM, Fig. 6) and, for a subset of the materials, small angle X-ray scattering (SAXS).| Name (number) | R | Phase transition temperatures (°C) (from DSC) |
|---|---|---|
| All transition temperatures obtained by DSC while cooling from isotropic at 5 K min−1.a Additional SAXS data is available in ESI.b No low temperature transition was observed by monitoring the transmission of polarized 632 nm light through a sample while it was cooled in a cryostat.c A glass structure (G) can be obtained by cooling from the mesophase at several K min−1. More information can be found in the ESI. | ||
| HAT538–40 | K 69 Colh 122 I | |
| HAT61 | K 69.5 Colh 99.5 I | |
| NO2-HAT541 | (T < −40) Colh 136.1 I | |
| NO2-HAT623,27,42 | (T < −20) Colh 137 I | |
| 1F-HAT643 | K 39 Colh 116 I | |
| 1,2,3-Trifluoro-TAT544 | K 139 Colh 154 I | |
| 1,2,3-Trifluoro-TAT645 | K 130 Colh 143 I | |
| 2F-HAT5a (10a) | R1= C5H11 | K −12.5 Colh 142.1 I |
| R2= C5H11 | ||
2F-HAT620![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (10b) b (10b) |
R1= C6H13 | (T < −150) Colh 116.5 I |
| R2= C6H13 | ||
| 2F-HAT(6/12)a (10c-i) | R1 = C6H13 | K 39.6 Colh 42.4 I |
| R2 = C12H25 | ||
| 2F-HAT(12/6)a (10c-ii) | R1 = C12H25 | K 39.5 K′ 54.9 Colh 65.6 I |
| R2 = C6H13 | ||
| 3F-PAT5 (16a) | R1= C5H11 | K 7.7 Colh 171.5 I |
| R2 = C5H11 | ||
| 3F-PAT6a,c (16b) | R1 = C6H13 | G −12.7 K′ 25.5 K 34.7 Colh 160.1 I |
| R2 = C6H13 | ||
| 4F-TAT5a (9a) | R1 = C5H11 | K 109.5 Colh 200.5 I |
| 4F-TAT6a (9b) | R1 = C6H13 | K 97.7 Colh 183.0 I |
| 4F-TAT12 (9c) | R1 = C12H25 | K 91.6 Colh 113.5 I |
| 4F-TAT1a (7) | R1 = CH3 | K 197.6 Colh 310.3 I |
The discotic behavior of the new fluorinated compounds is interesting as compared to the parent compounds HAT5 and HAT6, as well as to other triphenylene based molecules with dipolar substituents (Table 1). We observe a clear trend in the isotropic transition temperature, TCol-I, that consistently increases as more fluorine atoms are added and as tails are removed. As the majority of the molecular mass in these molecules is derived from the long alkoxy tails, this leads to lower mass molecules having higher isotropic transition temperatures. The lower limit of the columnar mesophase, TK-Col, shows a more complicated behavior (Fig. 2). Relative to their respective HATn precursors, TK-Col is lowered in all of the materials except for the 4F-TATn compounds. These compounds differ from the rest not only because they have an additional fluorine, but more importantly, because they completely lack tails on their fluorinated ring. It is noteworthy that, except for 4F-TAT1 (7), the 4F-TATn compounds (9a, 9b and 9c) all have very similar TK-Col while having a substantial difference in TCol-I, indicating that the crystallization transition in these compounds is more influenced by the core–core interaction rather than those involving the tails. These core–core interactions are especially relevant in the precursor compound 7, which has only methoxy groups in lieu of conventional longer alkoxy tails (Fig. 3). Both of the transition temperatures of this nearly tail free compound 7 are increased by about 100 degrees above their longer tailed counterparts, while the isotropic transition occurs close to that of the non-fluorinated, but non-mesogenic, counterpart HAT1.
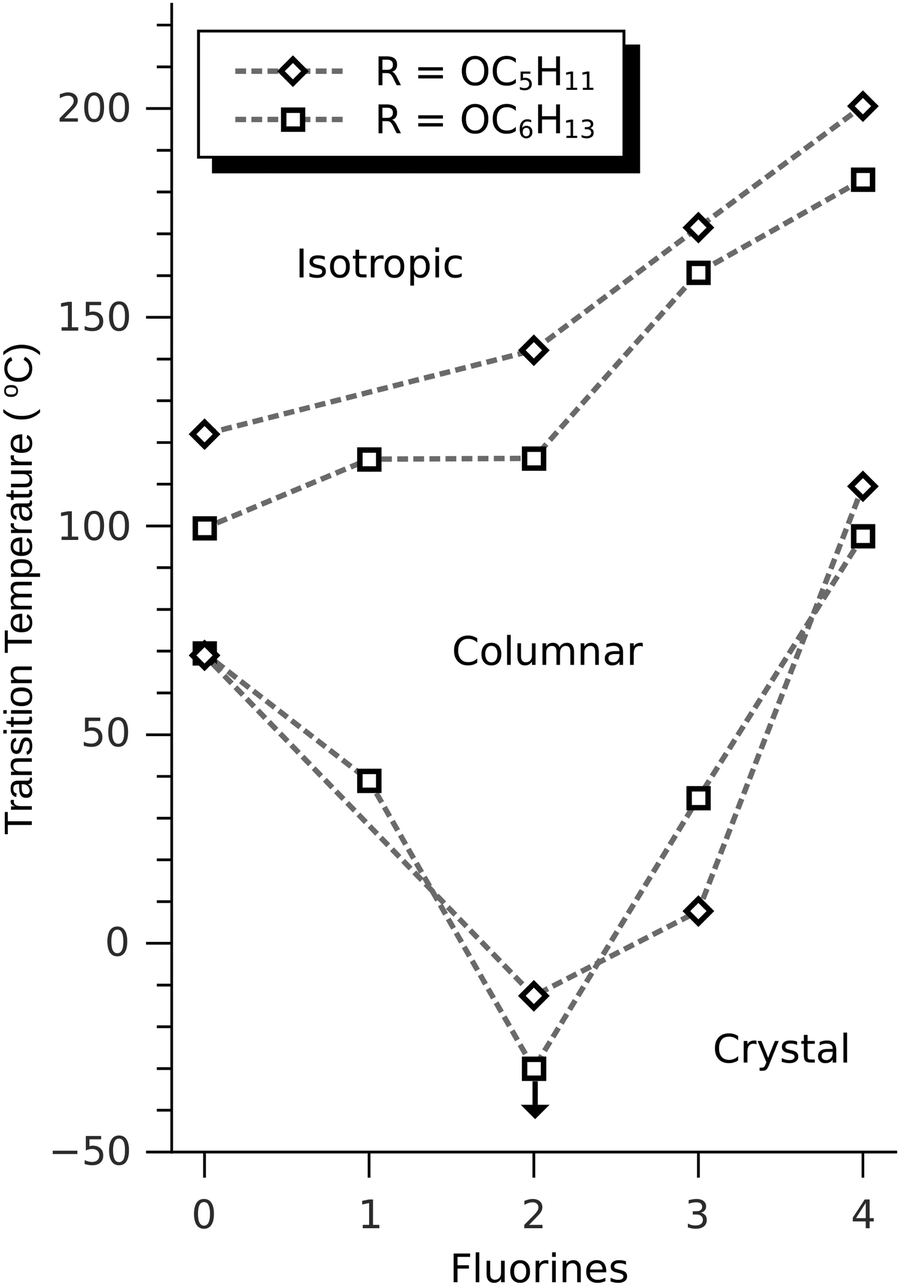 | ||
