
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExternal complexation of BODIPYs by CB[7] improves in-cell fluorescence imaging†
Mehmet Menaf
Ayhan
 *a,
Emrah
Özcan
ab,
Fahri
Alkan
c,
Metin
Çetin
d,
İlker
Ün
e,
David
Bardelang
f and
Bünyemin
Çoşut
*a
*a,
Emrah
Özcan
ab,
Fahri
Alkan
c,
Metin
Çetin
d,
İlker
Ün
e,
David
Bardelang
f and
Bünyemin
Çoşut
*a
aDepartment of Chemistry, Gebze Technical University, Gebze, Kocaeli, Turkey
bInstitute of Physics, Faculty of Science, University of South Bohemia, Branišovská 1760, 370 05, České Budějovice, Czech Republic. E-mail: bcosut@gtu.edu.tr; menafayhan@gtu.edu.tr
cDepartment of Nanotechnology Engineering, Abdullah Gül University, Kayseri, Turkey
dDepartment of Molecular Biology and Genetics, Gebze Technical University, Gebze, Kocaeli, Turkey
eChemistry Group Laboratories, TÜBİTAK UME, Gebze, Kocaeli, Turkey
fAix Marseille Univ, CNRS, ICR, Marseille, France
First published on 8th November 2021
Abstract
Organic luminescent compounds with high emission properties play a crucial role in fluorescence labelling and optoelectronic devices. In this work, we prepared three water soluble BODIPY derivatives (B-4, B-5, and B-6) which are weakly fluorescent due to non-radiative relaxation pathways (charge transfer: CT or heavy atom effect). However, CB[7] significantly improves BODIPY fluorescence by ∼10 fold for B-4, and by ∼3 fold for B-5. The (TD)DFT analyses suggest that for B-4 and B-5, the CT state is blue-shifted as a result of the external binding of CB[7] near the pyridinium groups. This effect favoured a radiative decay through a locally-excited (LE) π→π* transition state of BODIPYs resulting in a CB[7]-induced emission increase in solution (and in the solid state), without compromising singlet-to-triplet intersystem crossing (ISC). The improved emission of the BODIPY·CB[7] complexes was used for the fluorescence imaging of U87 cells illustrating the relevance of this approach. These results suggest that BODIPY·CB[7] complexes could be used as theragnostic agents by combining fluorescence imaging and treatment by photodynamic therapy.
Introduction
Boron dipyrromethene (BODIPY) derivatives are one of the most actively studied groups of fluorophores due to the ease of synthesis, excellent photostability, and tunability of their photophysical and photochemical properties. These organic fluorophores are attracting a great deal of attention because they have been used in solar cells,1 photodynamic therapy (PDT),2,3 organic fluorescent emitting devices (OFEDs),4,5 semiconductors,6 bioimaging,7–9 and chemo-sensors.10 According to the Jablonski diagram, the excitation of a BODIPY chromophore leads to three competitive pathways of energy emission corresponding to different photo-functions: fluorescence emission (S1 → S0 radiative decay), intersystem crossing or ISC (S1 → T1 non-radiative transition), and other nonradiative decay pathways (e.g. rotation, distortion, vibration, or charge transfer (CT)). Optimization of a given photo-function can be performed by suppressing undesired decay pathways. Moreover, BODIPYs often tend to aggregate in