
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
δ C–H (hetero)arylation via Cu-catalyzed radical relay†
Zuxiao
Zhang
 ,
Leah M.
Stateman
and
David A.
Nagib
*
,
Leah M.
Stateman
and
David A.
Nagib
*
The Ohio State University, Department of Chemistry and Biochemistry, Columbus, OH 43210, USA. E-mail: nagib.1@osu.edu
First published on 9th November 2018
Abstract
A Cu-catalyzed strategy has been developed that harnesses a radical relay mechanism to intercept a distal C-centered radical for C–C bond formation. This approach enables selective δ C–H (hetero)arylation of sulfonamides via intramolecular hydrogen atom transfer (HAT) by an N-centered radical. The radical relay is both initiated and terminated by a Cu catalyst, which enables incorporation of arenes and heteroarenes by cross-coupling with boronic acids. The broad scope and utility of this catalytic method for δ C–H arylation is shown, along with mechanistic probes for selectivity of the HAT mechanism. A catalytic, asymmetric variant is also presented, as well as a method for accessing 1,1-diaryl-pyrrolidines via iterative δ C–H functionalizations.
Introduction
The remote C–H functionalization of amines via intramolecular hydrogen atom transfer (HAT) has enabled a distinct approach to the synthesis of pyrrolidines for over a century.1,2 Yet, while this formal δ C–H amination has been interrupted to afford distal halogenation and oxygenation, it has rarely enabled δ C–C bond formation.3–5 A mechanistic explanation is that initiation of this radical rearrangement requires homolysis of an N-halo amide to generate the N-centered radical (Fig. 1a). Following selective 1,5-HAT, the translocated δ C˙ rapidly combines with the solvent-caged halide radical (X˙). Finally, intramolecular displacement of the resultant δ C–X bond is then spontaneous if X = I (or requires a strong base if X = Br, Cl). Notably, radical recombination to form C–X is rapid; and we have exclusively observed δ halogenation – even when this reaction is performed with a radical trap (e.g. acrylonitrile) as solvent. Given this challenge, the first examples of intercepting this N˙ to C˙ relay for C–C bond formation were only reported recently.4 Notably, these solutions (mostly entailing δ addition to acrylates) forgo the intermediacy of X˙ entirely; and instead, the N˙ is generated from an N–H or N–O bond.4,5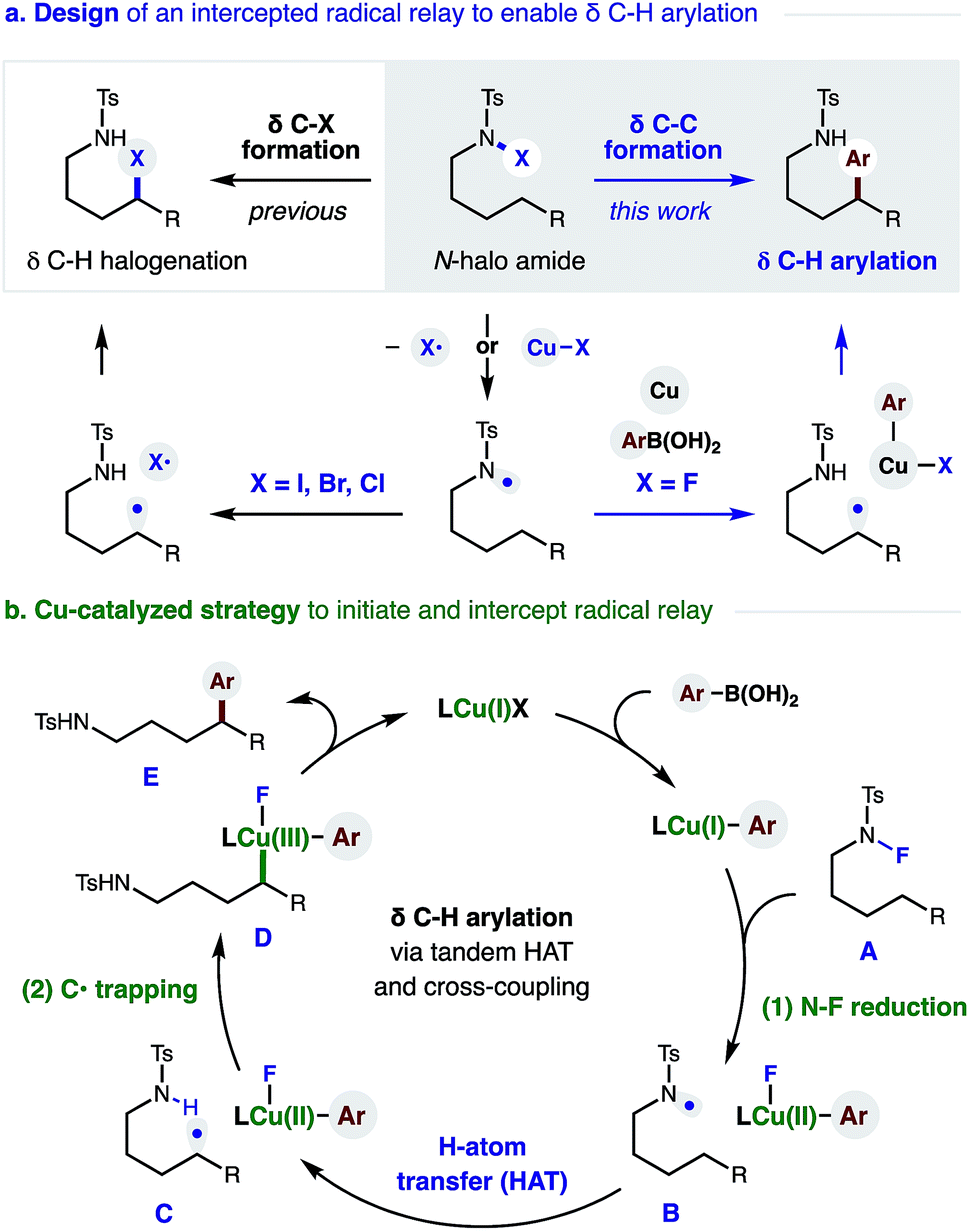 | ||
