
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of Na+ and K+ on the cucurbituril-mediated hydrolysis of a phenyl acetate†
Nazar
Rad
 * and
Volodymyr
Sashuk
*
* and
Volodymyr
Sashuk
*
Institute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: nrad@ichf.edu.pl
First published on 1st April 2022
Abstract
The environment around the active site affects the catalytic activity of enzymes. Studying the cucurbit[7]uril-promoted acid hydrolysis of a cationic phenyl acetate derivative, we found that the hydrophobic cavity of the macrocycle screens the reaction centre from the positively charged neighbouring group. Moreover, the chelation of alkali metal cations with the cucurbit[7]uril portal and acetyl group of the substrate reduces the hydrolysis rate of the encapsulated ester in an aqueous solution. This type of inhibition corresponds to a rare uncompetitive model in contrast to the more common competitive model that relies on substrate displacement.
Sodium and potassium are the most abundant cations in living cells. Due to the ion-specific proteins embedded into the membrane, the concentration of potassium is much higher in the cell than in seawater (140 mM vs. 5 mM), while the sodium concentration is lower (30 mM vs. 140 mM). The cell utilizes this imbalance to regulate cell transport, volume maintenance and signal transduction. Sodium and potassium also play a specific role in the activation of some enzymes. These cations attract the substrate or directly affect the catalytic process as a cofactor, and optimize the conformation of the enzyme.1,2 Alkali metal cations can also inhibit enzymatic processes.3 When the enzyme is highly negatively charged (such as acetylcholinesterase), it is difficult to distinguish the ionic strength effect from the specific binding of the cation to the enzyme.4 Therefore, the mechanism of cation action is not always clear.
Alkali metal cations can regulate the catalytic activity of a number of artificial enzymes.5 These enzyme mimics contain a crown-ether moiety responsible for cation coordination. The attachment of the cation induces conformational changes that affect catalytic activity.6–8 Notably, the developed artificial enzymes operate in non-aqueous media because of the low solubility of crown-ether in water. The ion-specific effect of the cation on the enzyme functionality in an aqueous medium is attenuated by solvation.9,10 Here we demonstrate for the first time that the coordination of alkali metal cations (Li+, Na+, K+ and Cs+) can affect the activity of an artificial enzyme even in water.
In our study, we used a water-soluble cucurbit[7]uril macrocycle (CB7). Like natural enzymes, cucurbiturils create stable binary complexes with certain organic guests and accelerate their chemical transformation.11 The portals of these macrocycles also attract metal cations.12 The coordination of a cation can either prevent the substrate encapsulation13 or lead to the formation of a ternary complex14 altering the position of the encapsulated guest inside the macrocyclic cavity (Na+ and Ag+).18,19 Transition metal cations coordinated to cucurbiturils promote desilylation (Ag+),15 and increase the chemoselectivity of deazotation (Ag+)16 and the enantioselectivity of Diels–Alder reaction (Cu2+).17
The catalytic activity of neat cucurbiturils can be rationalized by the excess electronic density on the two portals that accumulate hydroxonium ions around the reaction centre.18 These macrocycles promote the hydrolytic cleavage of amides,19 benzoyl chlorides,20 esters,21 ethers,18 triazenes,22 oximes,19 and Schiff bases,23 as well as the formation of hydrazones.24
As a model reaction we choose the acid hydrolysis of esters (Scheme 1). This reaction is slow enough for observation at low pH. The rate of hydrolysis depends on the concentration of the tetrahedral intermediate TI, which increases with increasing concentration of ester conjugated acid PhAc·H.25,26 We expected CB7 to promote hydrolysis by enhancing the basicity of the encapsulated substrate.27,28 Although ester hydrolysis is a reversible process, the excess of water molecules almost completely shifts the equilibrium towards product formation. The employed acetate derivative (PhAc) is reminiscent of the neurotransmitter acetylcholine, the concentration of which at the synapse is controlled by the hydrolytic enzyme acetylcholinesterase. As in the case of acetylcholine, a positively charged ammonium group was expected to facilitate complexation.
