
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold-catalyzed (4+3)-annulations of 2-alkenyl-1-alkynylbenzenes with anthranils with alkyne-dependent chemoselectivity: skeletal rearrangement versus non-rearrangement†
RahulKumar Rajmani
Singh
a,
Manisha
Skaria
 a,
Liang-Yu
Chen
b,
Mu-Jeng
Cheng
*b and
Rai-Shung
Liu
*a
a,
Liang-Yu
Chen
b,
Mu-Jeng
Cheng
*b and
Rai-Shung
Liu
*a
aFrontier Research Centers on Fundamental and Applied Science of Materials, Department of Chemistry, National Tsing-Hua University, Hsinchu, Taiwan, Republic of China. E-mail: rsliu@mx.nthu.edu.tw
bDepartment of Chemistry, National Cheng Kung University, Tainan 701, Taiwan. E-mail: mjcheng@mail.ncku.edu.tw
First published on 12th November 2018
Abstract
Two distinct (4+3)-nitroxy annulations between 1,5-enynes and anthranils have been developed to access tetrahydro-1H-benzo[b]azepine derivatives; the chemoselectivity varies with the types of alkynes. Terminal alkyne substrates deliver benzo[b]azepine derivatives via a novel skeletal rearrangement while internal 1,5-enynes afford products without a rearrangement process. To elucidate the mechanism of rearrangement, we performed 13C- and 2H-labeling experiments to identify the gold-containing isobenzofulvene intermediates, but their formation relies on the presence of anthranils.
Introduction
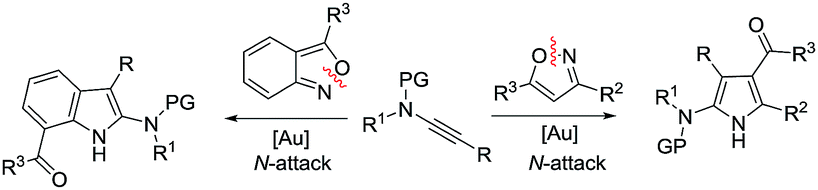
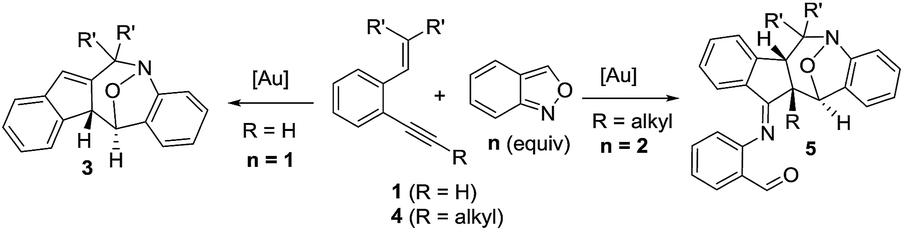
Cyclic nitroxy species (N–O) are widespread functionalities in numerous bioactive molecules and natural products.1 Tetrahydro-1H-benzo[b]azepines bearing a hydroxyl (I–IV) represent a family of privileged seven-membered azacycles,2 possessing potent activities in antiparasitic disease, antidiuretic hormone receptors and β2 adrenergic agonists.3 Synthetic procedures for compounds I–IV are generally long and tedious.2 A short route to construct tetrahydrobenzo[b]azepine cores involves the development of stereoselective (4+3)-annulations between anthranils and all-carbon 1,3-dipoles (eqn (1)), but only donor–acceptor cyclopropanes were shown to be applicable substrates.4 We are aware of no π-bond motifs that can serve as effective 1,3-dipoles.5Synthetic interest in isoxazoles and anthranils is rapidly growing in Au- and Pt-catalysis because of their various annulations with alkynes.6,7 Nevertheless, these hetero-aromatics serve as nucleophiles that attack π-alkynes via a N- or O-attack route, inevitably cleaving the N–O bonds; selected examples are provided in eqn (2) and (3). We sought the first (4+3)-nitroxy annulations using alkyne-based 1,3-dipoles and anthranils. This work reports two distinct (4+3)-annulations of 1,5-enynes with anthranils; interestingly, the chemoselectivity varies with the alkynes. Terminal 1,5-enynes 1 (R = H) afford seven-membered nitroxy heterocycles 3via an unprecedented rearrangement in gold catalysis;8 the mechanism of this novel rearrangement has been elucidated. Annulation products 5 derived from internal alkynes 4 are not skeletally rearranged, but are elaborated into various benzo[b]azepine frameworks (Fig. 1).