| Fig. 1 Cu-catalyzed radical relay enables δ C–H arylation. | ||
We proposed the interruption of this century-old X˙ rearrangement could also be facilitated by use of an N–F precursor. Since F˙ is highly unstable,6 N–F homolysis (and ensuing radical recombination with δ C˙) seemed unfavorable.7 In fact, a recent example of δ fluorination by HAT required an Fe-catalyzed protocol.8 Instead, we anticipated N–F reduction could be mediated by a Cu catalyst that would also enable Suzuki–Miyaura coupling of the distal organocopper with aryl boronic acids.9 A Cu-mediated pathway for C˙ formation and subsequent arylation also appeared viable – based on pioneering work by Liu and Stahl, who incorporated these elementary steps in their arylation of benzylic C–H bonds.10
In designing a strategy to enable the first δ C–H arylation via a radical relay mechanism,11–14 we proposed a Cu catalyst could serve the dual roles of radical initiation and aryl trapping of the distal C˙ (Fig. 1b). In our proposed mechanism, an in situ generated Cu(I) complex undergoes transmetallation with a (hetero)aryl boronic acid to afford a Cu(I)Ar species. We expected this more electron-rich Cu complex to be well-suited to initiate reduction of the N–F bond of amide Avia either a single-electron-transfer or atom-transfer mechanism. The resultant N-centered radical B would then undergo selective 1,5-HAT to afford δ C˙ amide C. In the second vital role of the Cu catalyst, an oxidized Cu(II)Ar complex could combine with C˙ in the mechanism described by Kochi,15 as well as Liu.10e The highly oxidized organometallic D should then reductively eliminate Cu(I) and δ aryl amide E. This final step simultaneously affords turnover of the catalytic cycle and sp3–sp2 C–C coupling. Importantly, we were intrigued by the possibility that ligand tunability could enable control of both reactivity and stereoselectivity in this δ C–H arylation.