| Fig. 2 Phase behavior of fluorinated alkoxytriphenylenes. The isotropic transition temperature increases with additional fluorination, even as tails (and associated entropy) are removed, as in 3F-PATn and 4F-TATn. The freezing point is reduced in all but the tetrafluoro compounds. This reduction in the freezing point is particularly noteworthy for 2F-HATn and 3F-PATn, where it extends the mesophase near room temperature. The mesophase of 2F-HAT6 has no detectable lower limit above −150 °C. Lines are a guide for the eyes. | ||
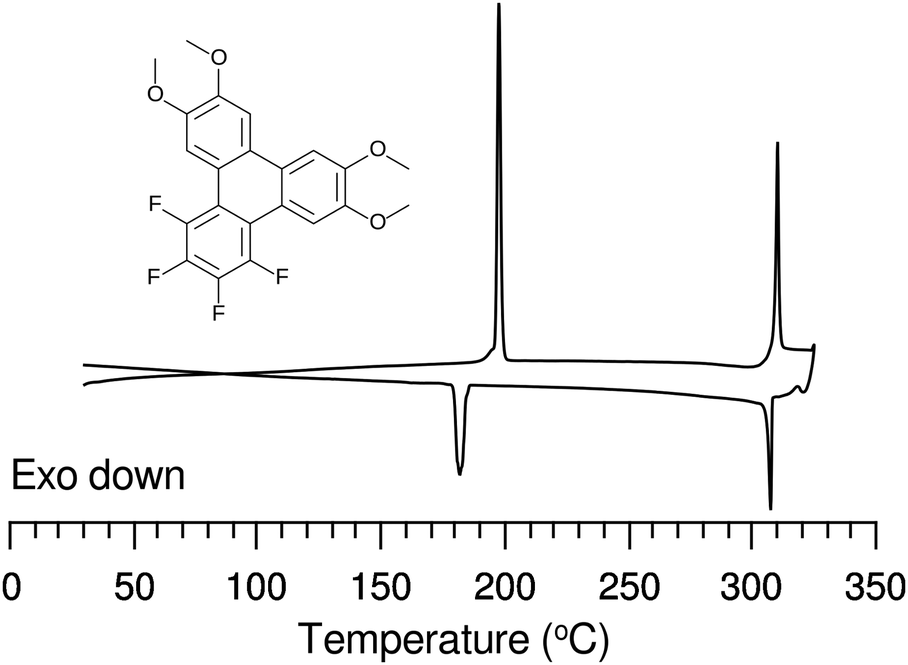 | ||
| Fig. 3 DSC curve of 4F-TAT1 (7) taken after an initial heating cycle, and with a scan rate of 5 K min−1. Despite the lack of long tails, this material possesses a broad columnar mesophase at elevated temperature (K 197.6 Colh 310.3 I). The material is sufficiently stable to allow multiple DSC cycles to be obtained, as demonstrated here where limited degradation is evident on the second cycle of the DSC. However, prolonged exposure to such high temperatures will result in severe thermal degradation. | ||
The discotic behavior of 4F-TAT1 (7) is remarkable. The DSC and POM for this substance are shown in Fig. 3 and 6 respectively. Note that nonfluorinated HAT1, HAT2 and HAT3 are not mesomorphic and that discotic activity in this nonfluorinated series of hexaalkoxytriphenylenes only begins in HAT4 with butoxy tails.45 Discotic compounds with four pentyloxy or longer tails and a tail-free ring bearing electronegative substituent groups (including fluorine) have been reported elsewhere44,46 where enhanced core–core interactions and nanosegregation of the tails is thought to lead to dimerization and stabilization of the columnar mesophase. Our results and others show that discotic behavior may be induced with single-carbon tails,47 or even no tails at all.48 Other unconventional approaches are possible, e.g., “flying seed” molecules where the introduction of tails containing bulky rings49 has been shown to support mesogenic behavior. This behavior in compounds with limited tail content points to important interactions between the core moieties. In our case, the introduction of fluorine modifies the (long range) electrostatic and (short range) dispersive forces, especially for relatively short tailed systems where the intercolumnar spacing is small. To better understand these interactions, ground state geometries and corresponding multipole moments were calculated for methoxy analogues of the studied compounds, using DFT at the B97D3/6-31g* level. The dipole moments of 2F-HAT1, 3F-PAT1 and 4F-TAT1 were found to be 2.62 D, 2.80 D and 3.68 D, respectively. These dipole moments are strongly influenced by the position of the oxygen atoms in the alkoxy groups (i.e., the conformation of the alkoxy groups). Specifically, O–F steric interactions can force the nearby oxygen atoms out of the molecular plane, substantially changing the molecular dipole moment magnitude and direction. This is especially the case in 2F-HATn (which has the widest mesophase range in this study) because the two oxygens on the fluorinated ring may sit either on the same side of the molecule or on opposite sides (Fig. 4), resulting in either a significant out-of-plane dipole, or a small but non-zero in-plane dipole, respectively. We find that these configurations are nearly degenerate in energy, indicating that fluctuations between these conformations is possible, especially in the liquid crystalline phase. In the crystalline phase of 1,4-difluoro-2,3-dimethoxytriphenylene, intermolecular interactions result in a symmetric disposition of the syn-methoxy groups.