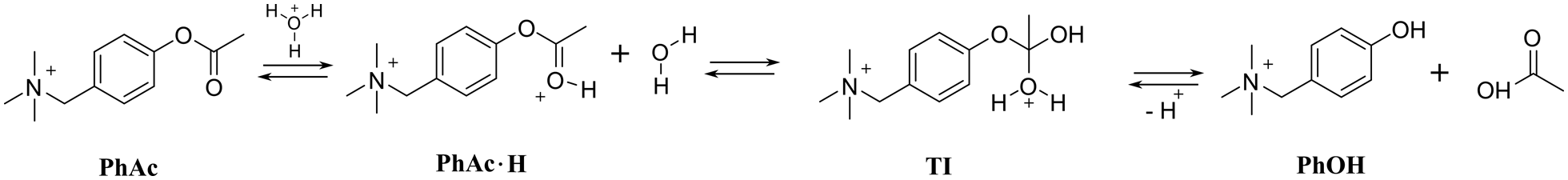 | ||
| Scheme 1 Acid hydrolysis of PhAc. | ||
Initial experiments were performed in an aqueous solution of acid in the absence of any metal cations to avoid the competition of cations with the substrate. The binding constant of PhAc with CB7 determined by 1H NMR titration is equal to (1.5 ± 0.42) × 105 M−1. The addition of CB7 to PhAc shifts all proton signals upfield (ESI† Fig. S2). The most shifted signal belongs to aromatic proton 3c in the meta-position to the acetate group. Significantly shifted are also the proton resonances of ammonium group 5c. The less shifted signal corresponds to the proton of acetate group 1c. Based on this, we can conclude that the phenyl ring and ammonium group are buried deep inside the cavity, and the acetate group is localised close to one of the CB7 rims (Fig. 1a).
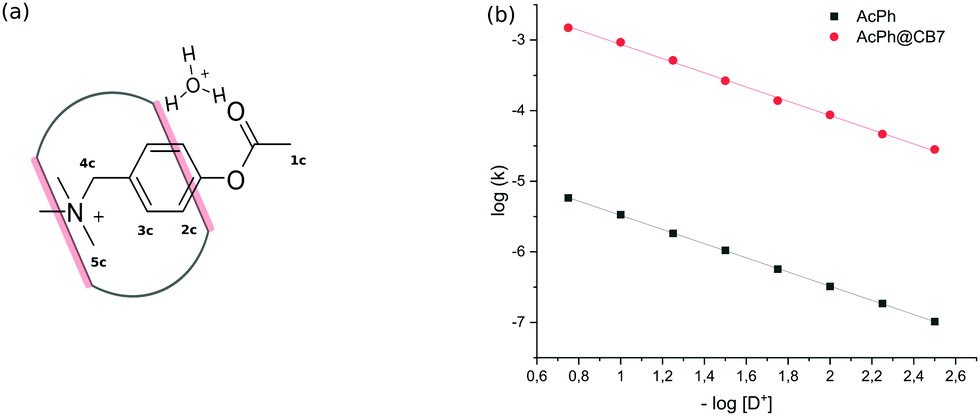 | ||
| Fig. 1 (a) Schematic depiction of transition state stabilization by CB7 portals. (b) pD-profiles of acid hydrolysis of PhAc (1.5 mM) and PhAc@CB7 (1.5 mM). | ||
In the salt-free solution, the hydrolysis rate of PhAckf depends linearly on the concentration of H3O+ ions in the range of pH between 0.75 and 2.5 (Fig. 1b). Since both PhAc and H3O+ involved in the formation of the transition state are positively charged species, kf depends also on the ionic strength of the solution (eqn (1)). A similar positive salt effect has already been noted in the hydrolysis of acetylcholine in acid solution.29
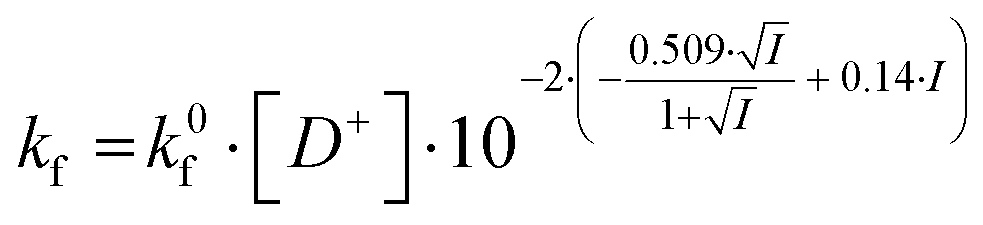 | (1) |
Adding 1.1 eq of CB7 to PhAc accelerates the reaction by more than two orders of magnitude. In this case, the rate of hydrolysis kb increases linearly (Fig. 1b) as pH decreases following eqn (2).