Results and discussion
To test our hypothesis, we combined p-F-phenylboronic acid, an N-fluoro-tosylamide, and 5% Cu(OTf)2 (Fig. 2). Upon optimization of ligand, base, and solvent mixtures (all crucial factors to enable maximum reaction efficiency, see ESI† for details), we were pleased to find the radical relay mechanism could indeed be interrupted to afford δ arylation (1, 79%). Control experiments reveal that both bisoxazoline ligand (±) L1 and an aryl boronic acid are necessary for efficient amide consumption, suggesting the Cu complex requires both donating ligands to reduce the N–F.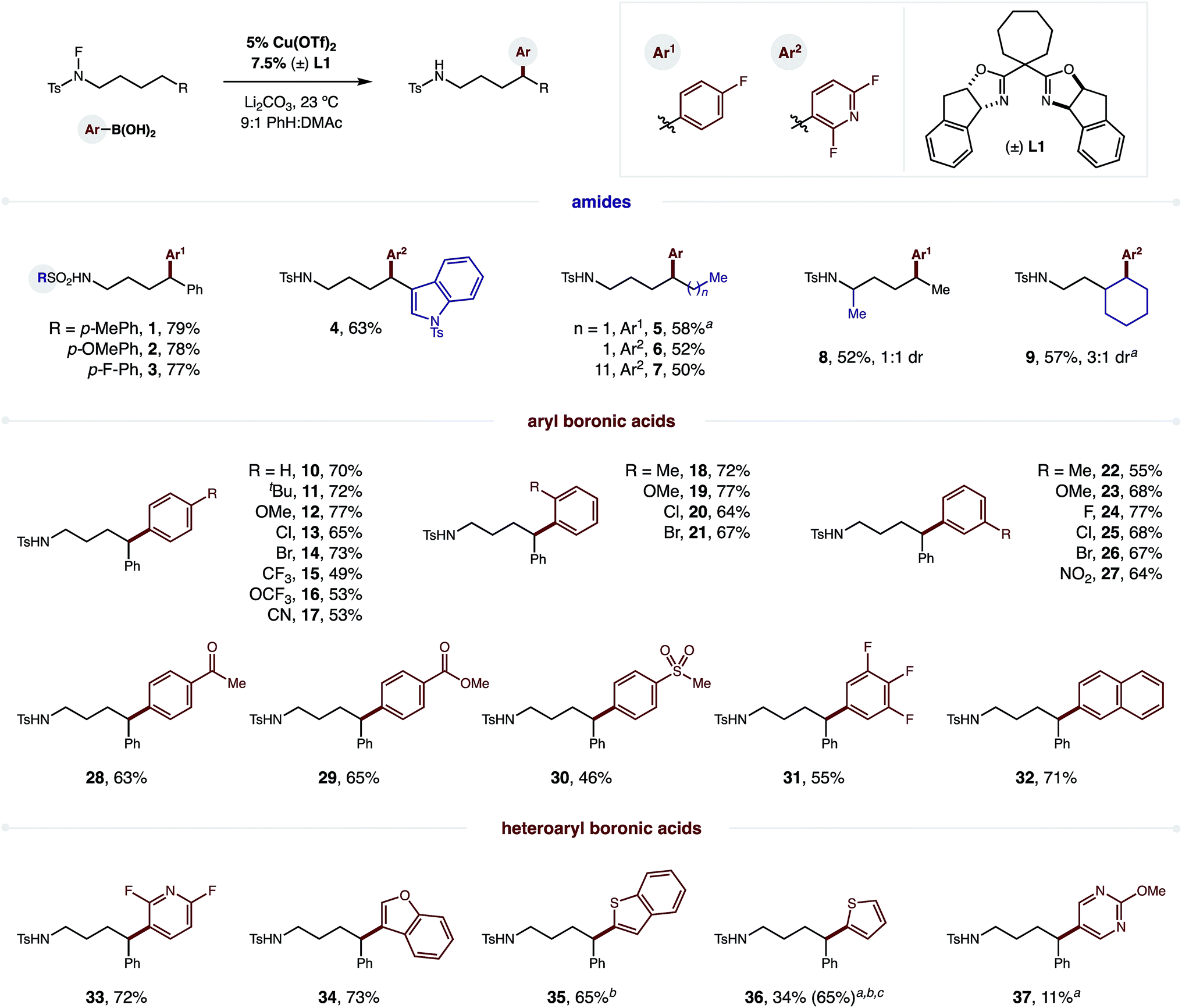 | ||
Fig. 2 Scope of δ C–H (hetero)arylation by Cu-catalyzed radical relay and coupling with aryl boronic acids. Conditions: sulfonamide (0.2 mmol), arylboronic acid (2 equiv.), Li2CO3 (1 equiv.), 5% Cu(OTf)2, 7.5% (±) L1, PhH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMAc (4 mL; 9:1), r.t. isolated yields. aNMR yield. b10% Cu(OTf)2, 15% (±) L1.cBased on recovered starting material. :DMAc (4 mL; 9:1), r.t. isolated yields. aNMR yield. b10% Cu(OTf)2, 15% (±) L1.cBased on recovered starting material. | ||
We next turned our attention to investigating the scope and generality of this δ C–H arylation (Fig. 2). Interestingly, electronic variation of the sulfonamide does not greatly affect reaction efficiency (1–3). Heteroarenes, such as indole, can be incorporated on the amide (4; without 6-exo-trig cyclization), or within the aryl boronic acid (Ar2). In the latter case, we found the difluoropyridyl arene products to be particularly easy to isolate. Notably, we found that it is not necessary to abstract the H-atom from a benzylic position. In fact, secondary C–H bonds are readily arylated (5–9), in accordance with our previous work on δ C–H aminations.16 α-Branched amides are also viable (8), although they afford lower diastereoselectivity (1:1) than γ substitution (9, 3:1).
