
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reduction of organic azides by indyl-anions. Isolation and reactivity studies of indium–nitrogen multiple bonds†
Mathew D.
Anker
 ,
Matthias
Lein
and
Martyn P.
Coles
*
,
Matthias
Lein
and
Martyn P.
Coles
*
School of Chemical and Physical Sciences, Victoria University of Wellington, P. O. Box 600, Wellington, New Zealand. E-mail: martyn.coles@vuw.ac.nz
First published on 12th November 2018
Abstract
The synthesis of a new potassium–indyl complex, K[In(NONAr)] (NONAr = [O(SiMe2NAr)2]2−, Ar = 2,6-iPr2C6H3) and its reactivity with organic azides RN3 is reported. When R = 2,6-bis(diphenylmethyl)-4-tBu-phenyl, a dianionic alkyl-amide ligand is formed via C–H activation across a transient In–Nimide bond. Reducing the size of the R-group to 2,4,6-trimethylphenyl (mesityl, Mes) enables oxidation of the indium and elimination of dinitrogen to afford the imide species, K[In(NONAr)(NMes)]. The anion contains a short In–Nimide bond, shown computationally to contain appreciable multiple bond character. Reaction of isolated imides with an additional equivalent of azide (R = Mes, SiMe3) generates tetrazenido-indium compounds K[In(NONAr){κ-N,N′-N4(Mes)(R)-1,4}], shown by X-ray crystallography to contain planar InN4 heterocycles in the anion.
Introduction
Low valent compounds of the group 13 elements aluminium, gallium and indium exhibit a wide range of chemical reactivity.1 When present in the +1 oxidation-state,2 the electron-configuration of the metallic element implies the presence of a lone-pair of electrons in an ns orbital, prompting comparisons with neutral group 14 carbenoid species.3 Consequently, a rich area of coordination chemistry has developed, particularly focussed on the Ga(I) compounds.4 In addition to the ability of these compounds to behave as ligands, the lighter homologues are potent reducing agents, readily giving up two electrons to attain a more stable +3 oxidation-state. This reactivity has been harnessed in a wide range of chemical reactions,5 many of which are unique to this class of compound.6The most common members of this class of compound are represented by the general formula M(X), where the charge on the metal is balanced by a mono-anionic ligand, [X]−. These ligands are typically bulky, a requirement to limit (or prevent) aggregation and protect the metal from unwanted redox chemistry. This concept is best illustrated in the context of this work by the series M(BDIAr) (BDI = β-diketiminate, [HC(CMeNAr)]−, Ar = 2,6-iPr2C6H3), for which mono-metallic Al,5f Ga5d and In5a compounds are known.
A recent development in the chemistry of mono-valent aluminium and indium is to employ a dianionic ligand [X2]2− to support M(I) metal centres, generating an overall negative charge on the metal-containing species, [M(X2)]−. Whilst this class of compound has been well studied for gallium,4a,4b,7 the corresponding aluminyl-8 and indyl-9 anions have only been recently isolated (Fig. 1), and hence the chemistry of these compounds is in its infancy.10 Initial studies of [Al(X2)]− and [In(X2)]− systems indicate significant lone-pair character at the metal (from DFT calculations), with preliminary reactivity consistent with an Al(I) or In(I) nucleophile. We report in this contribution an investigation of the reducing potential of a new potassium indyl compound towards organic azides. This class of substrate was selected to target synthetically challenging indium imide species.