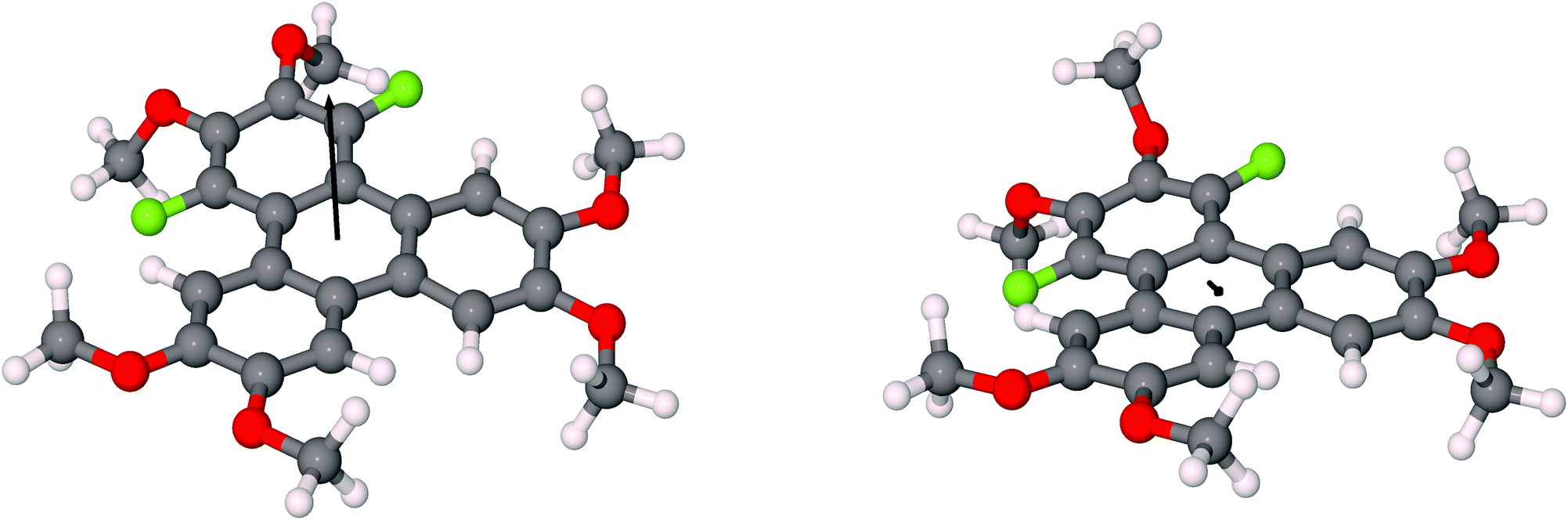 | ||
| Fig. 4 The conformers in the fluorinated ring and corresponding dipole moments of 1,4 difluoro-2,3,6,7,10,11-hexamethoxytriphenylene in (right) anti-configuration and (left) syn-configuration. The black arrow indicates the dipole moment vector. | ||
It must be emphasized, however, that the intermolecular electrostatic interaction energies cannot be quantitatively described by dipole–dipole interactions alone since the molecules are substantially closer to each other than a characteristic size of the molecular wave function. Instead, the actual electrostatic potential and charge distributions should be used, along with a more detailed accounting of the intermolecular interactions. A more complete analysis based on ab initio calculations and molecular dynamics simulations is in preparation.
SAXS characterization of mesophases
We have used SAXS in order to confirm the presence of liquid crystal mesophases, and to unambiguously identify their structures. SAXS experiments were carried out on a representative subset of compounds using a Rigaku Screen Machine, using a Cu K-α X-ray tube; with samples held in thin walled capillary tubes placed in a hot stage. Alignment of the sample was enhanced using permanent magnets close to the capillary, which created a field roughly orthogonal to both the capillary and X-ray beam. With the exception of 4F-TAT1, samples were heated into their isotropic phase with data collected on cooling. Due to the high clearing temperature of 4F-TAT1 (310 °C), diffraction patterns were obtained by heating a sample into the lower limits of the mesophase and collecting data on heating. The sample was not aligned, since it was not brought into the isotropic phase while within the magnetic field, and the resulting powder diffraction pattern reflects a texture containing many domains. Additional experimental details can be found in the ESI.† The mesophases were identified by assigning Miller indices, hkl, to the diffraction peaks. For the hexagonal mesophase, the d-spacing ratios for the 100, 110 and 200 peaks are expected to be 1:1/√3:1/2. In addition to these peaks corresponding to intercolumn order, we observed broad 001 peaks resulting from the disordered columns. The stacking distance within the columns is approximately 3.5 Å. A lack of correlations between intracolumn spacings in adjacent columns is indicated by the absence of peaks with simultaneously non-zero h or k and l indices, which confirms that the structures are liquid crystalline as opposed to, e.g., plastic crystals. Additionally, a number of samples were sufficiently well aligned so that the diffraction pattern showed a clear six-fold symmetry, further supporting the identification of the Colh mesophase. All samples studied were found to be hexagonal columnar mesogens, identified either by the above stated ratio of d-spacings, or, for weak scatterers which did not produce observable high order reflections, by the symmetry of the 100 peaks. A summary of SAXS results has been collected in Table 2.