To showcase the synthetic utility of employing aryl boronic acids within this remote Cu-catalyzed cross-coupling, we embarked on an exhaustive investigation of this component. As shown in Fig. 2, this δ arylation is amenable to electronic perturbation of the ArB(OH)2 – with both donating and accepting groups tolerated (10–31) – ranging from –OMe to –NO2 and including orthogonal functional handles, such as bromides, ketones, esters, and sulfones. Similarly, both ortho and meta substitution, as well as polyaromatics (32), readily participate in this reaction. As further demonstration of the likely utility of this approach to the synthesis of medicinal agents, we employed a family of heteroaryl boronic acids, including those containing N, O, S, and F, and all are amenable to this δ C–H arylation (33–37).
The robust δ regioselectivity observed in these reactions, including an arylation of a single 1-of-15 methylenes (7), led us to further investigate the limits of the selectivity in the key HAT step (Fig. 3). With this question in mind, we subjected an amide with two benzylic C–H bonds at δ and ε positions to these catalytic conditions. Selective δ arylation of this probe (38; 14:1) confirms 1,5-HAT is highly favored over 1,6-HAT in this reaction. However, this kinetic preference can be thermodynamically overridden if a weaker, benzylic ε C–H is pitted against a secondary δ C–H. In this case, 1,6-HAT is slightly favored (39; 1.7:1). Importantly, and as a complement to previous work on α C–H arylation,10–12 when a molecule containing a distal benzylic C–H bond is employed, no undirected arylation is observed (40; > 20:1 δ). Finally, in a three-way competition between δ/ε secondary C–H bonds and a ζ benzylic C–H bond, only the product of 1,5-HAT is observed, rather than either 1,6 or 1,7 HAT (41–42; > 20:1 δ).