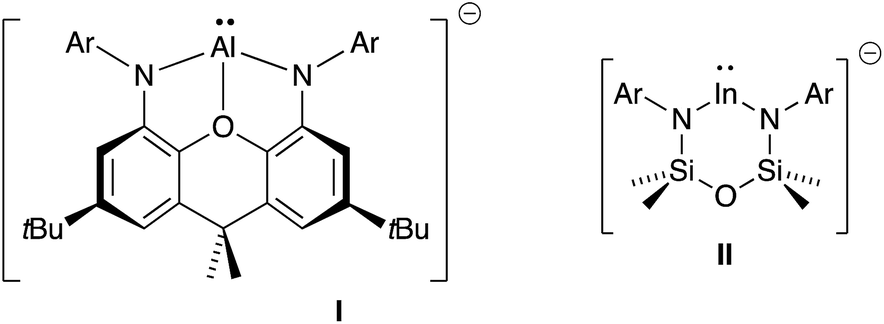 | ||
| Fig. 1 The first reported aluminyl- and indyl-anions (Ar = 2,6-iPr2C6H3). | ||
Monomeric group 13 metal imides [M(X)(NR)]n (X = ancillary ligand; M = Al, Ga or In; R = organic fragment; n = 1) are of academic and practical interest in several research areas. They formally contain metal–nitrogen multiple-bonds,11 and are implicated as intermediates in the formation of electronically important AlN, GaN and InN materials.12 Isolation of these compounds remains, however, synthetically challenging and only five examples have been crystallographically characterized since the first structural report in 2001 (III–VI, Fig. 2).13 Furthermore, these unusual compounds are restricted to a single example of an indium imide (VIb).13d Structural characterization showed that the In–N bond distance in VIb (1.928(3) Å) was significantly shorter than the range observed for monomeric In amides (2.05–2.09 Å), and that the C–In–N–C core adopted a trans-bent geometry. These data were consistent with In–N multiple bond character.
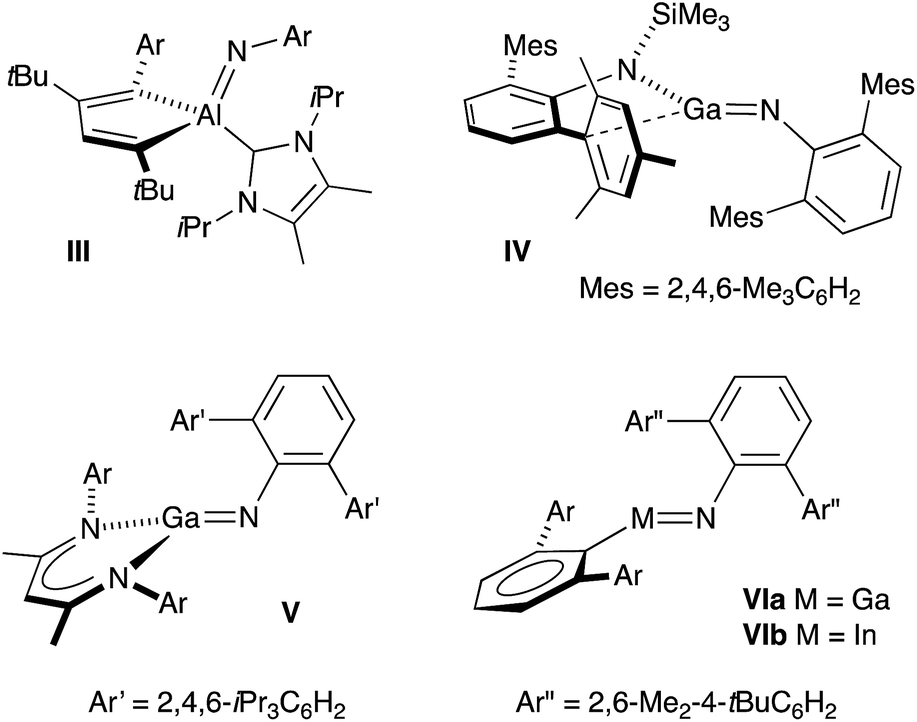 | ||
| Fig. 2 Structurally characterized monomeric compounds of Al, Ga and In containing unsupported M–Nimide bonds. | ||
The isolation of III–VI was achieved through kinetic stabilization of the M–Nimide bonds using sterically demanding ligands that prevent formation of ring- and cage-structures containing μ2- and μ3-NR ligands.14 A major limitation of this approach is that the bulk required to protect the imide bond from intermolecular aggregation renders it inaccessible to potential substrates, preventing any coherent study of its reactivity. It is encouraging to note, however, that in the absence of external substrates, intramolecular activation of ligand substituents can occur. This strongly suggests that once formed, the M–Nimide functional group is highly reactive.15
A general synthetic methodology to group 13 imides is the reduction of organic azides by a monovalent M(I) metal complexes (Scheme 1).13b–d,16 These reactions proceed with elimination of N2 and oxidation of the metal M(III), which occurs with a concurrent increase in the coordination number of the metal. It is of note that, if insufficient steric protection is provided during synthesis, in situ addition of unreacted azide in the reaction mixture to the transient ‘M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR’ bonds can occur (Scheme 1).17 For Al and Ga, this has enabled the isolation of metallotetrazenes VII (also referred to as metal-containing tetrazoles),17,18 which are rationalized as the product of a (2 + 3)-cycloaddition. This chemistry has not been extended to indium.