polar solvents causing decrease in emission and thereby limiting their use in biological applications. In the past decade, significant efforts were devoted to develop strategies aiming to address these drawbacks and improve the photophysical features of BODIPYs.11 Current approaches mainly focus on chemically modifying BODIPYs to suppress aggregation and nonradiative decay pathways, but require specific structural designs and sizable synthetic efforts.12 Besides, aggregation-induced emission (AIE) has recently emerged as another way to obtain useful fluorescence; however, this method is limited by the small number of AIE chromophores, the challenge of controlling aggregation,11,13–16 and the concerns of colloidal stability questioning safety for biomedical applications. Alternatively, supramolecular chemistry could afford low-cost strategies to tune the photophysical properties of BODIPYs by forming host–guest inclusion complexes.17–21 For biological applications, water-soluble host molecules are required. Among macrocyclic compounds,22,23 cucurbiturils (CB[n]: n = 5–8, 10)24–26 occupy a special position due to the availability of various cavity sizes, their rigid hydrophobic cavity lined by two carbonyl fringed portals and their low toxicity.25,27–30 Among the CB[n] macrocycles, CB[7] (Chart 1) has received a lot of attention due to its reasonably good water solubility, appropriate cavity size for most therapeutic agents and conventional chromophores,31–36 and low toxicity.37 Several researchers have helped to push forward the field of fluorescent dyes by making key contributions on supramolecular complexes involving macrocycles, in particular CB[n].18 Several classes of dyes have thus received intense attention and often fluorescence increases are noted after CB[n] encapsulation due to disfavored non radiative decay processes, deaggregation, and dyes experiencing both low polarity and low polarizability cavities.38 However, today, there are only a handful of studies about the effect of cucurbiturils on the photophysical and photochemical properties of BODIPY molecules.39–46 In most cases, cucurbituril encapsulation increases fluorescence by either preventing aggregation or manipulating the guest pKa values. In this work, we used CB[7] to inhibit the nonradiative decay processes of excited BODIPYs by external complexation enabling to improve guest emission for fluorescence imaging. The “supra-chromophore” approach enabled the number of synthetic steps to be dramatically reduced while allowing switchable photophysical properties to be introduced. | ||
| Chart 1 Structures of BODIPYs and CB[7] used in this work (R1, R2 = H or I). | ||
Results and discussion
Experimental
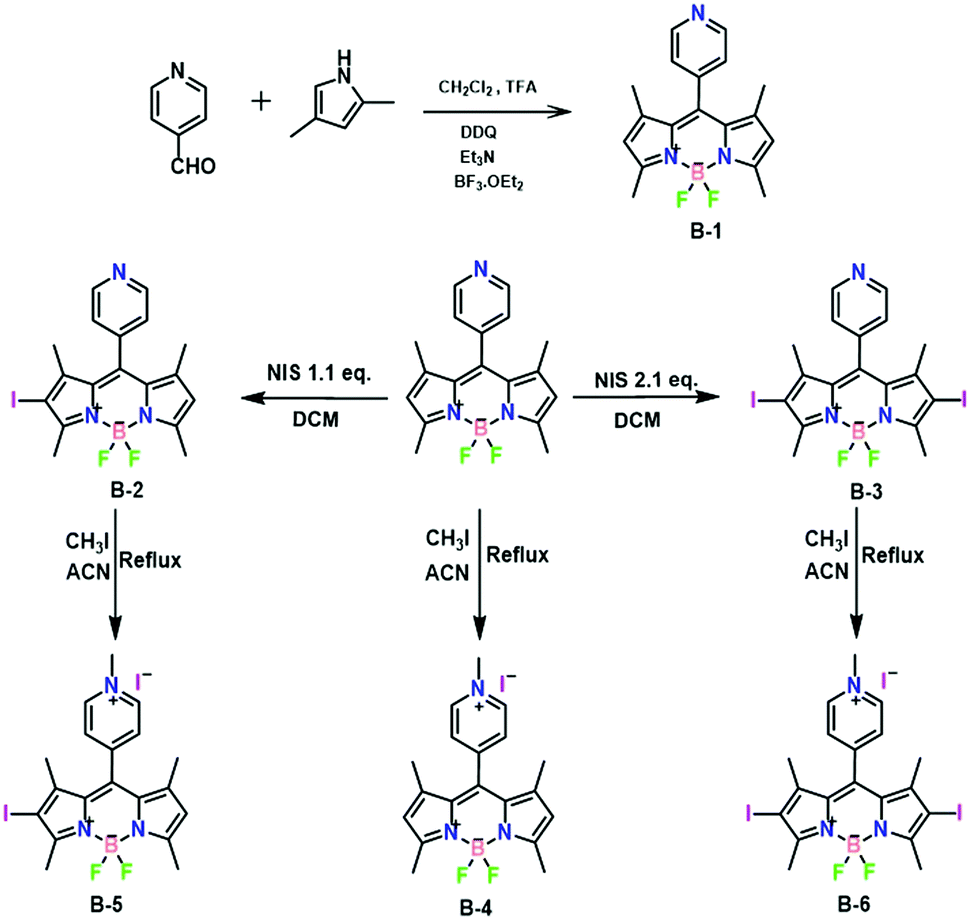 | ||
| Scheme 1 Synthetic route to BODIPYs B-4, B-5, and B-6. | ||