| kb = k0b·[D+] | (2) |
The slope of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kbversus log[D+] is equal to unity (Fig. S6b, ESI†). Thus, the rate of CB7-mediated hydrolysis is insensitive to the ionic strength of the solution. The lack of the primary salt effect for encapsulated PhAc is explained by the presence of the hydrophobic cavity that screens the reaction centre (acetate group) from the charged anchor (ammonium group).
kbversus log[D+] is equal to unity (Fig. S6b, ESI†). Thus, the rate of CB7-mediated hydrolysis is insensitive to the ionic strength of the solution. The lack of the primary salt effect for encapsulated PhAc is explained by the presence of the hydrophobic cavity that screens the reaction centre (acetate group) from the charged anchor (ammonium group).
The efficiency of the macrocycle as a catalyst can be assessed by the acceleration factor α. The acceleration factor, calculated as the ratio of the hydrolysis rate constants of the encapsulated and free PhAc (eqn (3)), decreases as the ionic strength of the solution increases. That is, if the substrate is charged, the ionic-strength-independent acceleration factor α0 should be given instead. For CB7-promoted PhAc acid hydrolysis, α0 is equal to 263 ± 11.
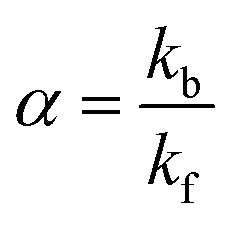 | (3) |
Assuming that the acceleration effect of CB7 is due to an increase in substrate basicity, the concentration of PhAc@CB7·H should be 263 times higher than the concentration of PhAc·H at the same pH. Since almost all substrate (96%) is encapsulated by CB7 and the concentration of protonated ester is very low (pKa is equal about −7), the concentrations of free (PhAc) and encapsulated substrate (PhAc@CB7) should be practically the same under reaction conditions. Thus, the found α0 should correspond to the ratio between the basicities of the free and encapsulated substrate (eqn (4)).
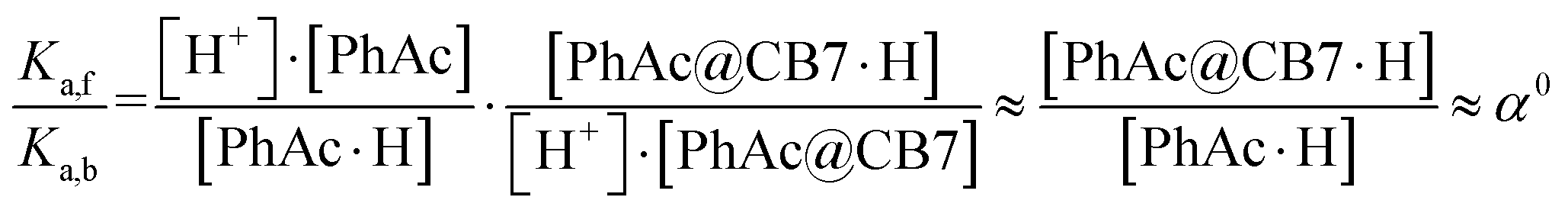 | (4) |
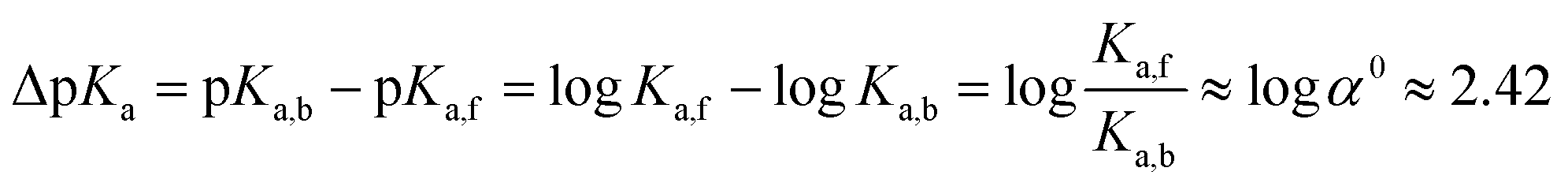 | (5) |
The logarithm of α0 is equal to 2.42 (eqn (5)) which is in the range of typical CB7-induced pKa shifts (2–3).27,28 This confirms our assumption that the stabilization of the ester conjugated acid by CB7 is the main reason for accelerated hydrolysis.