| Name (number) | Temp. (°C) | h k l | d spacings (Å) | |
|---|---|---|---|---|
| Obs. | Calc. | |||
| 2F-HAT5 (10a) | 30 | 100 | 22.9 | 22.9 |
| 110 | 13.2 | 13.3 | ||
| 200 | 11.5 | 11.5 | ||
| 001 | 3.5 | 3.5 | ||
| 3F-PAT6 (16b) | 100 | 100 | 17.6 | 17.6 |
| 110 | 10.0 | 10.2 | ||
| 001 | 3.5 | 3.5 | ||
| 4F-TAT5 (9a) | 190 | 100 | 15.8 | 15.8 |
| 001 | 3.5 | 3.5 | ||
| 4F-TAT6 (9b) | 180 | 100 | 16.8 | 16.8 |
| 001 | 3.6 | 3.6 | ||
| 4F-TAT1 (7) | 220 | 100 | 11.0 | 11.0 |
| 110 | 6.4 | 6.4 | ||
| 200 | 5.5 | 5.5 | ||
| 001 | 3.6 | 3.6 | ||
Given the unusual properties of 4F-TAT1, it is of interest to compare its structure to that of a more conventional discotic liquid crystal with lengthy tails. Fig. 5 contains diffraction patterns of 3F-PAT6 and 4F-TAT1, clearly illustrating the effects that the reduced tail content has on the mesophase structure. Both compounds show sharp peaks (long-range order) reflecting the hexagonal column packing. However, 4F-TAT1 has much more closely packed columns since there is very little to prevent the cores from approaching each other. The broad diffuse scattering associated with the tails in 3F-PAT6 is lost. The diffuse peaks reflecting the disordered stacking of molecules within columns, however, is almost identical between the compounds: the columnar structure is the same. Therefore, the tails play little if any role in determining the distance between cores in a column, which is crucial to electronic transport.
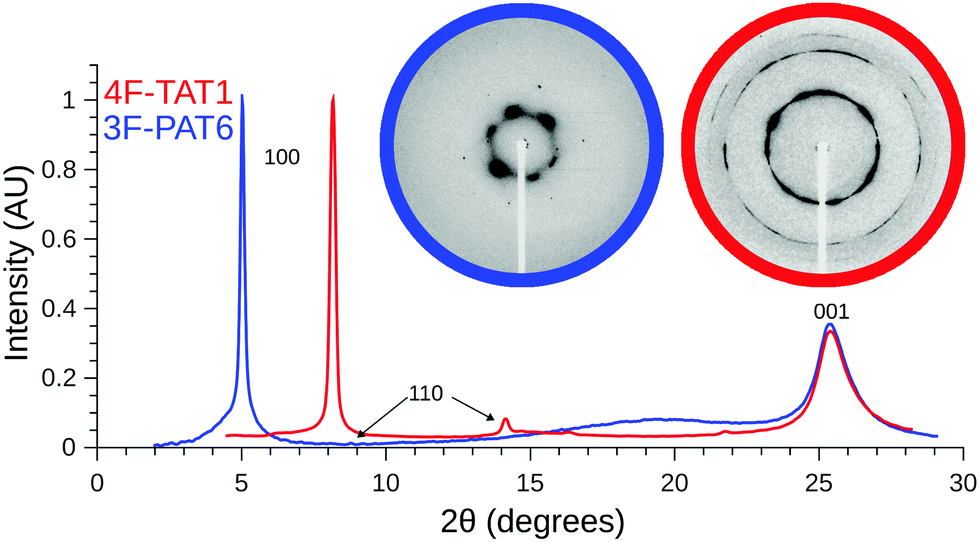 | ||
| Fig. 5 SAXS diffraction patterns collected from 4F-TAT1 at 220 °C and 3F-PAT6 at 100 °C. A portion of the small angle region is shown in the inset, with an emphasis on the 100 and 110 scattering. 3F-TAT1's lack of long tails results in a much shorter intercolumn spacing and a lack of the broad diffuse scattering associated with the tails. It also results in a greater degree of in-plane order resulting in a number of peaks which are suppressed in 3F-PAT6. | ||
 | ||
| Fig. 6 POM texture for selected compounds. Top row from left to right: 2F-HAT5, 2F-HAT6, 2F-HAT(12/6). Middle row from left to right: 3F-HAT5, 3F-HAT6, 2F-HAT (6/12). (All at 30 °C.) Bottom row from left to right: 4F-TAT5 at 150 °C, 4F-TAT6 at 150 °C, and 4F-TAT1 at 300 °C. All images were taken with crossed polarizers. The scale bar found in the lower left corner applies to all images. | ||
Charge transport properties
In order to understand the effect of fluorination on electronic transport, the hole mobilities throughout the range of the columnar mesophase in 2F-HAT5, 3F-HAT6, 4F-TAT5 and 4F-TAT6 have been measured. The presence of a static molecular dipole moment can potentially have a number of competing effects on the bulk charge mobility. The relatively long range of dipole interactions (compared to van der Waals and quadrupolar interactions) can increase correlation lengths along the column, potentially increasing order and the mobility.22 Neighboring molecules can be expected to orient themselves in order to cancel out their net dipole moment, which will govern the preferred orientation of molecules within the column (subject to the aforementioned caveats concerning higher moments). This promotes a coplanar arrangement which, depending on the structure of the HOMO (LUMO) level, may be