B-1, B-2, and B-3 were synthesized according to the literature procedures.47 Next, B-1, B-2, and B-3 underwent a reaction with 20 equivalents of methyl iodide in acetonitrile at 40 °C. The reaction proceeded smoothly in 24 h with full conversion and afforded the targets B-4, B-5, and B-6 in ∼75% isolated yield. Incorporation of the methyl group was supported by the presence of a diagnostic peak at 4.77 ppm in their 1H NMR spectra (Fig. S1, S3, and S5, ESI†). 13C NMR spectra and mass spectra confirmed this result (Fig. S2, S4, S6, S7, S8 and S9, ESI†). Additionally, the molecular structure of B-4 was further established by single-crystal X-ray diffraction (vide infra, Fig. 4a and Table S1, ESI†).
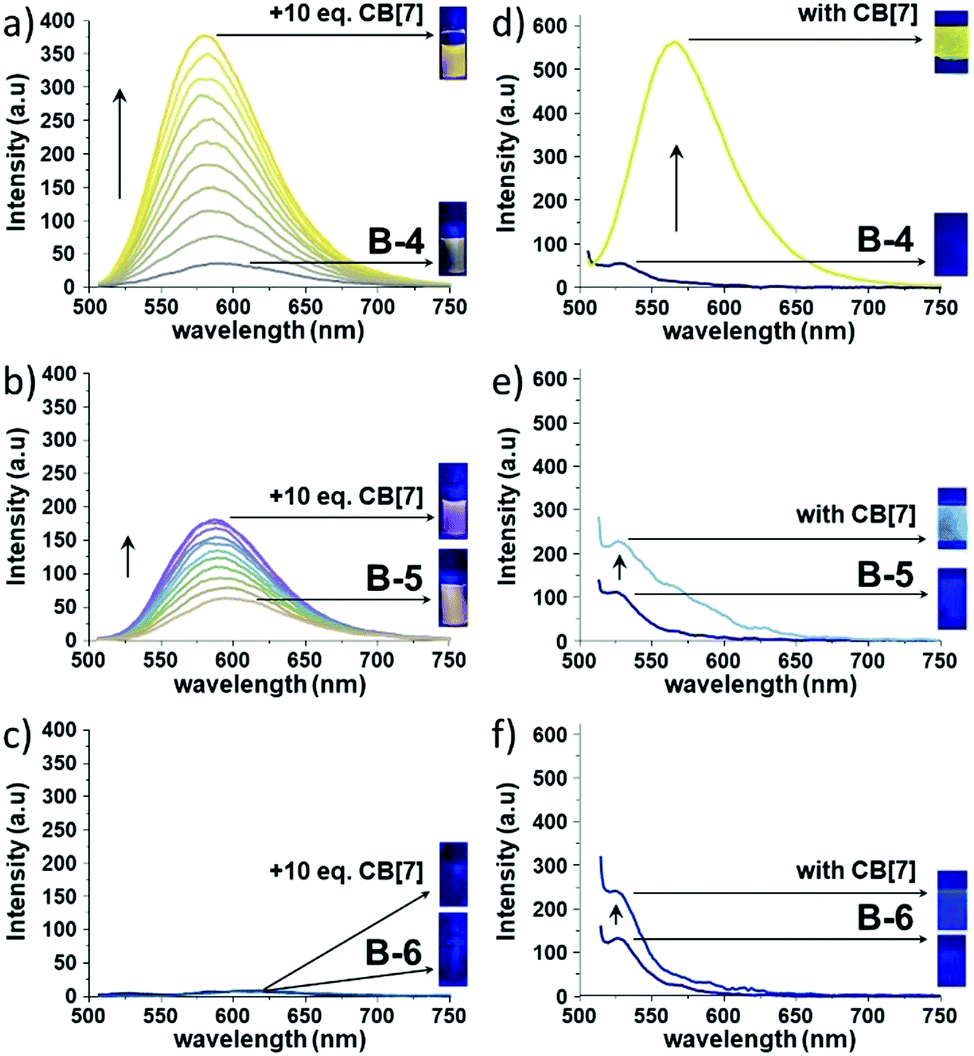 | ||
| Fig. 1 Fluorescence spectra of (a) B-4, (b) B-5, and (c) B-6 (1.0 × 10−5 M each, excitation wavelength = 490 nm) in water upon the addition of CB[7] and solid state fluorescence spectra of (d) B-4, (e) B-5, and (f) B-6 without and with CB[7]. | ||
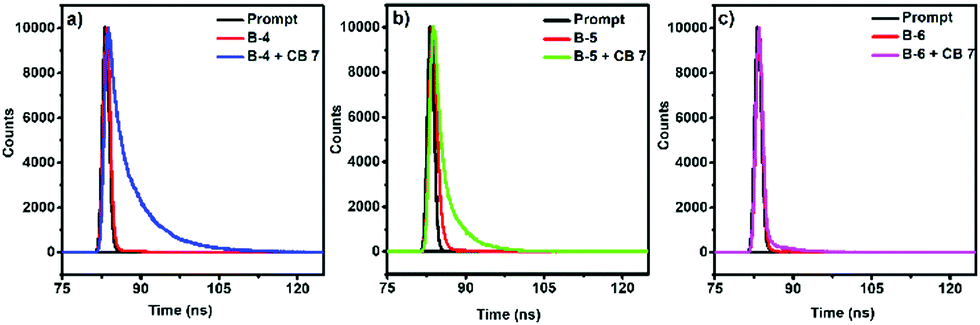 | ||
| Fig. 2 Fluorescence decay profiles of (a) B-4 without and with CB[7], (b) B-5 without and with CB[7], and (c) B-6 (10-5 M) without and with CB[7] using laser excitation at 390 nm. | ||
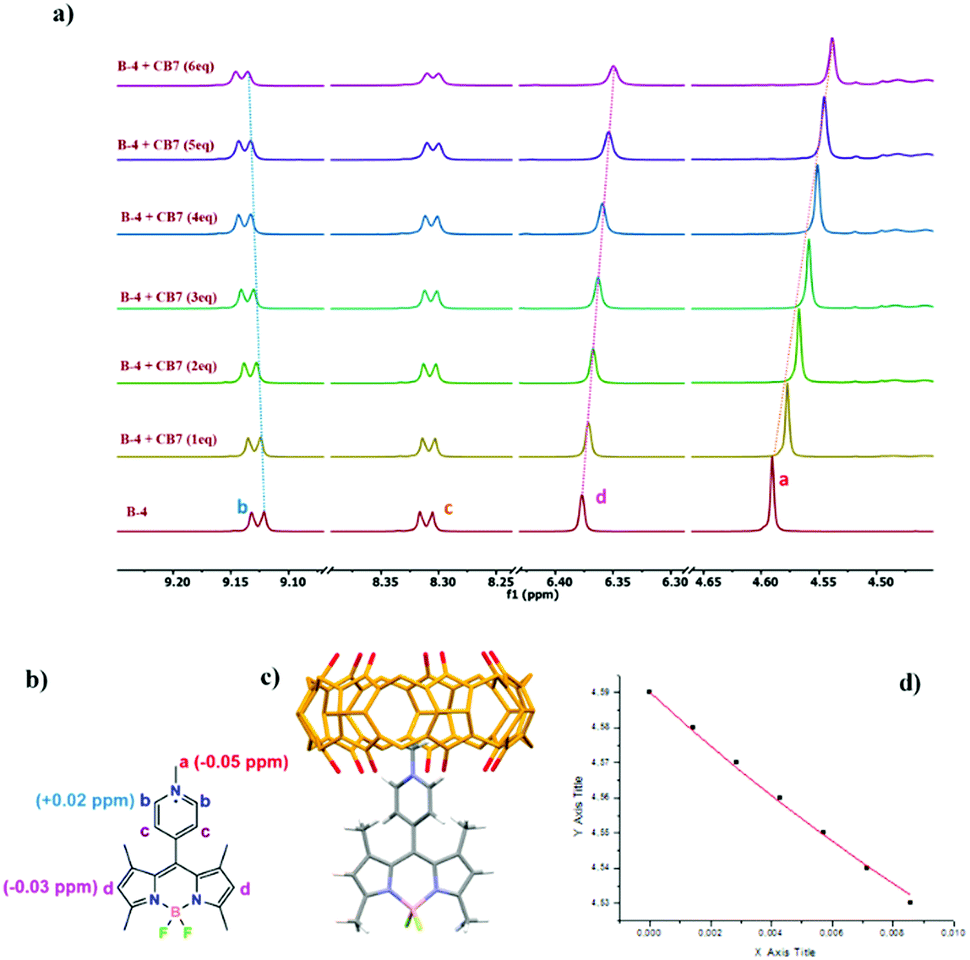 | ||
Fig. 3 (a) 1H-NMR titration of B-4 (0.5 mM) with CB [7] in D2O/CD3CN (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) solution, (b) assignment of the corresponding proton chemical shifts and (c) lowest energy DFT minimized structure of the 1:1 inclusion complex of B-4 + CB [7]. (d) The chemical shift of guest molecule Ha versus CB[7] concentration and the non-linear curve fit in red. :20) solution, (b) assignment of the corresponding proton chemical shifts and (c) lowest energy DFT minimized structure of the 1:1 inclusion complex of B-4 + CB [7]. (d) The chemical shift of guest molecule Ha versus CB[7] concentration and the non-linear curve fit in red. | ||
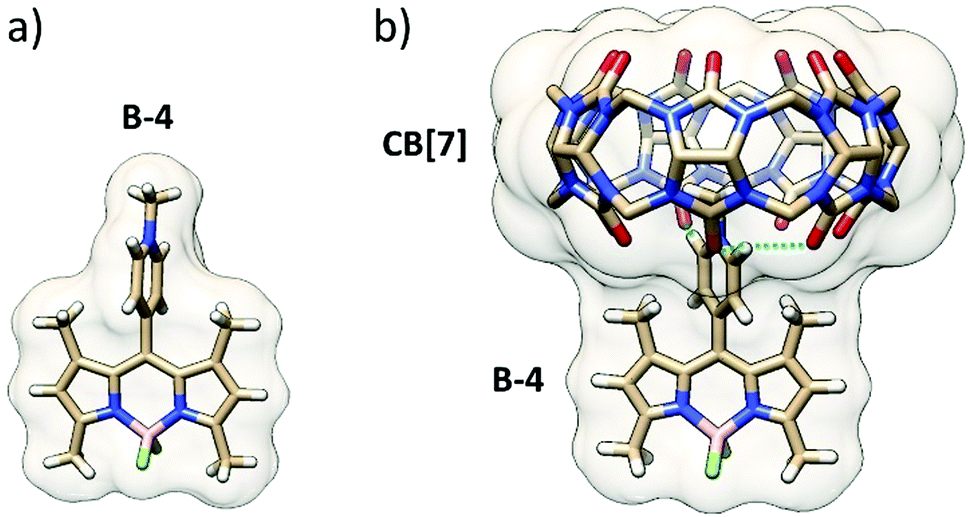 | ||
| Fig. 4 X-ray structure of B-4 and DFT minimized structure of the B-4·CB[7] complex (hydrogen bonds: dashed lines, hydrogens of CB[7] removed for clarity). | ||
Upon CB[7] addition, the absorption profiles of B-4, B-5, and B-6 remained nearly identical (Δλmax ∼ 3–4 nm) in line with the absence of CB[7]-induced deaggregation and thus suggesting that the BODIPYs are soluble at μM concentrations (Fig. S10, ESI†). In stark contrast, the corresponding fluorescence spectra showed dramatic changes after CB[7] addition (Fig. 1, left column and Fig. S11, ESI†). The fluorescence intensity of B-4 was enhanced by ∼10 fold and blue-shifted by 13 nm in the presence of 10 equivalents of CB[7]. Similarly, the fluorescence intensity of B-5 increased by ∼3 fold, with a slight blue shift by 8 nm in emission maximum with CB[7]. However, the addition of CB[7] to an aqueous solution of B-6 did not cause significant changes in guest emission intensity. We should note that, at 10−6 M concentration of B-4, B-5, and B-6, the corresponding fluorescence spectra showed only small changes after CB[7] addition probably due to weak binding interactions (Fig. S12, ESI†). Furthermore, the solid-state emission properties of B-4, B-5, and B-6 without and with CB[7] were also explored. Solid films obtained on glass slides by slow solvent evaporation of aqueous solutions of B-4, B-5, and B-6, without and with CB[7], were analyzed by solid state