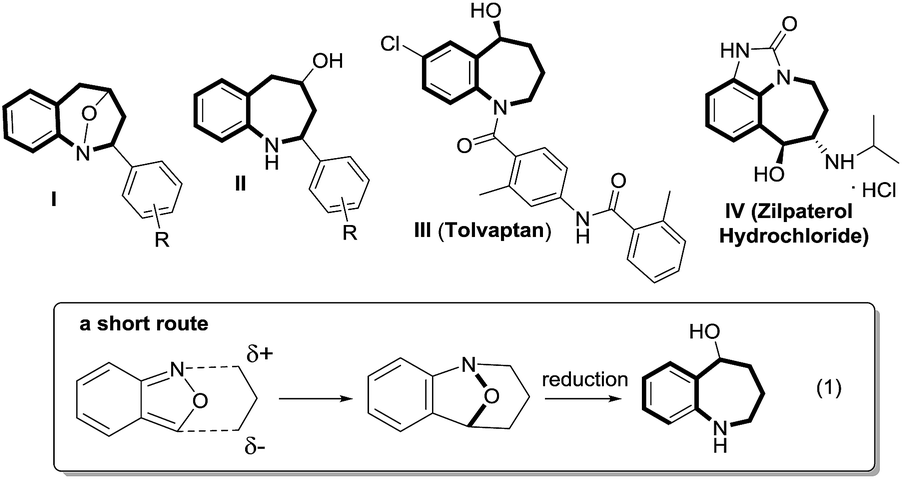 | ||
| Fig. 1 Representative molecules and a postulated short route. | ||
Annulations with N–O cleavages
 | (2) |
 | (3) |
This work: (4+3)-nitroxy annulations
 | (4) |
Results and discussion
We optimized the reactions of terminal 1,5-enyne 1a with anthranil 2a (1.2 equiv.) using various gold catalysts; the results are shown in Table 1. Operations in dry dichloroethane (DCE, 25 °C) with L′AuCl/AgNTf2 (L′ = P(t-Bu)2(o-biphenyl), IPr, PPh3) afforded seven-membered nitroxy product 3a in 8–68% yield (entries 1–3), with P(t-Bu)2(o-biphenyl)AuCl/AgNTf2 being the most effective. To our delight, (PhO)3PAuCl/AgNTf2 increased the yield of the desired 3a up to 73% (entry 4); different silver salts as those in (PhO)3AuCl/AgX (X = SbF6 and OTf) delivered compound 3a in relatively low yields (35–42%, entries 5 and 6). With (PhO)3PAuCl/AgNTf2, the yields of compound 3a in different solvents were as follows: DCM (62%), acetonitrile (30%) and MeNO2 (0%, entries 7–9). AgNTf2 alone was completely inactive (entry 10). The molecular structure of compound 3a was characterized by X-ray diffraction9 to reveal a (4+3)-annulation with an intact N–O bond. In the absence of anthranil 2a, 1,5-enyne 1a was isomerized by a gold catalyst to afford 1′-methylvinyl-1H-indene 1a′, which was structurally unrelated to our target 3a. Anthranil 2a is obviously indispensable to enabling the (4+3)-annulations with structural rearrangement.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalysta (mol %) | 2a n equiv. | Solvent | Time (h) | Temp (t °C) | Yieldsb (%) | ||
| 1a | 3a | 1a′ | ||||||
| a 1a (0.20 M), 2a (1.2 equiv.). b Product yields are given after purification on a silica gel column, L = P(t-Bu)2(o-biphenyl), IPr = 1,3-bis(diisopropylphenyl)imidazol-2-ylidene). | ||||||||
| 1 | LAuCl/AgNTf2 | 1.2 | DCE | 5 | 25 | — | 68 | — |
| 2 | IPrAuCl/AgNtf2 | 1.2 | DCE | 15 | 25 | 25 | 8 | — |
| 3 | Ph3PAuCl/AgNtf2 | 1.2 | DCE | 12 | 25 | — | 35 | — |
| 4 | (PhO)3PAuCl/AgNtf2 | 1.2 | DCE | 4 | 25 | — | 73 | — |
| 5 | (PhO)3PAuCl/AgSbF6 | 1.2 | DCE | 10 | 25 | 10 | 35 | — |
| 6 | (PhO)3PAuCl/AgOTf | 1.2 | DCE | 2 | 60 | — | 42 | — |
| 7 | (PhO)3PAuCl/AgNtf2 | 1.2 | DCE | 10 | 25 | — | 62 | — |
| 8 | (PhO)3PAuCl/AgNtf2 | 1.2 | MeCN | 10 | 25 | — | 30 | — |
| 9 | (PhO)3PAuCl/AgNtf2 | 1.2 | MeNO2 | 20 | 25 | 80 | — | — |
| 10 | AgNtf2 | 1.2 | DCE | 24 | 25 | 85 | >5 | — |
| 11 | (PhO)3PAuCl/AgNtf2 | 0 | DCE | 4 | 25 | — | — | 65 |

Under these optimized conditions, we assess the generality of these new annulations with various terminal 1,5-enynes and anthranils. The results are provided in Table 2; only a single diastereomeric product was obtained for all instances. In several instances, vinyl-1H-indene 1a′ was present as a byproduct in a minor proportion (5–15%). The annulations of anthranil 2a (1.2 equiv.) with terminal 1,5-enynes 1b–1d bearing various 4-phenyl substituents (X = Me, Cl, and F) proceeded smoothly to yield 3b–3d in 68–77% yields (entries 2–4). For their 5-phenyl analogues 1e–1g, the resulting annulation products 3e–3g (Y = Me, Cl and F) were obtained in 65–74% yields (entries 5–7). Variations of the olefin substituents as those in species 1h–1j (R, R = cyclopentyl, cyclohexyl and dipropyl) were still compatible with these new N–O annulations to afford compounds 3h–3j in 55–67% yields (entries 8–10). We have also prepared a terminal alkyne such as 1-ethynyl-2-styrylbenzene 1k that gave a recovery yield (>95%) of two reactants under the standard conditions.