beneficial to hole (electron) transfer. At the same time, disordered permanent dipoles can be expected to affect the energetic disorder50 and charge trapping within the liquid crystalline system, which might create an energetic barrier against charge transport. In order to study the net effect of these competing factors, we have employed the time of flight (TOF) technique to measure the mobilities of holes along the columns for a representative subset of the compounds.The mechanism of charge transport within these mesogens is thermally assisted hopping conduction, where a collection of charges “hop” between localized states on the constituent molecules, taking a random walk across the sample driven by an applied potential. These moving charges create a transient photocurrent which is recorded and used to determine the transit time for charges across the cell. The mobility is given by μ = d2/Vτ, where d is the thickness of the sample, V is the applied voltage, and τ is the time of flight. The charges are photogenerated using a pulsed laser at a wavelength selected based on the absorption spectra of each compound, such that the penetration depth is much smaller than the cell thickness, and at an intensity such that space charge effects are absent. Data were collected after heating the samples into the isotropic state and slowly cooling them into the columnar mesophase. All of the times of flight were extracted from linear-linear plots of current vs. time. The mobilities were analyzed according to a one-dimensional hopping model with Gaussian disorder as used to study similar compounds,27,40,51 which predicts a mobility of the form
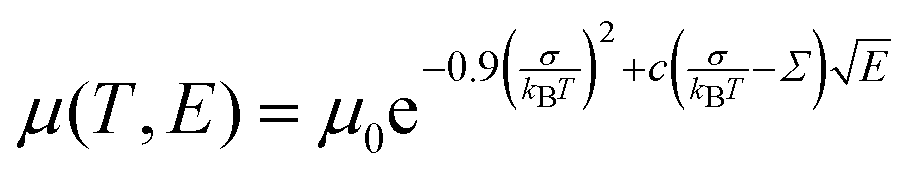
In all cases studied, the mobility is monotonically dependent on temperature (Fig. 7 and 8). This is expected for hopping transport, where the intermolecular hop is assisted by the thermal energy available in the system. The mobilities themselves are also rather close to each other in all of the studied samples, with typical values between 10−4 and 10−3 cm2 V−1 s−1 (Table 4). These values are comparable to the mobilities of the unsubstituted HATn compounds,22,43 so any differences in parameters governing transport, such as intermolecular orientation, polaron formation, columnar disorder, or a proclivity for multiple trapping, must either be small or canceled out by a complimentary effect. While it is not surprising that 2F-HAT5 and 2F-HAT6 have similar properties, it is significant that we can break molecular symmetry, as in 3F-PATn, or even remove all the tails from a ring, as in 4F-TATn, without seriously affecting the charge transport characteristics (Fig. 8) (though, as noted above, the molecular stacking distance, at least, is similar for 4F-TAT1 and 3F-PAT6). This is in contrast to NO2-HAT6 which has mobilities lower than in HAT6 and a strong temperature dependence near room temperature.27 It is also notable that, while 1,4-difluoro-2,3-dimethoxytriphenylene has widely spaced molecules in the crystal, the hole mobilities of the corresponding liquid crystals with longer tails are comparable to those of HATn, perhaps indicating that equilibration of syn- and anti-isomers allowing the molecules to approach more closely in the discotic phase.
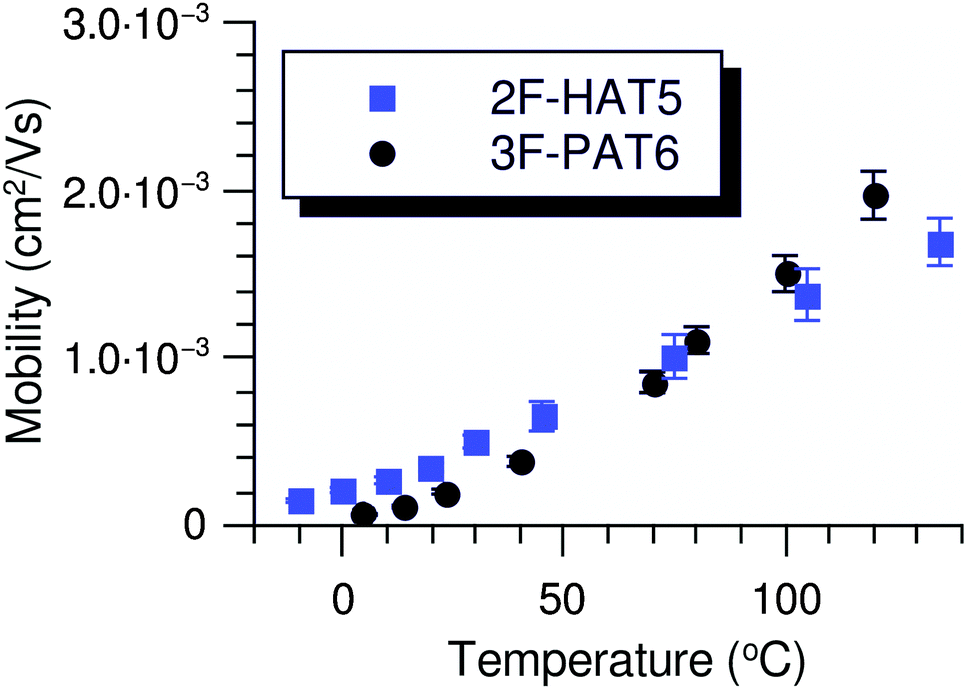 | ||