fluorescence spectroscopy. Very weak emissions were observed at 530 nm, blue-shifted compared solution-state emissions (Fig. 1, right column). The solid-state emission intensity of B-4 and B-5 was decreased compared to the emissions in solution due to aggregation-induced quenching combined with non-radiative pathways. However, CB[7] caused a significant increase in the solid-state emission of B-4 (∼9 fold that of free B-4) and a red shift of 36 nm. Nevertheless, the corresponding spectra for B-5 and B-6 only showed a small and negligible increase in emission intensity, respectively, at the same wavelength in the presence of CB[7]. Besides, CB[7] has been shown to increase fluorescence lifetimes and quantum yields of the guest molecules due to its low polar inner cavity. Fluorescence decay measurements of B-4, B-5, and B-6 in the absence and presence of CB[7] are shown in Fig. 2 and Table 1.
| Compound | λ abs (nm) | λ em (nm) | a 1 (%) | T 1 (ns) | a 2 (%) | T 2 (ns) | Φ F (%) |
|---|---|---|---|---|---|---|---|
| a Lifetime (a1/a2: mono-/bi-exponential fitting). b Fluorescence quantum yield. | |||||||
| B-4 | 505 | 592 | 100 | 0.18 | — | — | 0.6 |
| B-4·CB[7] | 505 | 579 | 25 | 1.15 | 75 | 6.21 | 6 |
| B-5 | 525 | 594 | 100 | 0.60 | — | — | 0.07 |
| B-5·CB[7] | 525 | 586 | 43 | 0.78 | 57 | 3.84 | 1 |
| B-6 | 545 | — | 100 | 0.09 | — | — | — |
| B-6·CB[7] | 545 | — | 100 | 0.17 | — | — | — |
Without CB[7], the fluorescence decay of B-4 and B-5 was mono-exponential, corresponding to fluorescence lifetimes of about 0.18 and 0.60 ns, respectively. However, in the presence of CB[7], the fluorescence decay rates decreased while showing multi-exponential behaviors with major contributions from a longer lifetime (τ2) component of 6.21 ns for B-4 and 3.84 ns for B-5. The presence of CB[7] has only a weak effect on the fluorescence decay of B-6, being mono-exponential in nature and corresponding to a fluorescence lifetime of ∼0.09–0.17 ns. As for the change in quantum yield, the fluorescence quantum yield (ΦF) measurements point to improved fluorescence characteristics with the addition of CB[7]. In the presence of CB[7], the fluorescence quantum yields of B-4 and B-5 has increased ∼10–13 fold compared to the absence of CB[7] (Table 1).
BODIPYs are considered as good photosensitizers for PDT. Singlet oxygen generation measurements for the three BODIPY dyes were then performed in the absence and in the presence of CB[7] (Fig. S13, ESI†). While B-4 did not show singlet oxygen generation, B-5 and B-6 efficiently produced singlet oxygen due to the heavy atom effect promoting singlet-to-triplet intersystem crossing (ISC). Surprisingly, CB[7] did not change these results with virtually identical responses with respect to each BODIPY alone. The observed fluorescence improvements without compromising the ISC processes make this supramolecular approach with CB[7] a promising strategy to develop smart agents combining biological imaging and photodynamic therapy. To clarify the mechanism by which CB[7] impacts the BODIPY emissive features, we next focused on the structural and theoretical studies.