We next examined anthranils 2b–2f bearing various C(5)-substituents (X′ = Me, Cl, Br, OMe and OCO2Et), yielding cyclic nitroxy species 3k–3o in 48–77% yields, with X′ = OMe becoming less efficient (entries 11–15). Methoxy-containing anthranil 2e renders the gold catalyst less reactive because of its high basicity. This gold catalysis worked well with additional anthranils 2g and 2h bearing C(6)-substituents (Y′ = Br and Me), yielding the desired 3p and 3q in 41% and 70% yields, respectively (entries 15 and 16). We also varied the C(3)-substituents of anthranils (R′ = Ph 2i; Me 2j) to yield the desired 3r and 3s in 35% and 63% yields, respectively (entries 18 and 19). An effective range of alkynes and anthranils manifests the practicability of these new nitroxy annulations.
This gold-catalyzed reaction was also extensible to an internal alkyne 4a, but led to a distinct (4+3)-annulation reaction without a skeletal rearrangement. Among various gold catalysts, P(OPh)3AuCl/AgSbF6 was superior to its NTf2 catalyst analogue, delivering a nitroxy product 5a with respective yields of 78% and 68%; a molar ratio of 4a/2a = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.1 was the optimized condition. The molecular structure of 5a was inferred from its 5b analogue (Table 3, entry 1).9
:2.1 was the optimized condition. The molecular structure of 5a was inferred from its 5b analogue (Table 3, entry 1).9
 | (5) |

We assess the scope of these nitroxy annulations with various internal 1,5-enynes 4 and anthranils 2; only one diastereomeric product was obtained without exception. Entries 1–6 show the compatibility of these reactions with 1,5-enynes 4b–4d and 4e–4g bearing 4- and 5-phenyl substituents (X = Me, F and Cl or Y = Me, F and Cl), delivering compounds 5b–5d and 5e–5g in 65–75% yields (entries 1–6). An X-ray diffraction study9 confirms the molecular structure of compound 5b showing no skeletal rearrangement. 1,5-Enynes 4h and 4i bearing varied trisubstituted alkenes were also suitable for the reactions, affording the desired nitroxy species 5h and 5i in 71–72% yields (entries 7 and 8). When the alkyl substituents R were a cyclopropyl or CH2OTBS group, the corresponding compounds 5j and 5k were obtained in 56% and 53% yields, respectively (entries 9 and 10). We tested the reactions of various anthranils 2b–2f bearing various C(5)-substituents (X′ = Me, Br, Cl, OMe and OCO2Et), giving the expected products 5l–5p in 55–75% yields with the methoxy substituent being less efficient (entries 11–15). For additional anthranils 2g and 2h bearing 6-substituents (Y′ = Br and Me), the resulting products 5q and 5r were obtained in 48% and 68% yields, respectively (entries 16 and 17).