NR’ bonds can occur (Scheme 1).17 For Al and Ga, this has enabled the isolation of metallotetrazenes VII (also referred to as metal-containing tetrazoles),17,18 which are rationalized as the product of a (2 + 3)-cycloaddition. This chemistry has not been extended to indium.
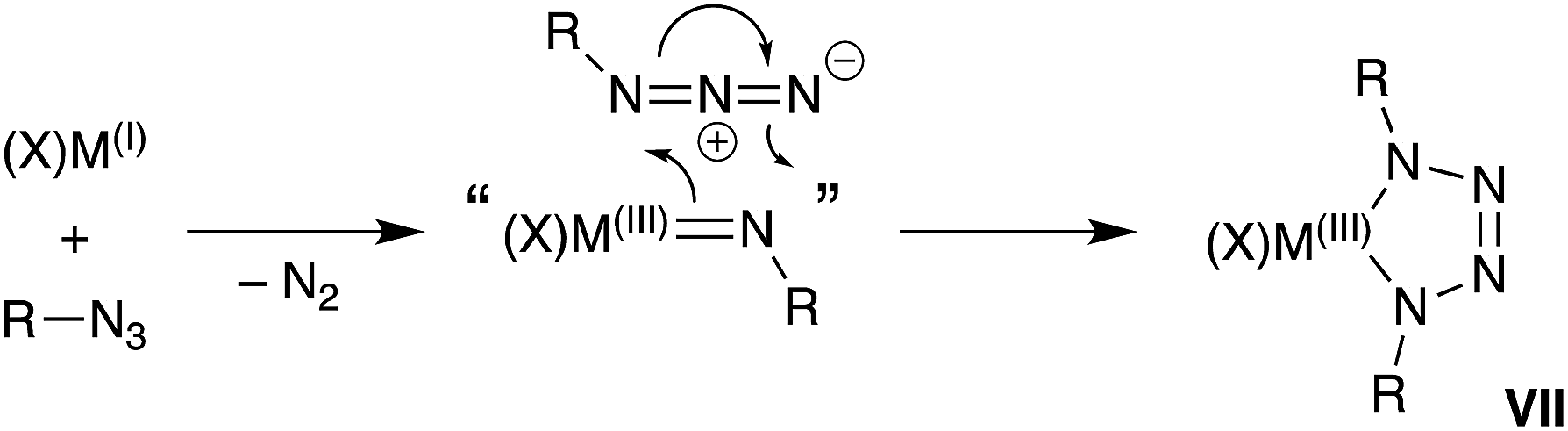 | ||
| Scheme 1 Synthesis of group 13 metal-imides from organic azides and proposed conversion to metallotetrazenes (VII). | ||
In this contribution we report a new potassium indyl salt and its controlled (stepwise) reactivity with organic azides. The initial products are characterised as a new class of anionic indium(III) imide, shown crystallographically and computationally to contain In–Nimide multiple bonds. Reaction of isolated examples with additional azide proceeds via a (2 + 3)-cycloaddition pathway to generate tetrazenido-indium salts, containing the first structurally characterized examples of the InN4-heterocycle.