:1 host:guest complexes for all compounds (Fig. S7, S8, S9, ESI†). We next used 1H NMR to get further insights into the kind of supramolecular complex keeping in mind that the BODIPY frame is likely too large to be engulfed in the host cavity. When a guest molecule is included in CB[7], the signals of the guest protons inside the cavity are shifted upfield (Δδ < 0) while the protons sitting next to the CB[7] carbonyl portals have their signals deshielded compared to those for the guest alone (Δδ > 0).27
To ensure full guest solubility at mM concentrations, we used a small percentage of CD3CN; this solvent is known to be only weakly competitive (Ka, ACN/CB[7] = 11 ± 1 M−1)51 but also a possible source of weaker complexations. 1H NMR titrations at 0.5 mM of guest and with increasing amounts of CB[7] in D2O/CD3CN (80:20 vol%) showed complexation-induced shifts (CIS) for methyl protons (Ha) of the meso pyridinium group of Δδ = −0.05, −0.06, and −0.07 ppm for B-4, B-5, and B-6, respectively (Fig. 3, Fig. S14, S15, ESI†). This suggests that the pyridinium group of each BODIPY is at least partly included in CB[7]. Small downfield shifts of pyridinium aromatic protons (Hb) support these results, presumably experiencing the deshielding effect of the nearby host carbonyl rim. Simultaneously, the singlets corresponding to pyrrolic hydrogen atoms (Hd) were weakly shielded, probably due to a redistribution of the electron density as signals of the methyl hydrogen atoms of the pyrroles are unchanged. Analysis of the fluorescence spectra of Fig. 1 suggested that the binding constants for equilibria involving the formation of a 1:1 complex could be of the order of 103 to 104 M−1. Non-linear curve fitting using data from NMR titrations52 afforded binding constants Ka, B-4/CB[7] = 22 ± 11 M−1, Ka, B-5/CB[7] = 67 ± 19 M−1, and Ka, B-6/CB[7] = 34 ± 16 M−1 (Fig. S14, S15, ESI†), which are rather weak but account for the competitive behavior of acetonitrile necessary to do the titrations. Indeed, even if this co-solvent is weakly competitive, 20 vol% represent a very large concentration. We hypothesize that binding constants in pure water should be one or two orders of magnitude higher.
To go further, BODIPY B-4 was crystallized, and its single-crystal X-ray diffraction structure was obtained. The structure showed that the pyridinium group is almost perpendicular to the BODIPY frame because of the restricted rotation imposed by the adjacent methyl groups (Fig. 4a).
While pyridiniums are largely accepted to be good guest fragments for CB[7], the steric hindrance at the anchor point on the BODIPY frame prevents deep inclusion in the CB[7] cavity. Indeed, the DFT minimized structure (Fig. 4b), in line with mass spectrometry and NMR shows a shallow inclusion by the methyl-pyridinium moiety, two methyl groups of the BODIPY frame coming close to one carbonyl portal. Three guest:host strong, likely charge-assisted, H-bonds are noticed: one singly engaged N+–C–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (1.90 Å) and a bifurcated N+–C–H⋯OC bond (2.38 and 2.47 Å). In this geometry, the pyridinium group is slightly rotated off the perpendicular position with respect to the BODIPY frame.
C bond (1.90 Å) and a bifurcated N+–C–H⋯OC bond (2.38 and 2.47 Å). In this geometry, the pyridinium group is slightly rotated off the perpendicular position with respect to the BODIPY frame.
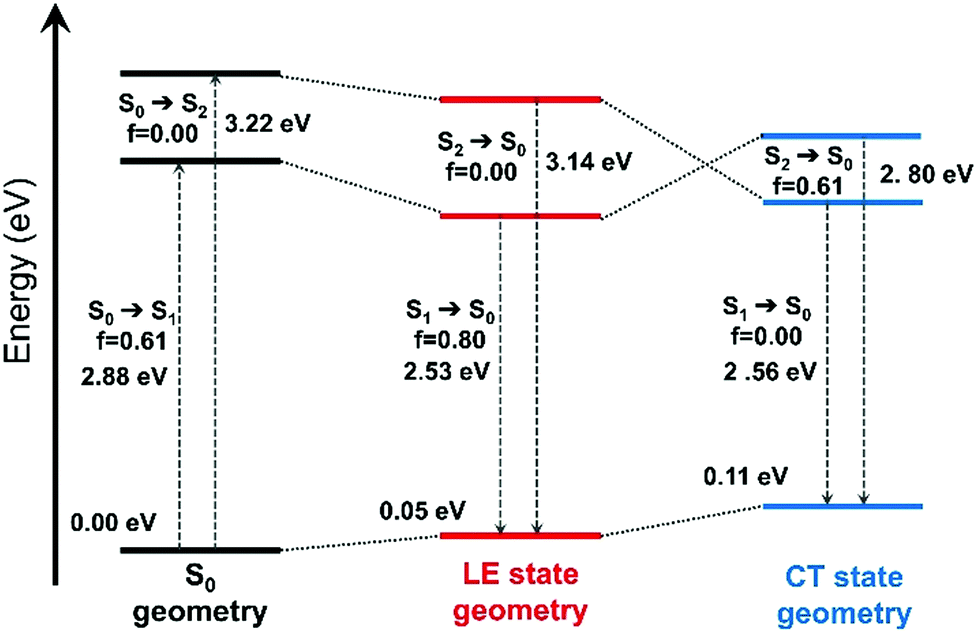 | ||
| Fig. 5 Evolution of the ground-state and excited-state energies, and of oscillator strengths with respect to different optimized geometries of the ground-state (S0) and the excited-states (LE and CT states) of B-4. The interaction type and MOs involved in these transitions are shown in Fig. S16 (ESI†). | ||
The effect of excited-state geometry relaxation on the transition energy of the CT state is even larger (0.66 eV). Overall, both the excited states exhibit similar total energies for their respective minimum. Besides, when the torsion angle (θ) between the BODIPY and the 4-aryl moieties was changed, pure CT and LE states showed significant mixing to a hybridized state (Fig. S17, ESI†) pointing to a possible LE→CT conversion path. These results suggest that the fluorescence quenching observed for B-4 most likely arises from non-radiative relaxation driven by this interplay between the dark CT and bright LE states.53 We also note that there is still some overlap between pyridinium and BODIPY-based orbitals for the particle wavefunction as shown in Fig. S17 (ESI†), which makes complete charge separation and quenching through photoinduced electron transfer (PET) mechanism unlikely for the investigated systems.