In the next step, we studied the effect of alkali metal ions. The addition of sodium chloride sped up the hydrolysis of free PhAc due to the increase of ionic strength and reduced the activity of CB7. The obtained kinetic data do not agree with competitive inhibition, where the sodium cation competes with the substrate for the macrocycle (Fig. 2b).
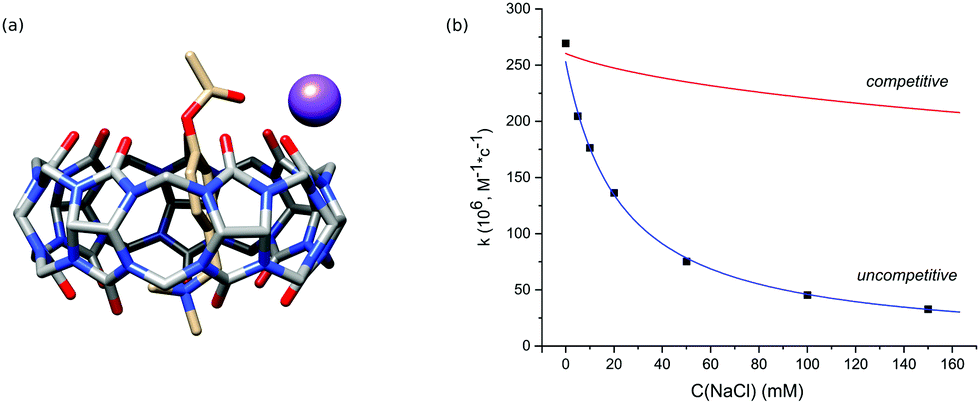 | ||
| Fig. 2 (a) PM6-optimized structure of the PhAc@CB7·Na ternary complex. The sodium ion coordinates with oxygen atoms of two ureido groups of CB7 and the acetyl group of PhAc. (b) The sodium cation effect on the hydrolysis rate constant of PhAc@CB7. Experimental data fit the model of uncompetitive inhibition. | ||
Moreover, the competitive inhibition scenario does not explain the shifts in the 1H NMR spectrum. Upon adding salt, the signal of the 2c proton moves upfield, which can only be accounted for by the translocation of the phenyl ring deeper into the CB7 cavity (Fig. 3b).
 | ||
| Fig. 3 (a) NMR titration experiments of free PhAc (1.5 mM) with CB7 (0–1.65 mM) and (b) the PhAc@CB7 complex (2.0–1.4 mM) with NaCl (0–460 mM). The concentration of CB7 and NaCl increases from the bottom up. The signals of two aromatic hydrogen atoms 2c and 3c coalesce at the concentration of NaCl equal to 10 mM. | ||
To elucidate the source of inhibition, we recorded NMR spectra in the presence of alkali metal halides. The analysis has shown that the shielding of 2c proton signals depends strongly on the nature of the cation (ESI† Fig. S7) and does not change when varying the anion (ESI† Fig. S8). This observation is supported by acceleration factors αCB7, which differ markedly for four cations, and are of practically the same value for four anions (Table 1).