We performed the reductive N–O cleavage of compounds 3a and 5a to manifest their synthetic utility. Treatment of species 3a with Zn in AcOH/MeOH/H2O10 gave compound 6a in 89% yield while the reaction with Pd/H2 gave compound 6b efficiently. Alternatively, compound 5a was hydrolyzed with HCl/water to yield ketone derivative 7b that was convertible to 1-amino-5-ol 7c with Zn/AcOH reduction, and to the diol derivative 7d with Pd/H2 reduction. An imine reduction of species 5a was achieved with Pd/H2 to afford species 7a. Unexpectedly, Zn-reduction of species 5a in HOAc/MeOH/water led to a structural rearrangement to form compound 7e in 81% yield. The imine moiety of the initial 5a was incorporated into the structural skeleton of product 7e, but the mechanism is not clear at this stage. Molecular structures of compounds 7a and 7e were verified by X-ray diffraction.9 The mechanism for the transformation of 5a into 7e will be elucidated in a future study (Scheme 1).
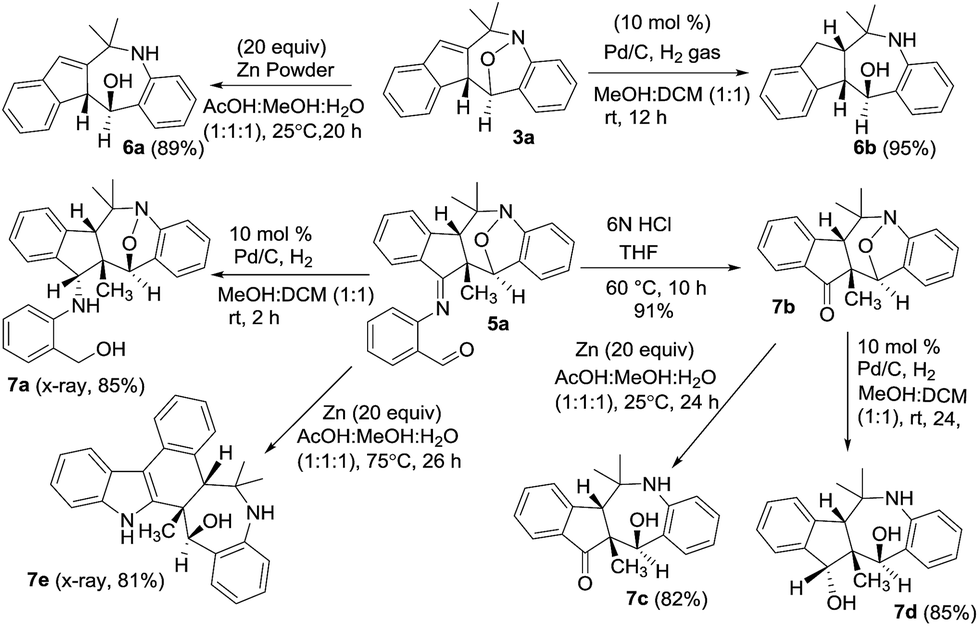 | ||
| Scheme 1 Reductive cleavage of the N–O bonds. | ||
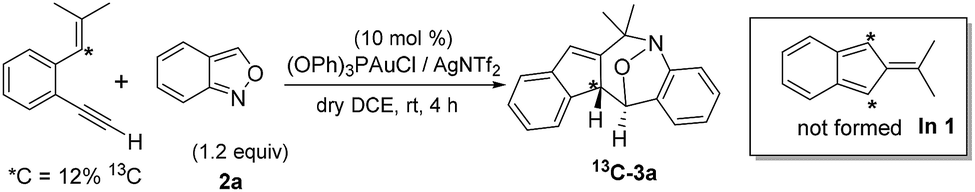
Among the two nitroxy annulations, the mechanism for terminal 1,5-enynes 1a is difficult to deduce because its cycloisomerization product 1a′ is not skeletally rearranged. We prepared 13C-1a containing 12% 13C at only the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H carbon, and its resulting product 3a contained the 13C-content only at the alkyl C–H carbon (eqn (6)). Isobenzofulvene species In 1 was unlikely to occur here although it was observed in a ruthenium-catalyzed cycloisomerization.11 In the presence of D2O, we found that the resulting d1-3a contained deuterium (X = 0.29D) only at its alkenyl C–H moiety (eqn (7)). Accordingly, gold-containing isobenzofulvene In 2 is compatible with these 13C and 2H-labeling experiments.