Results and discussion
Synthesis of a new potassium indyl salt
The NONAr-ligand (NONAr = [O(SiMe2NAr)2]2−; Ar = 2,6-iPr2C6H3) stabilizes anionic indyl species as the indyllithium complex In(NONAr)(Li{THF}3), or in the ion-separated salt [K(crypt-222)][In(NONAr)] (crypt-222 = [2.2.2]-cryptand).9 Reactivity studies of these species have been hampered by their inherent instability, prompting us to examine an alternative source of the indyl anion. A modified procedure was therefore developed that avoids lithium reagents, and does not require the use of expensive crypt-222 reagents to stabilize the salt.The dipotassium salt, K2[(NONAr)(THF)n] (1), is readily obtained from the reaction of KH with the ligand pre-cursor (NONAr)H2.19 NMR analysis of the freshly prepared salt is consistent with incorporation of a single molecule of THF (n = 1), and the reagent can be used without further purification. However, crystallization from THF affords the Tris–THF adduct (n = 3) which forms a dimer [1_{THF}3]2 in the solid-state (Fig. S3†). The potassium salt can also be isolated as the bis-diethylether adduct K2[(NONAr)(Et2O)2] which forms a polymer in the solid-state (Fig. S4†), or as the 18-crown-6 (18-c-6) adduct that crystallizes as the dimer [K2{(NONAr)(18-c-6)}]2 (Fig. S5†).
The reaction of 1 with InCl3 affords a new indium-containing complex, 2. The NMR spectra show a symmetrical environment for the ligand backbone with a single peak for the SiMe2 groups. Although this is consistent with the three-coordinate species ‘In(NONAr)Cl’, elemental analysis was inconsistent with this formula and the compounds was therefore analysed by single-crystal X-ray diffraction (Fig. 3 and Table 1).
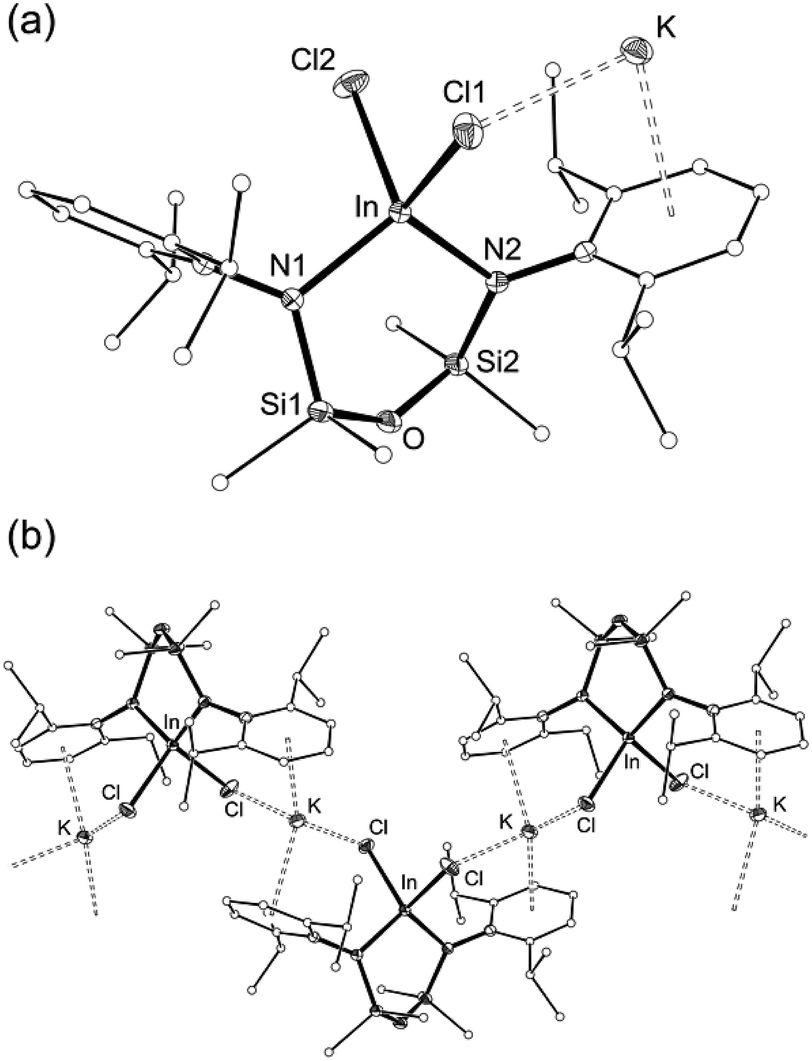 | ||
| Fig. 3 (a) Displacement ellipsoid plot (30% probability, carbon atoms reduced, hydrogen atoms omitted) of K[In(NONAr)Cl2] (2). (b) Segment of polymeric chain (⋯[2]3⋯), showing intermolecular K⋯Cl and K⋯C interactions. | ||
The asymmetric unit of 2 contains the four-coordinate indium anion [In(NONAr)Cl2]− (Fig. 3a). The charge is balanced by a potassium atom that forms π–aryl interactions with an Ar-groups of the diamide ligand, and has a close-contact with a chloride ligand. The crystal structure shows a 1-D polymer [2]n with additional interactions between the K-atom and aryl-/chloride groups from neighbouring molecules (Fig. 3b).