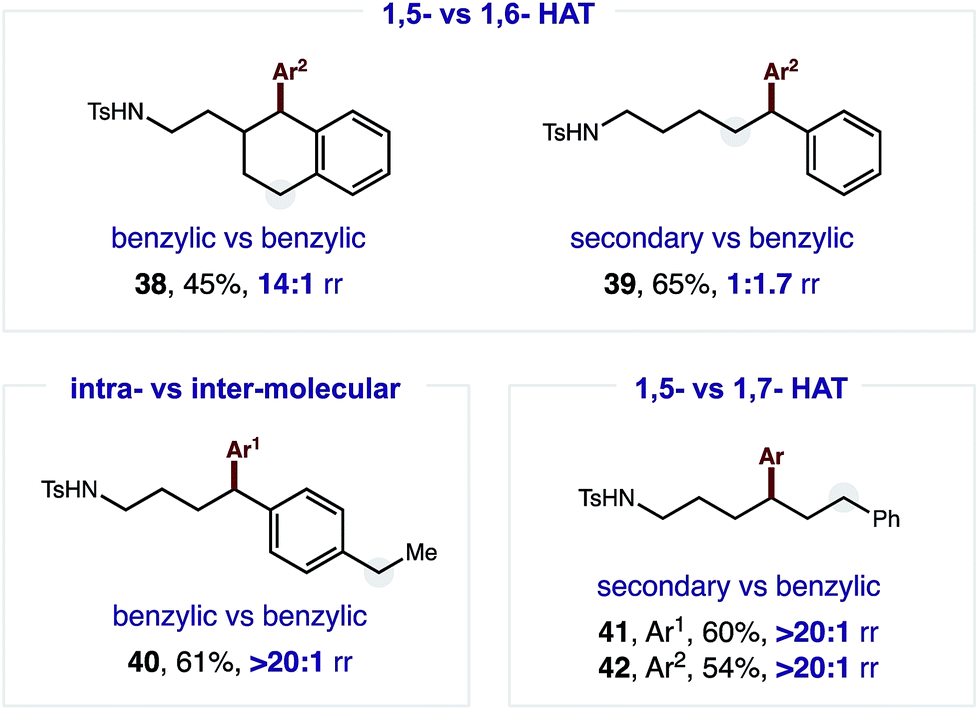 | ||
| Fig. 3 Regioselectivity probes of intramolecular HAT. | ||
Although this δ C–H arylation was enabled by interrupting the classic Hofmann–Löffler–Freytag (HLF) reaction, we questioned if these complementary transformations could be sequentially combined (Fig. 4). For example, we have demonstrated that this Cu-catalyzed δ arylation (I) yields 4-aryl-butyl amides (II), and subsequent AcOI-mediated δ amination affords α diaryl pyrrolidines (III), bearing a tetra-substituted carbon (43–46). We expect this two-step, double C–H functionalization sequence will provide a synthetically enabling route to access medicinally relevant pyrrolidines,17 in a rapid, modular, and non-classical fashion.
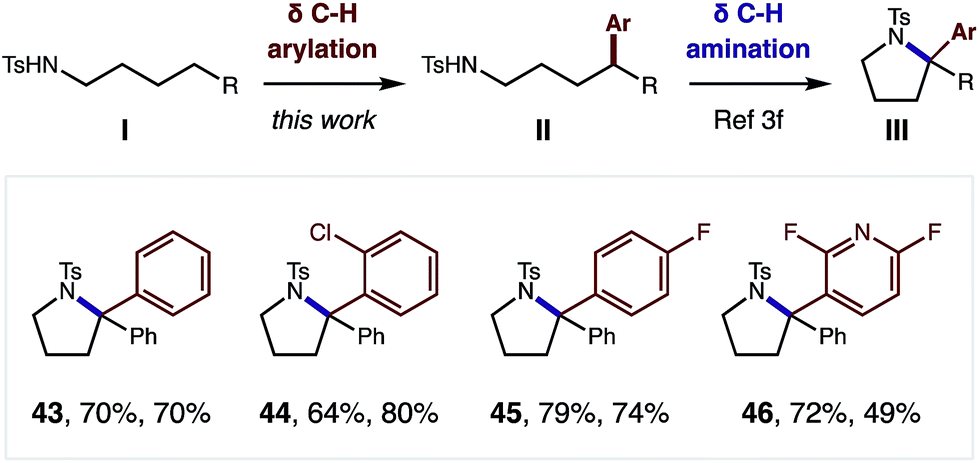 | ||
| Fig. 4 Pyrrolidine synthesis via iterative δ C–H arylation and δ C–H amination. | ||
Finally, given the strong dependence of reaction efficiency on ligand choice, as well as the ready availability of enantioenriched bisoxazoline ligands, we tested whether a catalytic, asymmetric version of this δ C–H arylation is possible (eqn (1)). To our delight, we found that employing enantiopure L118 affords enantioenriched diaryl amide (1, 48% ee). Moreover, decreasing the temperature to −4 °C (and employing a co-solvent with a lower mp) allows access to this 1,1-diaryl-product with 65% ee – affording the first example of an enantioselective, radical C–H arylation.