C–H carbon, and its resulting product 3a contained the 13C-content only at the alkyl C–H carbon (eqn (6)). Isobenzofulvene species In 1 was unlikely to occur here although it was observed in a ruthenium-catalyzed cycloisomerization.11 In the presence of D2O, we found that the resulting d1-3a contained deuterium (X = 0.29D) only at its alkenyl C–H moiety (eqn (7)). Accordingly, gold-containing isobenzofulvene In 2 is compatible with these 13C and 2H-labeling experiments.
 | (6) |
 | (7) |
Scheme 2 depicts the mechanisms of the two annulations. Internal 1,5-enynes 4 react with LAu+ to form cyclopropyl gold carbenes B (or B′) in two resonance forms; exo-(4+3)-annulations of species B′ with anthranils 2a likely yield gold-carbene species C that subsequently capture a second anthranil to yield products 5. This mechanism is essentially the same as that of their annulations with nitrosoarenes.12 Herein, a stepwise mechanism for the annulation of anthranils with 1,3-dipoles B/B′ is also likely to occur. Terminal 1,5-enyne 1a also generates cyclopropylgold carbene E because its cycloisomerization product 1a′ is also a 1-vinylindene derivative. We envisage that the cyclopropyl C–H proton of gold carbene E is acidic because of its proximity to the gold carbene functionality; the deprotonation with anthranil 2a generates cyclopropylidenylgold species F that undergoes a “methylenecyclopropane-trimethylenemethane” rearrangement,13 further generating gold-containing isobenzofulvene species In 2. An exo-(3+4)-annulation between fulvene In 2 and anthranil 2a affords the observed product 3a. The intermediacy of organogold species G is supported by 2H and 13C-labeling experiments.
 | ||
| Scheme 2 Plausible mechanisms for rearrangement and non-rearrangement. | ||
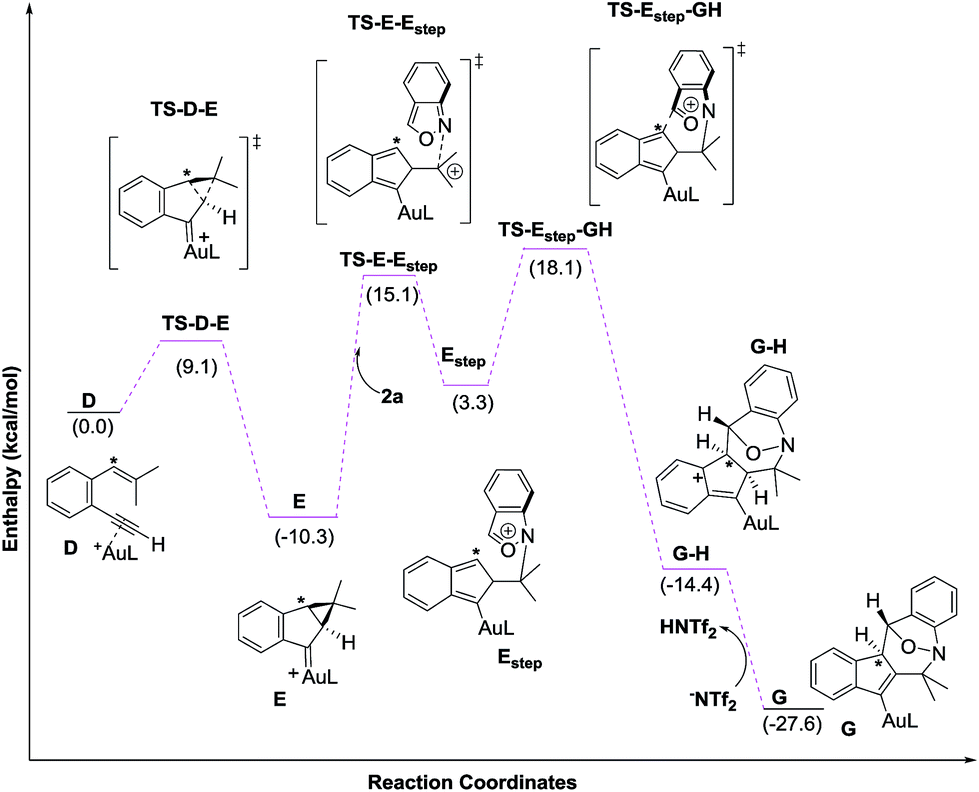
Density functional theory calculations were then performed to investigate the feasibility for the key steps D → G. Four possible paths 1–4 are considered; Path 1 is our proposed mechanism in Scheme 2. The energy profile is provided in Scheme 4. The formation of cyclopropylgold carbenes E from π-alkyne D has a low barrier of 9.1 kcal mol−1; the anion-promoted deprotonation of gold carbene E to form cyclopropylidenylgold species F is operable as the enthalpy cost is 16.9 kcal mol−1; the energy of species F is slightly higher than that of π-alkyne D by only 6.6 kcal mol−1. The remaining steps F → In 2 and In 2 → G are also operable as the transition states TS-F-In2 and TS-In2-G are close to π-alkyne D energy levels. One notable feature is that the enthalpy of transition state TS-F-ln2 is surprisingly smaller than that of species F by −0.3 kcal. This atypical case has similar precedents in the literature.14 This is because TS-F-In2 has less zero-point vibration energy than F, due to the loss of one degree of freedom in the transition state. This also means that F → In2 is a barrierless process.