The impact of CB[7] on the excited-state properties of B-4 was also investigated (Fig. 6). For the B-4·CB[7] complex, both the LE and CT states originate from the same transitions as discussed before, without significant contributions from the CB[7]-based energy levels. When the distance between B-4 and CB[7] is large (6 Å), the energy difference between the CT and LE states is predicted to be comparable to the case of the isolated system (Fig. 6c and d, respectively). However, as the distance between B-4 and CB[7] decreases, the CT state blueshifts due to ion–dipole interactions between the guest pyridinium moiety and CB[7], while the energy of the LE state is only marginally affected. For the S0 geometry, the energy difference between the LE and CT states increases to 0.65 eV compared to 0.34 eV for B-4 alone (Fig. S18, ESI†). For the CT state geometry, CB[7] complexation swaps energy levels, the LE state becomes the S1 state for distances smaller than 5 Å (Fig. 5d). Beyond the impact on excited-state energetics, CB[7] complexation is also expected to restrict the guest rotational and vibrational degrees of freedom, thereby limiting the LE → CT conversion channel. Consequently, radiative decay from the LE state becomes favorable again for the B-4·CB[7] complex resulting in a significant increase in emission intensity.
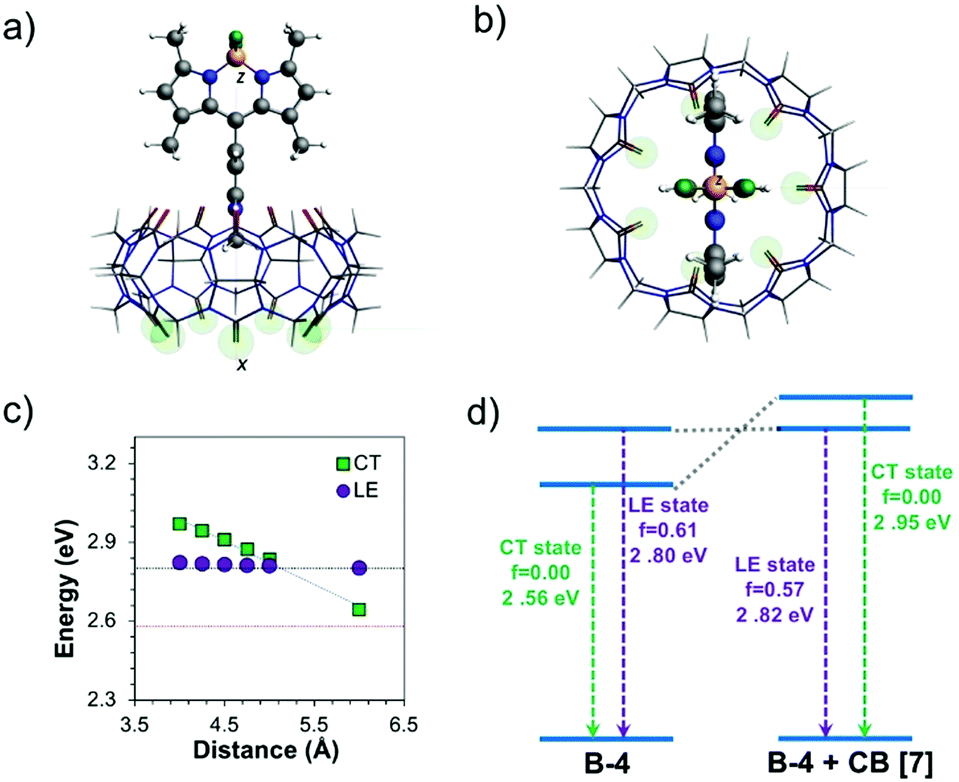 | ||
| Fig. 6 Different views (a, b) of the minimized B-4·CB[7] complex. The B-4–CB[7] distance is plotted in (c) against the energy of the CT and LE states in the optimized CT geometry of B-4. The red and blue dotted lines represent the energies of the CT and LE states, respectively, for isolated B-4 (the distance between CB[7] and B-4 is from the guest carbon atom of the methyl-pyridinium and from the barycenter of the host oxygen atoms; green circles). Energy diagram in (d) illustrates how the energy of the excited-state is impacted by CB[7] complexation and corresponding to a host–guest distance of 4.25 Å (lowest energy structure for the B-4·CB[7] complex). | ||
Similarly, the excited-state properties were also examined for B-5 and B-6 (Fig. S19, ESI†). For both BODIPYs, the LE state showed a redshift (∼0.1 eV and ∼0.2 eV respectively) in the ground-state geometry compared to that observed for B-4, in line with the experimental results. In comparison, the CT state showed a blueshift in the range of ∼0.25–0.30 eV in the same geometry. Due to LE-state stabilization and CT state destabilization when iodine atoms are introduced, the energy gap between the CT and LE states in both S0 and LE-state geometries increased by ∼ 0.3 eV for B-5 compared to B-4. More importantly, the energetic order of these states does not change for the CT-state geometry. While iodine substitution is expected to increase the fluorescence quenching of B-5 owing to the heavy-atom effect, the electronic structure and excited-states energetics could reduce non-radiative decay from the CT state. In the case of B-6, one would expect a further increase in the emission intensity based solely on excited state energetics as the energy gap between the LE and CT states is even larger. However, the balance becomes unfavorable due to the further iodination of B-6 increasing the ISC rate and resulting in efficient fluorescence quenching in line with previous reports.54
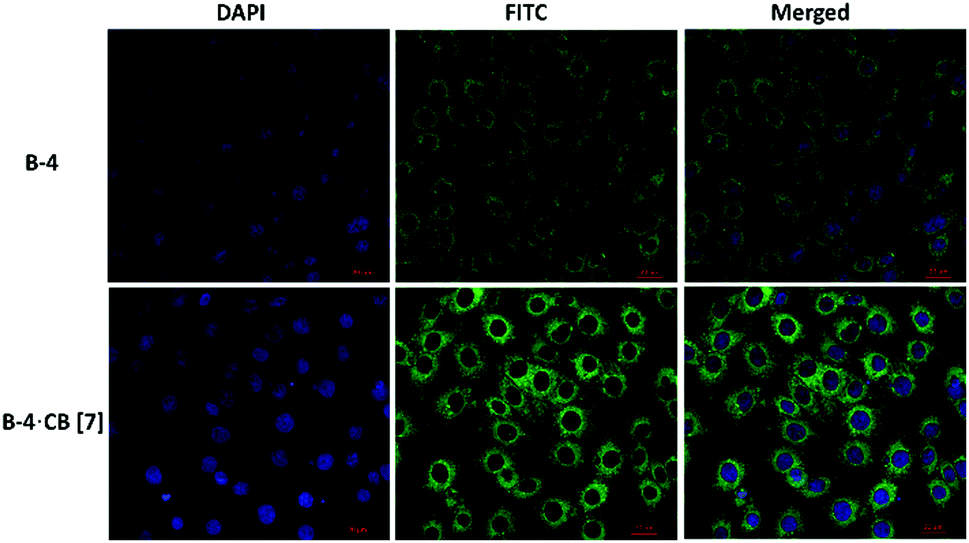 | ||
| Fig. 7 Confocal fluorescence microscopy images of U87 cells after incubation with B-4 without (top line), and with (bottom line) CB[7] for 4 hours. Cell nuclei were stained with DAPI after PFA fixation (DAPI, λEx/Em = 405/450 nm; FITC, λEx/Em = 488/520 nm). | ||
When U87 cells were incubated with B-4, B-5, or B-6 alone, only a weak fluorescence was observed. However, the fluorescence increased in the presence of CB[7] and especially when B-4 was used (Fig. 6); images show clear improvements in the green emission localized in the intracellular regions. These results indicate that CB[7] complexation is retained in vitro with a preserved inhibition of nonradiative decay pathways for B-4. More investigations are necessary to determine what are the mechanisms of cell uptake and whether the host and the guest are internalized together or separately. However, these experiments exceed the scope of this study. Nevertheless these results illustrate well the potential of CB[7] to improve the fluorescence properties of a dye, even by external complexation.