| Fig. 7 Mobilities of 2F-HAT5 and 3F-PAT6 throughout their mesophase range with an applied field of 62.2 kV cm−1. Each substance possesses a mobility on the order of 10−4 cm2 V−1 s−1 near room temperature. While the columnar phase of 2F-HAT5 persists to much lower temperatures than shown, charge transport becomes increasingly dispersive at lower temperatures due to trapping. The uncertainty in determination of the time of flight is reflected in the error bars. | ||
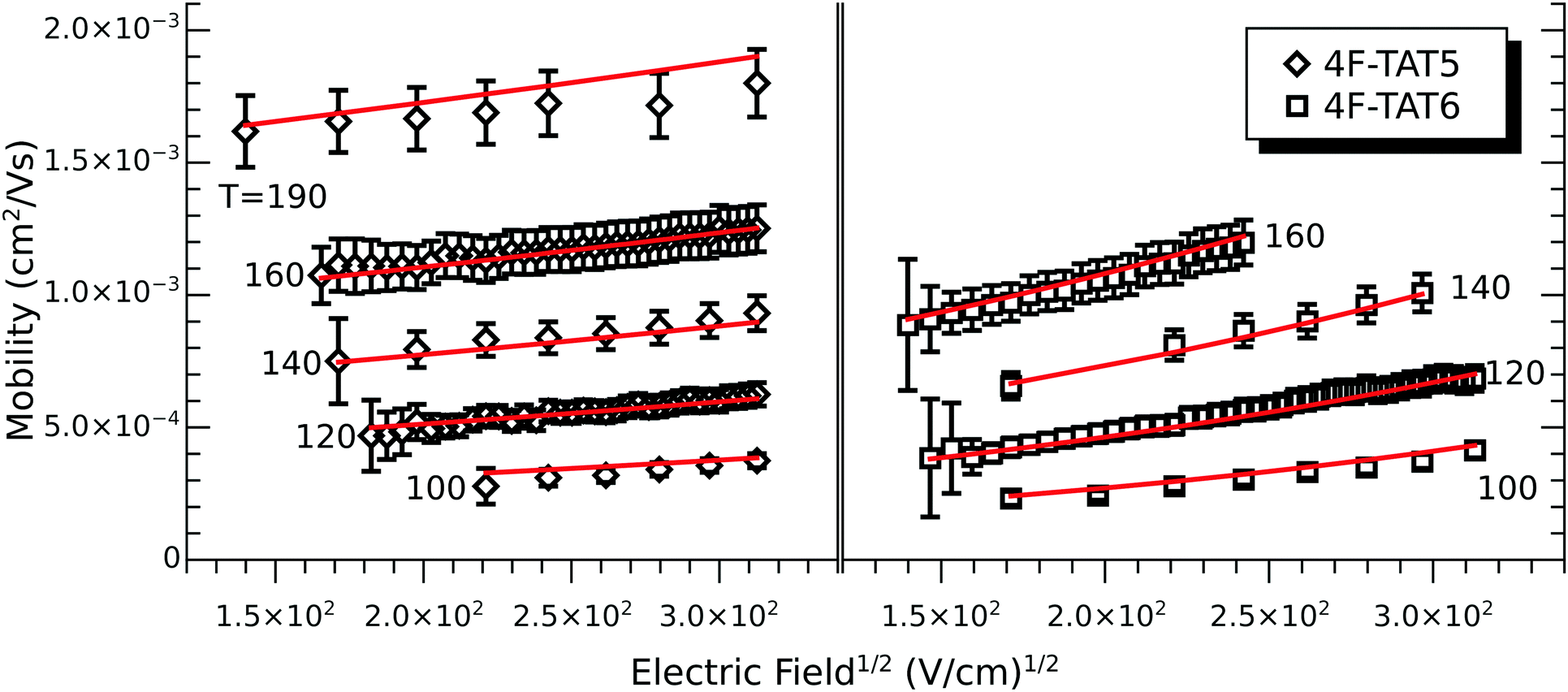 | ||
| Fig. 8 Mobilities for (left) 4F-TAT5 and (right) 4F-TAT6 throughout their columnar mesophase (temperatures are in Celsius). Gray lines are fits to the model. Removing the tails from the fluorinated ring not only preserves the mesomorphic behavior, but the resulting mobilities remain comparable to their tailed analogues. The large uncertainty at low field reflects the inherent difficulty of extracting a time of flight in this regime. | ||
Conclusions
A new and versatile route to fluorinated alkoxytriphenylene liquid crystals has been devised and the critical pair of PCDHF and SNAr synthesis steps involved were demonstrated to be useful and effective. In this route the alkoxy chains are installed late in the synthesis and a wide variety of hexaalkoxytriphenylene derivatives can be easily obtained. From the same intermediates this new methodology can also provide pentaalkoxytriphenylenes, which the earlier route is incapable of delivering, and tetraalkoxytriphenylenes as well. By selective fluorination of these materials, multiple series of substances have been produced with columnar mesophases, some with minimal (methoxy) tail content. Some of our compounds also have wide temperature ranges, which makes them of practical interest, especially since the charge transport properties remain on par with the parent HAT compounds. The chemistry described is a powerful technique for the synthesis of fluorinated liquid crystals with potentially valuable properties. In addition, and in contrast to conventional examples of triphenylene liquid crystals, compounds with a diminished number and even an absence of tails have been discovered which possess a columnar phase.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support of the National Science Foundation, grant #1809536.Notes and references
- M. Hird, Fluorinated liquid crystals - properties and applications, Chem. Soc. Rev., 2007, 36, 2070–2095 RSC.
- R. J. Bushby and O. R. Lozman, Discotic liquid crystals 25 years on, Curr. Opin. Colloid Interface Sci., 2002, 7, 343–354 CrossRef CAS.
- R. J. Bushby and K. Kawata, Liquid crystals that affected the world: discotic liquid crystals, Liq. Cryst., 2011, 38, 1415–1426 CrossRef CAS.
- S. Kumar, Discotic liquid crystal-nanoparticle hybrid systems, NPG Asia Mater., 2014, 6, 82 CrossRef.
- S. Chandrasekhar and G. S. Ranganath, Discotic liquid crystals, Rep. Prog. Phys., 1990, 53, 57–84 CrossRef CAS.
- T. Wöhrle, I. Wurzbach, J. Kirres, A. Kostidou, N. Kapernaum, J. Litterscheidt, J. C. Haenle, P. Staffeld, A. Baro, F. Giesselmann and S. Laschat, Discotic liquid crystals, Chem. Rev., 2016, 116, 1139–1241 CrossRef PubMed.
- S. Chandrasekhar, B. K. Sadashiva and K. A. Suresh, Liquid crystals of disc-like molecules, Pramana, 1977, 9, 471–480 CrossRef CAS.