Reduction of 2 with two equivalents of potassium yields the new indyl compound K[In(NONAr)] (3) as a hexane soluble, yellow solid. The 1H NMR spectrum of 3 displays a single resonance for the SiMe2 substituents, indicative of a symmetrical environment for the NONAr-ligand. There are no resonances attributable to In–H hydride ligands (Fig. S10†).20
The molecular structure of 3 (Fig. 4) is reminiscent of the recently reported aluminyl anion.8 The asymmetric unit consists of two [In(NONAr)]− anions linked by potassium cations that are involved in π–aryl interactions to flanking Ar groups (C⋯K distances 3.109(4)–3.346(3) Å). The In⋯In separation is 4.710(3) Å, with In–N bond lengths (2.182(3)–2.240(3) Å) consistent with anionic In(I) metal centres.9 There are no bonding interactions between the indium and the oxygen-atom of the backbone (In⋯O = 3.557(2) and 3.577(2) Å), consistent with a strictly two-coordinate indyl anion.
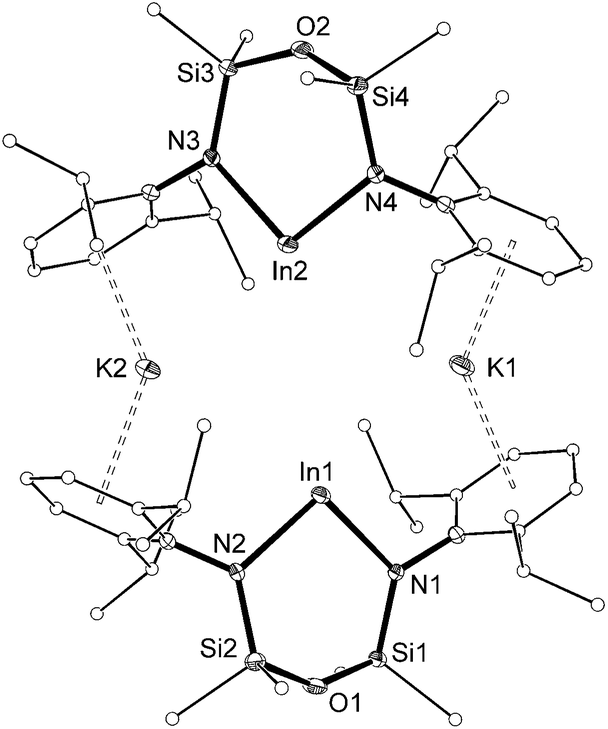 | ||
| Fig. 4 Displacement ellipsoid plot (30% probability, carbon atoms reduced, hydrogen atoms omitted) of [K{In(NONAr)}]2 ([3]2). | ||
Reactivity of the indyl anion with organic azides
Our initial attempt to isolate an imide from 3 was made using an equimolar amount of the sterically demanding 2,6-bis(diphenylmethyl)-4-tBu-phenyl azide (Ar‡N3, Scheme 2). The reagents were combined at −78 °C, allowed to warm to room temperature and stir for 1 h. The 1H NMR spectrum of colourless crystals 4 obtained on workup showed a loss symmetry for the NON-backbone (δH 0.58 and 0.45, 6H, SiMe2) and a reduction in the intensity of the CHPh2 resonance (δH 5.56, 1H). A new peak at 3.24 ppm (with no corresponding carbon resonance in HSQC experiments, Fig. S15†) is assigned to an NH functionality. | ||