 | (1) |
Conclusions
In summary, we have interrupted the HLF reaction to afford a δ C–H arylation. The regioselectively translocated C˙ of this classic mechanism was intercepted and employed in C–C bond formation by using a Cu catalyst. The two key roles of this catalyst are: (i) to generate N˙ without also forming a δ halogenating X˙, and (ii) to trap the translocated C˙ as an organometallic intermediate. This organocopper species enables cross-coupling with a range of (hetero)aryl boronic acids at the δ position of sulfonamides in a regioselective (and stereoselective) manner. We expect this strategy will facilitate further development of methods to interrupt the classic HLF reaction with valuable C–C bond formation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Institutes of Health (NIH R35 GM119812), National Science Foundation (NSF CAREER 1654656), and American Chemical Society Petroleum Research Fund for financial support. LMS is supported by an NSF Graduate Research Fellowship.Notes and references
- δ C–H functionalization via directed HAT. For reviews, see: (a) J. Robertson, J. Pillai and R. K. Lush, Chem. Soc. Rev., 2001, 30, 94–103 RSC; (b) S. Chiba and H. Chen, Org. Biomol. Chem., 2014, 12, 4051–4060 RSC; (c) L. M. Stateman, K. M. Nakafuku and D. A. Nagib, Synthesis, 2018, 50, 1569–1586 CrossRef CAS PubMed; (d) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS PubMed.
- Pioneering examples of δ C–H amination: (a) A. W. Hofmann, Chem. Ber., 1883, 16, 558–560 CrossRef; (b) K. Löffler and C. Freytag, Chem. Ber., 1909, 42, 3427–3431 CrossRef; (c) E. J. Corey and W. R. Hertler, J. Am. Chem. Soc., 1960, 82, 1657–1668 CrossRef CAS; (d) P. de Armas, R. Carrau, J. I. Concepción, C. G. Francisco, R. Hernández and E. Suárez, Tetrahedron Lett., 1985, 26, 2493–2496 CrossRef CAS; (e) C. G. Francisco, A. J. Herrera and E. Suárez, J. Org. Chem., 2003, 68, 1012–1017 CrossRef CAS PubMed; (f) J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092–4106 RSC.
- Recent, remote C–H functionalizations via directed HAT: (a) K. Chen, J. M. Richter and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 7247–7249 CrossRef CAS PubMed; (b) K. Chen and P. S. Baran, Nature, 2009, 459, 824–828 CrossRef CAS PubMed; (c) N. Yoshikai, A. Mieczkowski, A. Matsumoto, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2010, 132, 5568–5569 CrossRef CAS PubMed; (d) Y.-F. Wang, H. Chen, X. Zhu and S. Chiba, J. Am. Chem. Soc., 2012, 134, 11980–11983 CrossRef CAS PubMed; (e) M. Yang, B. Su, Y. Wang, K. Chen, X. Jiang, Y.-F. Zhang, X.-S. Zhang, G. Chen, Y. Cheng, Z. Cao, Q.-Y. Guo, L. Wang and Z.