- N. Boden, R. J. Bushby, J. Clements, M. V. Jesudason, P. F. Knowles and G. Williams, One-dimensional electronic conductivity in discotic liquid crystals, Chem. Phys. Lett., 1988, 152, 94–99 CrossRef CAS.
- N. Boden, R. J. Bushby, J. Clements, M. V. Jesudason, P. F. Knowles and G. Williams, Errata, Chem. Phys. Lett., 1989, 154, 613 CAS.
- N. Boden, R. J. Bushby, J. Clements, B. Movaghar, K. J. Donovan and T. Kreouzis, Mechanism of charge transport in discotic liquid crystals, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 13274–13280 CrossRef CAS.
- R. Termine and A. Golemme, Charge Mobility in Discotic Liquid Crystals, Int. J. Mol. Sci., 2021, 22, 877 CrossRef CAS.
- M. Kumar, S. Varshney and S. Kumar, Emerging nanoscience with discotic liquid crystals, Polym. J., 2021, 53, 283–297 CrossRef CAS.
- S. Laschat, A. Baro, N. Steinke, F. Giesselmann, C. Hägele, G. Scalia, R. Judele, E. Kapatsina, S. Sauer, A. Schreivogel and M. Tosoni, Discotic liquid crystals: From tailor-made synthesis to plastic electronics, Angew. Chem., Int. Ed., 2007, 46, 4832–4887 CrossRef CAS PubMed.
- S. Sergeyev, W. Pisula and Y. H. Geerts, Discotic liquid crystals: a new generation of organic semiconductors, Chem. Soc. Rev., 2007, 36, 1902–1929 RSC.
- K. Hirota, K. Tajima and K. Hashimoto, Physicochemical study of discotic liquid crystal decacyclene derivative and utilization in polymer photovoltaic devices, Synth. Met., 2007, 157, 290–296 CrossRef CAS.
- T. Hassheider, S. A. Benning, H.-S. Kitzerow, M.-F. Achard and H. Bock, Color-tuned electroluminescence from columnar liquid crystalline alkyl arenecarboxylates, Angew. Chem., Int. Ed., 2001, 40, 2060–2063 CrossRef CAS.
- M. Steinhart, S. Zimmermann, P. Göring, A. K. Schaper, U. Gösele, C. Weder and J. H. Wendorff, Liquid crystalline nanowires in porous alumina: Geometric confinement versus influence of pore walls, Nano Lett., 2004, 5, 429–434 CrossRef.
- D. Perez and E. Guitian, Selected strategies for the synthesis or triphenylenes, Chem. Soc. Rev., 2004, 33, 274–283 RSC.
- A. N. Cammidge and R. J. Bushby, in Handbook of Liquid Crystals, ed. D. Demus, J. Goodby, G. W. Gray, H.-W. Spiess and V. Vill, Wiley, 1998, ch. 7, pp. 693–748 Search PubMed.
- S. Kumar. Chemistry of Discotic Liquid Crystals: From Monomers to Polymers, CRC Press, Boca Raton, FL, 2011 Search PubMed.
- D. Demus, J. W. Goodby, G. W. Gray, H. W. Spiess and V. Vill, Handbook of Liquid Crystals, Low Molecular Weight Liquid Crystals 2b, Wiley, 2011 Search PubMed.
- R. J. Bushby, K. J. Donovan, T. Kreouzis and O. R. Lozman, Molecular engineering of triphenylene-based discotic liquid crystal conductors, Opto-Electron. Rev., 2005, 13, 269–279 CAS.
- R. J. Bushby, N. Boden, C. A. Kilner, O. R. Lozman, Z. Lu, Q. Liu and M. A. Thornton-Pett, Helical geometry and liquid crystalline properties of 2,3,6,7,10,11-hexaalkoxy-1-nitrotriphenylenes, J. Mater. Chem., 2003, 13, 470–474 RSC.
- S. Kumar, M. Manickam, V. S. K. Balagurusamy and H. Schonherr, Electrophilic aromatic substitution in triphenylene discotics: Synthesis of alkoxynitrotriphenylenes, Liq. Cryst., 1999, 26, 1455–1466 CrossRef CAS.
- S. Kumar and M. Manickam, Nitration of triphenylene discotics: Synthesis of mononitro-, dinitro- and trinitro-hexaalkoxy triphenylenes, Mol. Cryst. Liq. Cryst., 1998, 309, 291–295 CrossRef CAS.
- N. Boden, R. J. Bushby and A. N. Cammidge, Preliminary communications functionalization of discotic liquid crystals by direct substitution into the discogen ring α-nitration of triphenylene-based discogens, Liq. Cryst., 1995, 18, 673–676 CrossRef CAS.
- H. Iino, J. Hanna, R. J. Bushby, B. Movaghar and B. J. Whitaker, Hopping conduction in the columnar liquid crystal phase of a dipolar discogen, J. Appl. Phys., 2006, 100, 043716 CrossRef.
- Z. Li and R. Twieg, Photocyclodehydrofluorination, Chem. – Eur. J., 2015, 21, 15534–15539 CrossRef CAS.
- Z. Li, Photocyclodehydrofluorination (PCDHF)—a synthetic method for fluorinated polynuclear aromatic hydrocarbons, PhD thesis, Kent State University, 2015 Search PubMed.
- H. R. A. Golf, H.-U. Reissig and A. Wiehe, Regioselective nucleophilic aromatic substitution reaction of meso-pentafluorophenl-substituted porphyrinoids with alcohols, Eur. J. Org. Chem., 2015, 1548–1568 CrossRef CAS.
- D. Samaroo, M. Vinodu, X. Chen and C. M. Drain, mesotetra(pentafluorophenyl)porphyrin as an efficient platform for combinatorial synthesis and the selection of new photodynamic therapeutics using a cancer cell line, J. Comb. Chem., 2007, 9, 998–1011 CrossRef CAS PubMed.