Conclusions
In summary, we describe the synthesis of three water soluble cationic BODIPY derivatives (B-4, B-5, and B-6) and the effect of CB[7] complexation on their photophysical properties. UV-vis absorption and fluorescence measurements have shown that these BODIPYs do not aggregate in water in the concentration range studied and are weakly emissive mainly due to charge transfer for B-4 and heavy atom effect for B-5 and B-6. However, the addition of CB[7] improved guest fluorescence by ∼10 fold for B-4 and by ∼3 fold for B-5, while B-6 remained essentially non-emissive in the solution and in the solid state. Fluorescence lifetimes were observed to be increased from 0.18 to 6.21 ns for B-4 and from 0.60 to 3.84 ns for B-5 after CB[7] addition. The origin of these improved photophysical properties upon CB[7] binding was studied by (TD)DFT and assigned to (i) the formation of external BODIPY·CB[7] complexes increasing the energy of the CT state for B-4 and B-5 as a result of ion–dipole interactions between the guest pyridinium and the carbonyl groups of the host favoring radiative decay from the locally-excited (LE) π → π* transition state of BODIPY and (ii) the restrictions of rotational and vibrational motions upon host binding. With a preserved singlet oxygen generation, CB[7] complexation was shown to selectively inhibit CT but not ISC of BODIPYs in solution, in the solid state, and in vitro. These improvements allowed to use the BODIPY·CB[7] complexes for confocal fluorescence imaging of U87 cells. While B-4, B-5, and B-6 alone only showed marginal fluorescence in vitro, clear emission from the intracellular regions was evidenced after co-incubation with B-4 and CB[7], supporting the possible use of external cucurbituril complexes for fluorescence imaging. Pleasingly, singlet oxygen generation was shown to remain unaffected by CB[7] complexation for B-5 and B-6 supporting possible use for photodynamic therapy (PDT). These results suggest that new BODIPY·CB[n] complexes could be designed for combined applications like simultaneous fluorescence imaging and PDT in a theragnostic context.Conflicts of interest
There are no conflicts to declare.Acknowledgements
CNRS and Aix Marseille University are acknowledged for providing continuous support.Notes and references
- H. Klfout, A. Stewart, M. Elkhalifa and H. He, ACS Appl. Mater. Interfaces, 2017, 9, 39873–39889 CrossRef CAS.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC.
- X. Cui, C. Zhang, K. Xu and J. Zhao, J. Mater. Chem. C, 2015, 3, 8735–8759 RSC.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
- C. L. Liu, Y. Chen, D. P. Shelar, C. Li, G. Cheng and W. F. Fu, J. Mater. Chem. C, 2014, 2, 5471–5478 RSC.
- E. Ozcan, M. Ozdemir, D. Ho, Y. Zorlu, R. Ozdemir, C. Kim, H. Usta and B. Cosut, ChemPlusChem, 2019, 84, 1423–1431 CrossRef CAS PubMed.
- D. Pfeifer, A. Russegger, I. Klimant and S. M. Borisov, Sens. Actuators, B, 2020, 304, 127312 CrossRef CAS.
- J. M. Franke, B. K. Raliski, S. C. Boggess, D. V. Natesan, E. T. Koretsky, P. Zhang, R. U. Kulkarni, P. E. Deal and E. W. Miller, J. Am. Chem. Soc., 2019, 141, 12824–12831 CrossRef CAS PubMed.
- P. Kaur and K. Singh, J. Mater. Chem. C, 2019, 7, 11361–11405 RSC.
- L. Wang, L. Li and D. Cao, Sens. Actuators, B, 2017, 239, 1307–1317 CrossRef CAS.
- S. Kim, J. Bouffard and Y. Kim, Chem. – Eur. J., 2015, 21, 17459–17465 CrossRef CAS.
- H. Lu, Q. Wang, L. Gai, Z. Li, Y. Deng, X. Xiao, G. Lai and Z. Shen, Chem. – Eur. J., 2012, 18, 7852–7861 CrossRef CAS.
- D. Tian, F. Qi, H. Ma, X. Wang, Y. Pan, R. Chen, Z. Shen, Z. Liu, L. Huang and W. Huang, Nat. Commun., 2018, 9, 1–9 CrossRef CAS.
- S. Choi, J. Bouffard and Y. Kim, Chem. Sci., 2014, 5, 751–755 RSC.
- S. Xu, W. Wu, X. Cai, C. J. Zhang, Y. Yuan, J. Liang, G. Feng, P. Manghnani and B. Liu, Chem. Commun., 2017, 53, 8727–8730 RSC.
- Z. Liu, Z. Jiang, M. Yan and X. Wang, Front. Chem., 2019, 7, 1–16 CrossRef.
- E. Özcan, B. Dedeoglu, Y. Chumakov, A. G. Gürek, Y. Zorlu, B. Çoşut and M. Menaf Ayhan, Chem. – Eur. J., 2021, 27, 1603–1608 CrossRef.