We next examined the energy profiles in the (4+3) annulations (Path 2) between cyclopropyl gold carbenes E and anthranil 2a. The reaction proceeds in a stepwise manner. As shown in Scheme 5, the N-attack of anthranil 2a at gold carbene E produces species Estep by an endothermic process (H = 13.6 kcal mol−1); its activation energy is as high as 25.4 kcal mol−1. In the next step involving Estep → GH, the energy level of TS-Estep-GH is higher than that of 1,5-enyne D by 18.1 kcal mol−1. We conclude that Path 2 is not as feasible as Path 1 according to Scheme 5.
We also considered the remaining Paths 3 and 4, as depicted in Scheme 3. In Path 3, the deprotonation and ring rearrangement take place simultaneously (E → In2), in contrast to a stepwise process in Path 1 (E → F → In2). Despite multiple attempts, we were unable to locate the transition state for the direct E → In2 step, suggesting that Path 3 probably does not exist. In Path 4, a ring opening takes place initially (E → In2-H), followed by deprotonation (In2-H→In2). However, our calculations show that this pathway is unlikely to occur as we are unable to locate In2-H; all geometry optimizations lead to E.
 | ||
| Scheme 3 Four possible paths for the D → G transformation. | ||
 | ||
| Scheme 4 DFT calculation and energy profiles of Path 1. | ||
 | ||
| Scheme 5 DFT calculation and energy profiles of Path 2. | ||
Conclusions
Before this work, Au- and Pt-catalyzed annulations of anthranils with alkynes typically produced azacyclic products that cleaved the N–O bonds. To develop new (4+3)-annulations of alkyne-derived 1,3-dipoles15 with anthranils, we achieve stereoselective synthesis of two classes of tetrahydrobenzo[b]azepines using 1,5-enynes, anthranils and a gold catalyst. Internal 1,5-enynes deliver these cyclic nitroxy species without skeletal rearrangement while their terminal alkyne analogues afford distinct annulation products with skeletal rearrangement. To elucidate the mechanism of this rearrangement, 2H and 13C-labeling experiments were performed to identify the intermediates of gold-containing isobenzofulvene species, the formation of which is dependent on the presence of anthranils.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the Ministry of Education (MOE 106N506CE1) and Ministry of Science and Technology (MOST 107-3017-F-007- 002), Taiwan, for financial support of this work.Notes and references
- See selected reviews: (a) F. Hu and M. Szostak, Adv. Synth. Catal., 2015, 357, 2583–2614 CrossRef CAS; (b) P. Vitale and A. Scilimati, Curr. Org. Chem., 2013, 17, 1986–2000 CrossRef CAS; (c) A. L. Sukhorukov and S. L. Lorfe, Chem. Rev., 2011, 111, 5004–5041 CrossRef CAS PubMed; (d) P. Grunanger, P. Vita-Finzi and J. E. Dowling, in Chemistry of Heterocyclic Compounds, Part 2, ed. E. C. Taylor and P. Wipf, Wiley, New York, 1999, vol. 49, pp. 1–888 CrossRef; (e) P. Pevarello, R. Amici, M. G. Brasca, M. Villa and M. Virasi, Targets Heterocycl. Syst., 1999, 3, 301–339 CAS.