| Salts | k f,M × 106, M−1 s−1 | k b,M × 106, M−1 s−1 | k f,M/kf,0 | k b,M/kb,0 | α CB7 |
|---|---|---|---|---|---|
| k f,0 and kb,0 correspond to the PhAc and PhAc@CB7 hydrolysis rate constants under salt-free conditions.a Due to the buffering effect of NaF, the pD of the reaction mixture was higher. | |||||
| salt-free | 1.3 | 269.4 | 1.0 | 1.0 | 207 |
| Cation effect | |||||
| LiCl | 1.28 | 268.0 | 1.0 | 1.0 | 210 |
| NaCl | 1.25 | 180.9 | 1.0 | 0.7 | 145 |
| KCl | 1.27 | 183.7 | 1.0 | 0.7 | 145 |
| CsCl | 1.24 | 229.9 | 1.0 | 0.9 | 185 |
| Anion effect | |||||
| NaFa | 0.83 | 117.1 | 0.6 | 0.4 | 141 |
| NaCl | 1.25 | 180.9 | 1.0 | 0.7 | 145 |
| NaBr | 1.33 | 179.6 | 1.0 | 0.7 | 142 |
| NaI | 1.35 | 197.8 | 1.0 | 0.7 | 147 |
Thus, we consider the model of uncompetitive inhibition of CB7 by the cation. According to this model, the cation binds to the CB7 portal next to the reactive centre to afford a PhAc@CB7·Na ternary complex (Fig. 2a). The positively charged alkali cation decreases the concentration of conjugated acid of ester PhAc@CB7·H to slow down the acid hydrolysis. The formation of the ternary complex is supported by the mass analysis of the isolated PhAc@CB7 complex revealing a monosodium doubly charged molecular ion (ESI† Fig. S21).
The kinetic data of PhAc@CB7 hydrolysis in the presence of different amounts of sodium chloride (Fig. 2b) were employed to estimate the PhAc@CB7 affinity for the sodium cation according to the model of uncompetitive inhibition. The obtained binding constant is almost identical to the constant determined by NMR titration within the statistical error (Table 2). Thus, the deactivation of CB7 correlates with the cation-induced dislocation of the macrocycle toward the ester group.
| Cation | PhAc@CB7 kinetics data | PhAc@CB7 NMR shifts | PhOH@CB7 NMR shifts |
|---|---|---|---|
| Three binding constants for the cation were determined in one experiment. The numbers in parentheses show the relative mean deviation of the calculated data from the experimental ones. | |||
| Li+ | 1.8 (0.7%) | 1.1 (6.8%) | 0.9 (4.5%) |
| Na+ | 46 (2.0%) | 46 (4.8%) | 19 (3.6%) |
| K+ | 42 (2.1%) | 43 (6.7%) | 30 (8.8%) |
| Cs+ | 12 (2.3%) | 11 (4.8%) | 22 (11.5%) |
The binding constants calculated for other cations also yield very close values for both methods. The cation affinity for the PhAc@CB7 complex (Table 2) decreases in the same order as the inhibition efficiency of the cations: Na+ > K+ > Cs+ > Li+ (Fig. 4, ESI† Table S3).