- C. Chen, X. Ni, H. W. Tian, Q. Liu, D. S. Guo and D. Ding, Angew. Chem., Int. Ed., 2020, 59, 10008–10012 CrossRef CAS PubMed.
- X. Y. Lou, N. Song and Y. W. Yang, Chem. – Eur. J., 2019, 25, 11975–11982 CrossRef CAS.
- X. Yao, T. Li, J. Wang, X. Ma and H. Tian, Adv. Opt. Mater., 2016, 4, 1322–1349 CrossRef CAS.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS.
- F. Schibilla, L. Stegemann, C. A. Strassert, F. Rizzo and B. J. Ravoo, Photochem. Photobiol. Sci., 2016, 15, 235–243 CrossRef CAS.
- Y. L. Loukas, Analyst, 1997, 122, 377–381 RSC.
- J. W. Lee, S. Samal, N. Selvapalam, H. J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS.
- O. Reany, A. Li, M. Yefet, M. K. Gilson and E. Keinan, J. Am. Chem. Soc., 2017, 139, 8138–8145 CrossRef CAS.
- V. D. Uzunova, C. Cullinane, K. Brix, W. M. Nau and A. I. Day, Org. Biomol. Chem., 2010, 8, 2037–2042 RSC.
- D. Das, K. I. Assaf and W. M. Nau, Front. Chem., 2019, 7, 1–23 CrossRef.
- M. Sayed and H. Pal, J. Mater. Chem. C, 2016, 4, 2685–2706 RSC.
- H. Yin, F. Dumur, Y. Niu, M. M. Ayhan, O. Grauby, W. Liu, C. Wang, D. Siri, R. Rosas, A. Tonetto, D. Gigmes, R. Wang, D. Bardelang and O. Ouari, ACS Appl. Mater. Interfaces, 2017, 9, 33220–33228 CrossRef CAS.
- S. R. Wang, J. Q. Wang, G. H. Xu, L. Wei, B. S. Fu, L. Y. Wu, Y. Y. Song, X. R. Yang, C. Li, S. M. Liu and X. Zhou, Adv. Sci., 2018, 5, 1–9 CAS.
- C. Hu, L. Grimm, A. Prabodh, A. Baksi, A. Siennicka, P. A. Levkin, M. M. Kappes and F. Biedermann, Chem. Sci., 2020, 11, 11142–11153 RSC.
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed.
- A. Palma, M. Artelsmair, G. Wu, X. Lu, S. J. Barrow, N. Uddin, E. Rosta, E. Masson and O. A. Scherman, Angew. Chem., Int. Ed., 2017, 56, 15688–15692 CrossRef CAS PubMed.
- Z. Zeng, X. Zhang, G. Luo, Y. Meng, L. Zhang, W. Zhao, Z. Tao and Q. Zhang, J. Mater. Chem. C, 2021, 9, 6160–6165 RSC.
- X. Zhang, X. Xu, S. Li, L. H. Wang, J. Zhang and R. Wang, Sci. Rep., 2018, 8, 1–7 Search PubMed.
- W. M. Nau and J. Mohanty, Int. J. Photoenergy, 2005, 7, 133–141 CrossRef CAS.
- S. S. Thakare, G. Chakraborty, P. Krishnakumar, A. K. Ray, D. K. Maity, H. Pal and N. Sekar, J. Phys. Chem. B, 2016, 120, 11266–11278 CrossRef CAS.
- B. Yuan, H. Wu, H. Wang, B. Tang, J. F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2021, 60, 706–710 CrossRef CAS PubMed.
- J. F. Yin, Y. Hu, D. G. Wang, L. Yang, Z. Jin, Y. Zhang and G. C. Kuang, ACS Macro Lett., 2017, 6, 139–143 CrossRef CAS.
- M. A. Alnajjar, J. Bartelmeß, R. Hein, P. Ashokkumar, M. Nilam, W. M. Nau, K. Rurack and A. Hennig, Beilstein J. Org. Chem., 2018, 14, 1961–1971 CrossRef CAS PubMed.
- M. Gupta, K. Parvathi, S. Mula, D. K. Maity and A. K. Ray, Photochem. Photobiol. Sci., 2017, 16, 499–506 CrossRef CAS.
- O. Buyukcakir, F. T. Yasar, O. A. Bozdemir, B. Icli and E. U. Akkaya, Org. Lett., 2013, 15, 1012–1015 CrossRef CAS PubMed.
- A. Singh, W. T. Yip and R. L. Halterman, Org. Lett., 2012, 14, 4046–4049 CrossRef CAS.
- G. Chakraborty, M. K. Choudhary, M. Sundararajan, A. K. Ray, S. Mula and H. Pal, J. Phys. Chem. B, 2021, 125, 7946–7957 CrossRef CAS PubMed.
- M. M. Ayhan, E. Özcan, B. Dedeoglu, Y. Chumakov, Y. Zorlu and B. Coşut, CrystEngComm, 2021, 23, 268–272 RSC.
- J. Karolin, L. B. A. Johansson, L. Strandberg and T. Ny, J. Am. Chem. Soc., 1994, 116, 7801–7806 CrossRef CAS.
- A. Gorman, J. Killoran, C. O’Shea, T. Kenna, W. M. Gallagher and D. F. O’Shea, J. Am. Chem. Soc., 2004, 126, 10619–10631 CrossRef CAS PubMed.
- X. Miao, W. Hu, T. He, H. Tao, Q. Wang, R. Chen, L. Jin, H. Zhao, X. Lu, Q. Fan and W. Huang, Chem. Sci., 2019, 10, 3096–3102 RSC.
- I. W. Wyman and D. H. MacArtney, Org. Biomol. Chem., 2008, 6, 1796–1801 RSC.
- D. Bardelang, J. L. Clément, J. P. Finet, H. Karoui and P. Tordo, J. Phys. Chem. B, 2004, 108, 8054–8061 CrossRef CAS.
- Z. Lin, A. W. Kohn and T. Van Voorhis, J. Phys. Chem. C, 2020, 124, 3925–3938 CrossRef CAS.
- R. P. Sabatini, T. M. McCormick, T. Lazarides, K. C. Wilson, R. Eisenberg and D. W. McCamant, J. Phys. Chem. Lett., 2011, 2, 223–227 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2051939. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ma00931a |
| This journal is © The Royal Society of Chemistry 2022 |