- (a) S. Gómez-Ayala, J. A. Castrillón, A. Palma, S. M. Leal, P. Escobar and A. Bahsas, Bioorg. Med. Chem., 2010, 18, 4721–4739 CrossRef PubMed; (b) W. L. Wan, J. B. Wu, F. Lei, X. L. Li, L. Hai and Y. Wu, Chin. Chem. Lett., 2012, 23, 1343–1346 CrossRef CAS; (c) O. Krebs, P. Kuenti, C. Michlig, K. Reuter, US Pat. No. 8,343,956 B2, 2013.
- For biological activity, see selected papers: (a) M. B. Dixon and Y. H. Lien, Ther. Clin. Risk Manage., 2008, 4, 1149–1155 CrossRef CAS; (b) E. K. Miller, K. Y. Chung, J. P. Hutcheson, D. A. Yates, S. B. Smith and B. J. Johnson, J. Anim. Sci., 2012, 90, 1317–1327 CrossRef CAS PubMed; (c) G. Aperis and P. Alivanis, Rev. Recent Clin. Trials, 2011, 6, 177–188 CrossRef CAS PubMed; (d) M. Gheorghiade, M. A. Konstam and J. C. Burnett, JAMA, J. Am. Med. Assoc., 2007, 297, 1332–1343 CrossRef CAS PubMed; (e) R. W. Schrier, P. Gross and M. Gheorghiade, N. Engl. J. Med., 2006, 355, 2099–2112 CrossRef CAS PubMed.
- (a) O. A. Ivanova, E. M. Budynina, Y. K. Grishin, I. V. Trushkov and P. V. Verteletskii, Angew. Chem., Int. Ed., 2008, 47, 1107–1110 CrossRef CAS PubMed; (b) O. A. Ivanova, E. M. Budynina, Y. K. Grishin, I. V. Trushkov and P. V. Verteletskii, Eur. J. Org. Chem., 2008, 5329–5335 CrossRef CAS; (c) L. K. B. Garve, M. Pawliczek, J. Wallbaum, P. G. Jones and D. B. Werz, Chem.–Eur. J., 2016, 22, 521–525 CrossRef CAS PubMed; (d) Z.-H. Wang, H.-H. Zhang, D.-M. Wang, P.-F. Xu and Y.-C. Luo, Chem. Commun., 2017, 53, 8521–8524 RSC.
- See selected reviews: (a) N. De and E. J. Yoo, ACS Catal., 2018, 8, 48–58 CrossRef CAS; (b) D. Garayalde and C. Nevado, ACS Catal., 2012, 2, 1462–1479 CrossRef CAS; (c) D. B. Huple, S. Ghorpade and R.-S. Liu, Adv. Synth. Catal., 2016, 358, 1348–1367 CrossRef CAS.
- (a) A.-H. Zhou, Q. He, C. Shu, Y.-F. Yu, S. Liu, T. Zhao, W. Zhang, X. Lu and L.-W. Ye, Chem. Sci., 2015, 6, 1265–1271 RSC; (b) X.-Y. Xiao, A.-H. Zhou, C. Shu, F. Pan, T. Li and L.-W. Ye, Chem.–Asian J., 2015, 10, 1854–1858 CrossRef CAS PubMed; (c) R. L. Sahani and R.-S. Liu, Angew. Chem., Int. Ed., 2017, 56, 1026–1030 CrossRef CAS PubMed; (d) W.-B. Shen, X.-Y. Xiao, Q. Sun, B. Zhou, X.-Q. Zhu, J.-Z. Yan, X. Lu and L.-W. Ye, Angew. Chem., Int. Ed., 2017, 56, 605–609 CrossRef CAS PubMed; (e) S. S. Giri and R.-S. Liu, Chem. Sci., 2018, 9, 2991–2995 RSC; (f) L. Li, T.-D. Tan, Y.-Q. Zhang, X. Liu and L.-W. Ye, Org. Biomol. Chem., 2017, 15, 8483–8492 RSC.