-J. Shi, Nat. Commun., 2014, 5, 4707 CrossRef CAS PubMed; (f) C. Martínez and K. Muñiz, Angew. Chem., Int. Ed., 2015, 54, 8287–8291 CrossRef PubMed; (g) Q. Qin and S. Yu, Org. Lett., 2015, 17, 1894–1897 CrossRef CAS PubMed; (h) T. Liu, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 5871–5874 CrossRef CAS PubMed; (i) H. Lu, K. Lang, H. Jiang, L. Wojtas and X. P. Zhang, Chem. Sci., 2016, 7, 6934–6939 RSC; (j) M. Parasram, P. Chuentragool, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2016, 138, 6340–6343 CrossRef CAS PubMed; (k) J. Ozawa, M. Tashiro, J. Ni, K. Oisaki and M. Kanai, Chem. Sci., 2016, 7, 1904–1909 RSC; (l) R. Wang, Y. Li, R.-X. Jin and X.-S. Wang, Chem. Sci., 2017, 8, 3838–3842 RSC; (m) D. Meng, Y. Tang, J. Wei, X. Shi and M. Yang, Chem. Commun., 2017, 53, 5744–5747 RSC; (n) M. A. Short, J. M. Blackburn and J. L. Roizen, Angew. Chem., Int. Ed., 2018, 57, 296–299 CrossRef CAS PubMed; (o) E. A. Wappes, K. M. Nakafuku and D. A. Nagib, J. Am. Chem. Soc., 2017, 139, 10204–10207 CrossRef CAS PubMed; (p) M. Parasram, P. Chuentragool, Y. Wang, Y. Shi and V. Gevorgyan, J. Am. Chem. Soc., 2017, 139, 14857–14860 CrossRef CAS PubMed; (q) S. Sathyamoorthi, S. Banerjee, J. Du Bois, N. Z. Burns and R. N. Zare, Chem. Sci., 2018, 9, 100–104 RSC; (r) E. A. Wappes, A. Vanitcha and D. A. Nagib, Chem. Sci., 2018, 9, 4500–4504 RSC; (s) Y. Tang, Y. Qin, D. Meng, C. Li, J. Wei and M. Yang, Chem. Sci., 2018, 9, 6374–6378 RSC.
- Distal C–C bond formation via N˙ mediated HAT: (a) W. Shu and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1881–1884 CrossRef CAS PubMed; (b) H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 1692–1696 CrossRef CAS PubMed; (c) G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS PubMed; (d) J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272–275 CrossRef PubMed; (e) D.-F. Chen, J. C. K. Chu and T. Rovis, J. Am. Chem. Soc., 2017, 139, 14897–14900 CrossRef CAS PubMed; (f) Y. Xia, L. Wang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 12940–12944 CrossRef CAS PubMed; (g) S. P. Morcillo, E. M. Dauncey, J. H. Kim, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 12945–12949 CrossRef CAS PubMed; (h) X. Shen, J.-J. Zhao and S. Yu, Org. Lett., 2018, 20, 5523–5527 CrossRef CAS PubMed; (i) H. Chen, L. Guo and S. Yu, Org. Lett., 2018, 20, 6255–6259 CrossRef CAS PubMed.
- Distal C–H alkylation via O˙ radical relay: (a) Ž. Čeković, Tetrahedron, 2003, 59, 8073–8090 CrossRef; (b) J. Zhang, Y. Li, F. Zhang, C. Hu and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 1872–1875 CrossRef CAS PubMed; (c) C. Wang, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 13495–13498 CrossRef CAS PubMed.
- F˙ instability is exemplified by the high electronegativity and ionization energy of fluorine: B. E. Smart, in Organofluorine Chemistry: Principles and Commercial Applications, ed. R. E. Banks, B. E. Smart and J. C. Tatlow, Springer US, Boston, MA, 1994, pp. 57–88 Search PubMed.
- Radical chain propagation by intermolecular C˙ addition to N–F is not observed. For a key example of this mechanism, see: M. Rueda-Becerril, C. C. Sazepin, J. C. T. Leung, T. Okbinoglu, P. Kennepohl, J.-F. Paquin and G. M. Sammis, J. Am. Chem. Soc., 2012, 134, 4026–4029 CrossRef CAS PubMed.