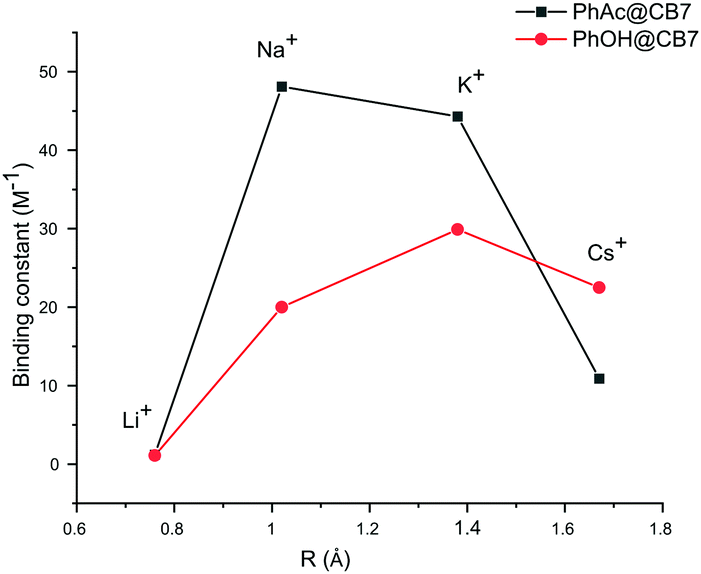 | ||
| Fig. 4 Plots of binding constants vs. cation radius for substrate PhAc@CB7 and product PhOH@CB7 complexes. | ||
The PhAc@CB7 complex binds sodium two times stronger than pure CB7 (K = 21 ± 2 M−1).30 This indicates that the organic guest reinforces the binding. The replacement of substrate PhAc by the product PhOH changes the affinity of the CB7-guest complex for cations. As with the substrate, the NMR shifts of the product PhOH@CB7 depend on the concentration of cations (ESI† Fig S11, S14, S16 and S18). The determined binding constants of PhOH@CB7 with cations decrease in the order K+ > Cs+ > Na+ > Li+ (Fig. 4). The similarity between the sodium affinity to free CB7 and PhOH@CB7 shows that, in contrast to the acetyl group, the hydroxyl group is not involved in the coordination of sodium cations. Thus, both cucurbit[7]uril and the substrate are involved in cation coordination.
To summarise, we scrutinised the effect of ionic strength on ester hydrolysis mediated by cucurbit[7]uril. In the absence of inorganic salts, the macrocycle screens the reaction centre from the cationic anchor making the substrate and the macrocycle insensitive to the ionic strength of the solution. However, after the addition of salts, the catalytic activity of the macrocycle is suppressed. The reason for this is the formation of a ternary complex with alkali metal cations that is less susceptible to hydronium ion attack. This type of inhibition can be described by the uncompetitive model in which the substrate remains bound to the macrocycle instead of being displaced by the inhibitor. This work shows that Na and K cations abundant in aqueous solutions can specifically modulate the operation of artificial enzymes. Moreover, the cation selectivity observed during the formation of ternary complexes can be the basis for the development of new cation receptors.
NR would like to thank the National Science Centre of Poland (grant MINIATURA 3 no 2019/03/X/ST4/01017) for funding the research and PLGrid Infrastructure for supporting the QM calculations.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. J. Page and E. Di Cera, Physiol. Rev., 2006, 86, 1049–1092 CrossRef CAS PubMed.
- M. Vasak and J. Schnabl, Met. Ions Life Sci., 2016, 16, 259–290 CAS.
- H. Zhao, J. Mol. Catal. B: Enzym., 2005, 37, 16–25 CrossRef CAS.
- V. Tougu and T. Kesvatera, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1996, 1298, 12–30 CrossRef.
- C. Yoo, H. M. Dodge and A. J. M. Miller, Chem. Commun., 2019, 55, 5047–5059 RSC.
- T. Tozawa, S. Tokita and Y. Kubo, Tetrahedron Lett., 2002, 43, 3455–3457 CrossRef CAS.
- G. H. Ouyang, Y. M. He, Y. Li, J. F. Xiang and Q. H. Fan, Angew. Chem., Int. Ed., 2015, 54, 4334–4337 CrossRef CAS PubMed.
- Y. J. Lee, K. S. Liu, C. C. Lai, Y. H. Liu, S. M. Peng, R. P. Cheng and S. H. Chiu, Chem. – Eur. J., 2017, 23, 9756–9760 CrossRef CAS PubMed.
- L. Escobar and P. Ballester, Chem. Rev., 2021, 121, 2445–2514 CrossRef CAS PubMed.