- A. S. Matharu, S. J. Cowling and G. Wright, laterally fluorinated liquid crystals containing the 2,2’-bithiophene moiety, Liq. Cryst., 2007, 34, 489–506 CrossRef CAS.
- X. Dai, L. Cai and J. Wen, The synthesis and mesopmorphic properties of 4-n-alkoxyphenyl 4-[(4-n-alkoxy-2,3,5,6-tetrafluorophenyl)ethyl]benzoates, Liq. Cryst., 2013, 40, 1146–1150 CrossRef CAS.
- S. Yamada, M. Morita, T. Agou, T. Kubota, T. Ichikawa and T. Konno, Thermoresponsive luminescence properties of polyfluorinated bistolanetype light-emitting liquid crystals, Org. Biomol. Chem., 2018, 31, 5609–5617 RSC.
- J. Lacour, D. Monchaud, G. Bernardinelli and F. Favarger, Synthesis, Enantiomeric conformations, and stereodynamics of aromatic orthosubstituted disulfones, Org. Lett., 2001, 3, 1407–1410 CrossRef CAS PubMed.
- R. Shang, Y. Fu, Y. Wang, Q. Xu, H.-Z. Yu and L. Liu, Copper-catalyzed decarboxylative cross-coupling of potassium polyfluorobenzoates with aryl iodides and bromides, Angew. Chem., Int. Ed., 2009, 48, 9350–9354 CrossRef CAS PubMed.
- H. Konishi, K. Aritomi, T. Okano and J. Kiji, A mild selective monobromination reagent system for alkoxybenzenes; n-bromosuccinimide-silica gel, Bull. Chem. Soc. Jpn., 1989, 62, 591–593 CrossRef CAS.
- P. Morales, J. Lagerwall, P. Vacca, S. Laschat and G. Scalia, Self-assembled ordered structures in thin films of HAT5 discotic liquid crystal, Beilstein J. Org. Chem., 2010, 6(51), 2010 Search PubMed.
- M. T. Allen, S. Diele, K. D. M. Harris, T. Hegmann, B. M. Kariuki, D. Lose, J. A. Preece and C. Tschierske, Intermolecular organisation of triphenylene based discotic mesogens by interdigitation of alkyl chains, J. Mater. Chem., 2001, 11, 302–311 RSC.
- V. Duzhko, A. Semyonov, R. J. Twieg and K. D. Singer, Correlated polaron transport in a quasi-one dimensional liquid crystal, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 064201 CrossRef.
- S. M. Naresh, Studies of electric transport in novel smectic and discotic liquid crystalline organic semiconductors, PhD thesis, Kent State University, 2010.
- K. Praefcke, A. Eckert and D. Blunk, Core-halogenated, helical-chiral triphenylene-based columnar liquid crystals, Liq. Cryst., 1997, 22, 113–119 CrossRef CAS.
- N. Boden, R. J. Bushby, A. N. Cammidge, S. Duckworth and G. Headdock, α-halogenation of triphenylene-based discotic liquid crystals: towards a chiral nucleus, J. Mater. Chem., 1997, 7, 601–605 RSC.
- K. Zhao, J. Du, X. Long, M. Jing, B. Wang, P. Hu, H. Monobe, B. Henrich and B. Donnio, Design of Janus triphenylene mesogens: Facile synthesis, mesomorphism, photoluminescence, and semiconductivity, Dyes Pigm., 2017, 143, 252–260 CrossRef CAS.
- C. Destrade, M. Mondon and J. Malthete, Hexasubstituted triphenylenes: a new mesomorphic order, J. Phys., Colloq., 1979, 40, 17–21 CrossRef.
- M. Ichihara, H. Suzuki, B. Mohr and K. Ohta, Different disk structures in the hexagonal columnar mesophases of 2,3-dicyano-6,7,10,11-tetraalkoxy1,4-diazatriphenylenes and 2,3-dicyano-6,7,10,11-tetraalkoxytriphenylenes, Liq. Cryst., 2007, 34, 401–410 CrossRef CAS.
- D. Pucci, I. Aiello, A. Aprea, A. Bellusci, A. Crispini and M. Gedini, Unsuspected mesomorphism in “tail-free” cyclopalladated 3,5-disubstituted-2-(2’-pyridyl)pyrroles, Chem. Commun., 2009, 1550–1552 RSC.
- S. Basurto, S. Garcia, A. G. Neo, T. Torroba, C. F. Marco, D. Miguel, J. Barbera, M. B. Ros and M. R. Fuente, Indene and psuedoazulene discotic liquid crystals: A synthetic and structural study, Chem. – Eur. J., 2005, 11, 5362–5376 CrossRef CAS PubMed.
- H. Geng, K. Luo, G. Zou, L. Zhao, H. Wang, Q. Li and H. Ni, Have ambipolar carrier transmission property based on novel platinum(II) complexes: Synthesis, photophysical properties, liquid crystalline characteristics, polarized luminescence, Dyes Pigm., 2018, 149, 82–91 CrossRef CAS.
- P. E. Parris, D. H. Dunlap and V. M. Kenkre, Energetic disorder, spatial correlations, and the high field mobility of injected charge carriers in organic solids, Phys. Status Solidi B, 2000, 218, 47–53 CrossRef CAS.
- I. Bleyl, C. Erdelen, H.-W. Schmidt and D. Haarer, One-dimensional hopping transport in a columnar discotic liquid-crystalline glass, Philos. Mag. B, 1999, 79, 463–475 CrossRef CAS.
- S. D. Baranovskii, Theoretical description of charge transport in disordered organic semiconductors, Phys. Status Solidi B, 2014, 251, 487–525 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Including synthesis details of compounds and precursors, DSC data, XRD data, details on TOF measurement, and crystal structure data for 1,4-difluoro-2,3-methoxytriphenylene. CCDC 1456337. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ma00606a |
| ‡ Presently at Advenchen Laboratories, LLC, 887 Patriot Dr A, Moorpark, CA 93021, USA. |
| This journal is © The Royal Society of Chemistry 2022 |