- (a) H. Jin, L. Huang, J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 794–797 CrossRef CAS PubMed; (b) H. Jin, B. Tian, X. Song, J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 12688–12692 CrossRef CAS PubMed; (c) R. L. Sahani and R.-S. Liu, Angew. Chem., Int. Ed., 2017, 56, 12736–12740 CrossRef CAS PubMed; (d) B. D. Mokar, P. D. Jadhav, Y. B. Pandit and R.-S. Liu, Chem. Sci., 2018, 9, 4488–4492 RSC.
- For gold-catalyzed enyne cycloisomerizations, see: (a) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed; (b) E. J. Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef PubMed; (c) A. S. K. Hashmi and M. Rudolph, Chem. Soc. Rev., 2008, 37, 1766–1775 RSC; (d) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449 CrossRef PubMed; (e) L. Zhang, j. Sun and S. A. Kozmin, Adv. Synth. Catal., 2006, 348, 2271–2296 CrossRef CAS.
- Crystallographic data of compounds 3a, 5b, 7a and 7e were deposited at the Cambridge Crystallographic Center; 3a, CCDC 1853703; 5b, CCDC 1853704; 7a, CCDC 1853705 and 7e, CCDC 1853706.†.
- (a) R. K. Kawade and R.-S. Liu, Angew. Chem., Int. Ed., 2017, 56, 2035–2039 CrossRef CAS PubMed; (b) P. Sharma, P. D. Jadhav, M. Skaria and R.-S. Liu, Org. Biomol. Chem., 2017, 15, 9389–9397 RSC.
- Isobenzofulvene In was the intermediate in the cycloisomerization of terminal 1,5-enyne 1a involving ruthenium vinylidene as the initial species. See: R. J. Madhushaw, C.-Y. Lo, C.-W. Hwang, M.-D. Su, H.-C. Shen, S. Pal, I. R. Shaikh and R.-S. Liu, J. Am. Chem. Soc., 2004, 126, 15560–15565 CrossRef CAS PubMed.
- For 1,n-enynes as 1,n-dipoles, see: (a) C.-H. Chen, Y.-C. Tsai and R.-S. Liu, Angew. Chem., Int. Ed., 2013, 52, 4599–4603 CrossRef CAS PubMed; (b) S. A. Gawade, S. Bhunia and R.-S. Liu, Angew. Chem., Int. Ed., 2012, 51, 7835–7838 CrossRef CAS PubMed; (c) A. Escribano-Cuesta, V. Lopez-Carrillo, D. Janssen and A. M. Echavarren, Chem.–Eur. J., 2009, 15, 5646–5650 CrossRef CAS PubMed; (d) D. B. Huple and R.-S. Liu, Chem. Commun., 2012, 48, 10975–10977 RSC.
- (a) M. Rule, R. F. Salinaro, D. R. Pratt and J. A. Berson, J. Am. Chem. Soc., 1982, 104, 2223–2228 CrossRef CAS; (b) R. F. Salinaro and J. A. Berson, J. Am. Chem. Soc., 1982, 104, 2228–2232 CrossRef CAS.
- (a) S. Wolfe, S. Hoz, C. K. Kim and K. Y. Yang, J. Am. Chem. Soc., 1990, 112, 4186–4191 CrossRef CAS; (b) W. C. Chen and C. H. Yu, Chem. Phys. Lett., 1997, 277, 245–251 CrossRef CAS; (c) H. F. Su, R. I. Kaiser and A. H. H. Chang, J. Chem. Phys., 2005, 122, 074320 CrossRef PubMed.
- For Au-containing all-carbon 1,3-dipoles, see selected examples: (a) G. Zhang and L. Zhang, J. Am. Chem. Soc., 2008, 130, 12598–12599 CrossRef CAS PubMed; (b) X. Huang and L. Zhang, J. Am. Chem. Soc., 2007, 129, 6398–6399 CrossRef CAS PubMed; (c) M. Schelweis, A. L. Dempwolff, F. Rominger and G. Helmchen, Angew. Chem., Int. Ed., 2007, 46, 5598–5601 CrossRef CAS PubMed; (d) T.-M. Teng, A. Das, D. B. Huple and R.-S. Liu, J. Am. Chem. Soc., 2010, 132, 12565–12567 CrossRef CAS PubMed; (e) T.-M. Teng and R.-S. Liu, J. Am. Chem. Soc., 2010, 132, 9298–9300 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1853703–1853706. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03619e |
| This journal is © The Royal Society of Chemistry 2019 |