- P. S. Cremer, A. H. Flood, B. C. Gibb and D. L. Mobley, Nat. Chem., 2018, 10, 8–16 CrossRef CAS PubMed.
- B. H. Tang, J. T. Zhao, J. F. Xu and X. Zhang, Chem. – Eur. J., 2020, 26, 15446–15460 CrossRef CAS PubMed.
- S. Zhang, L. Grimm, Z. Miskolczy, L. Biczok, F. Biedermann and W. M. Nau, Chem. Commun., 2019, 55, 14131–14134 RSC.
- C. Marquez, R. R. Hudgins and W. M. Nau, J. Am. Chem. Soc., 2004, 126, 5806–5816 CrossRef CAS PubMed.
- Z. Miskolczy, M. Megyesi, L. Biczok, A. Prabodh and F. Biedermann, Chem. – Eur. J., 2020, 26, 7433–7441 CrossRef CAS PubMed.
- X. Y. Lu and E. Masson, Org. Lett., 2010, 12, 2310–2313 CrossRef CAS PubMed.
- A. L. Koner, C. Marquez, M. H. Dickman and W. M. Nau, Angew. Chem., Int. Ed., 2011, 50, 545–548 CrossRef CAS PubMed.
- L. F. Zheng, S. Sonzini, M. Ambarwati, E. Rosta, O. A. Scherman and A. Herrmann, Angew. Chem., Int. Ed., 2015, 54, 13007–13011 CrossRef CAS PubMed.
- L. Scorsin, J. A. Roehrs, R. R. Campedelli, G. F. Caramori, A. O. Ortolan, R. L. T. Parreira, H. D. Fiedler, A. Acuna, L. Garcia-Rio and F. Nome, ACS Catal., 2018, 8, 12067–12079 CrossRef CAS.
- C. Klock, R. N. Dsouza and W. M. Nau, Org. Lett., 2009, 11, 2595–2598 CrossRef PubMed.
- N. Basilio, L. Garcia-Rio, J. A. Moreira and M. Pessego, J. Org. Chem., 2010, 75, 848–855 CrossRef CAS PubMed.
- A. Fierro, L. Garcia-Rio, S. Arancibia-Opazo, J. J. Alcazar, J. G. Santos and M. E. Aliaga, J. Org. Chem., 2021, 86, 2023–2027 CrossRef CAS PubMed.
- V. Sashuk, H. Butkiewicz, M. Fialkowski and O. Danylyuk, Chem. Commun., 2016, 52, 4191–4194 RSC.
- J. J. Alcazar, N. Geue, V. Valladares, A. Canete, E. G. Perez, L. Garcia-Rio, J. G. Santos and M. E. Aliaga, ACS Omega, 2021, 6, 10333–10342 CrossRef CAS PubMed.
- N. Rad, O. Danylyuk and V. Sashuk, Angew. Chem., Int. Ed., 2019, 58, 11340–11343 CrossRef CAS PubMed.
- Z. Said and J. G. Tillett, J. Org. Chem., 1981, 46, 2586–2588 CrossRef CAS.
- C. A. Bunton, J. H. Crabtree and L. Robinson, J. Am. Chem. Soc., 1968, 90, 1258 CrossRef CAS.
- N. Basilio, S. Gago, A. J. Parola and F. Pina, ACS Omega, 2017, 2, 70–75 CrossRef CAS PubMed.
- I. Ghosh and W. M. Nau, Adv. Drug Delivery Rev., 2012, 64, 764–783 CrossRef CAS PubMed.
- R. P. Bell and M. Robson, Trans. Faraday Soc., 1964, 60, 893 RSC.
- H. Tang, D. Fuentealba, Y. H. Ko, N. Selvapalam, K. Kim and C. Bohne, J. Am. Chem. Soc., 2011, 133, 20623–20633 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures, spectral data, and analytical data. See https://doi.org/10.1039/d2cc00772j |
| This journal is © The Royal Society of Chemistry 2022 |