- B. J. Groendyke, D. I. Abusalim and S. P. Cook, J. Am. Chem. Soc., 2016, 138, 12771–12774 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- Cu-catalyzed α C–H functionalization via HAT: (a) W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014–1018 CrossRef CAS PubMed; (b) A. Vasilopoulos, S. L. Zultanski and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 7705–7708 CrossRef CAS PubMed; (c) W. Zhang, P. Chen and G. Liu, J. Am. Chem. Soc., 2017, 139, 7709–7712 CrossRef CAS PubMed; (d) K. W. Bentley, K. A. Dummit and J. F. Van Humbeck, Chem. Sci., 2018, 9, 6440–6445 RSC; (e) F. Wang, P. Chen and G. Liu, Acc. Chem. Res., 2018, 51, 2036–2046 CrossRef CAS PubMed.
- Ni-catalyzed α C–H arylation via HAT: (a) M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. MacMillan, Science, 2016, 352, 1304–1308 CrossRef CAS PubMed; (b) D. R. Heitz, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 12715–12718 CrossRef CAS PubMed; (c) B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed; (d) D. T. Ahneman and A. G. Doyle, Chem. Sci., 2016, 7, 7002–7006 RSC.
- Photoredox α-amino C–H (hetero)arylation: (a) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed; (b) T. Hoshikawa and M. Inoue, Chem. Sci., 2013, 4, 3118–3123 RSC; (c) C. K. Prier and D. W. C. MacMillan, Chem. Sci., 2014, 5, 4173–4178 RSC; (d) T. Ide, J. P. Barham, M. Fujita, Y. Kawato, H. Egami and Y. Hamashima, Chem. Sci., 2018, 9, 8453–8460 RSC.
- During submission of this manuscript, a related δ C–H arylation – with complementary scope, efficiency, and utility – was published: Z. Li, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2018, 57, 13288–13292 CrossRef CAS PubMed.
- Non-radical, directed (γ) C–H arylations: (a) W. Cui, S. Chen, J.-Q. Wu, X. Zhao, W. Hu and H. Wang, Org. Lett., 2014, 16, 4288–4291 CrossRef CAS PubMed; (b) K. S. L. Chan, M. Wasa, L. Chu, B. N. Laforteza, M. Miura and J.-Q. Yu, Nat. Chem., 2014, 6, 146–150 CrossRef CAS PubMed; (c) K. S. L. Chan, H. Y. Fu and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 2042–2046 CrossRef CAS PubMed; (d) Y. Wu, Y. Q. Chen, T. Liu, M. D. Eastgate and J. Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554–14557 CrossRef CAS PubMed; (e) R.-Y. Zhu, Z.-Q. Li, H. S. Park, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 3564–3568 CrossRef CAS PubMed; (f) Y. Liu and H. Ge, Nat. Chem., 2017, 9, 26–32 CAS; (g) J. J. Topczewski, P. J. Cabrera, N. I. Saper and M. S. Sanford, Nature, 2016, 531, 220–224 CrossRef CAS PubMed; (h) P. J. Cabrera, M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2018, 140, 5599–5606 CrossRef CAS PubMed; (i) G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, J. Am. Chem. Soc., 2012, 134, 3–6 CrossRef CAS PubMed; (j) E. T. Nadres and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 7–10 CrossRef CAS PubMed; (k) S.-Y. Zhang, G. He, W. A. Nack, Y. Zhao, Q. Li and G. Chen, J. Am. Chem. Soc., 2013, 135, 2124–2127 CrossRef CAS PubMed; (l) J.-W. Xu, Z.-Z. Zhang, W.-H. Rao and B.-F. Shi, J. Am. Chem. Soc., 2016, 138, 10750–10753 CrossRef CAS PubMed; (m) B.-B. Zhan, Y. Li, J.-W. Xu, X.-L. Nie, J. Fan, L. Jin and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 5858–5862 CrossRef CAS PubMed.
- J. K. Kochi, Acc. Chem. Res., 1974, 7, 351–360 CrossRef CAS.
- E. A. Wappes, S. C. Fosu, T. C. Chopko and D. A. Nagib, Angew. Chem., Int. Ed., 2016, 55, 9974–9978 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- L. Wu, F. Wang, X. Wan, D. Wang, P. Chen and G. Liu, J. Am. Chem. Soc., 2017, 139, 2904–2907 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04366c |
| This journal is © The Royal Society of Chemistry 2019 |