| Scheme 2 Structurally characterized monomeric complexes of Al, Ga and In containing unsupported M–Nimide bonds. | ||
The structure of 4 was determined by X-ray diffraction and shows a κ2-C,N–N(H){C6H2(CPh2)(CHPh2)-tBu-2,6,4} ligand chelating to a four-coordinate, anionic indium(III) centre (Fig. 5 and Table 2). The potassium counterion is located between two aryl-substituents of the alkyl-amido ligand (C⋯K distances 3.148(2)–3.581(2) Å). The In–N3 distance (2.1855(14) Å) is consistent with a single bond to an amide nitrogen, and the location of electron density assigned to H1x in the difference map further supports this conclusion. Similar intramolecular activation has been observed at Al21 and Sn22 amido derivatives of the Ar‡ group, although in these instances the mechanism leading to the products are not known. We propose that formation of 4 occurs via intramolecular addition of a methine CHPh2 across the reactive ‘InNAr‡’ bond of a transient indium imide.15b
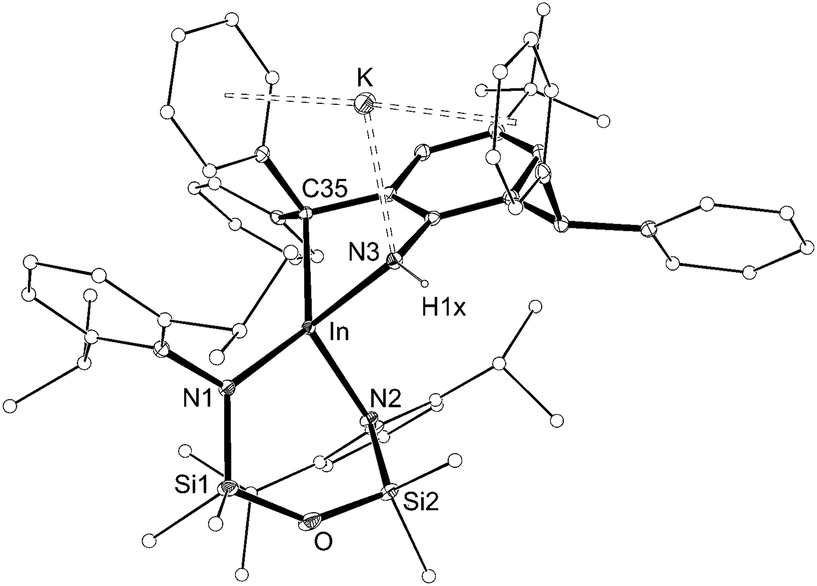 | ||
| Fig. 5 Displacement ellipsoid plot (30% probability, carbon atoms reduced, hydrogen atoms omitted) of K[In(NONAr)(κ2-C,N–N(H){C6H2(CPh2)(CHPh2)-tBu-2,6,4})] (4). | ||
| In–N1 | 2.1674(14) | In–N2 | 2.1676(14) |
| In–N3 | 2.1855(14) | In–C35 | 2.2875(16) |
| N1–In–N2 | 102.24(5) | N1–In–N3 | 112.64(5) |
| N2–In–N3 | 106.25(5) | N1–In–C35 | 132.97(6) |
| N2–In–C35 | 119.53(6) | N3–In–C35 | 77.12(6) |
To mitigate complications from ligand activation, the reaction was repeated with the sterically less intrusive 2,4,6-trimethylphenyl azide (mesityl azide, MesN3) under the conditions described above (Scheme 2). Concentration of the resulting solution and storage at −30 °C gave deep orange crystals (5). The 1H NMR spectrum is consistent with a symmetrical NON-backbone (δH 0.33, 12H, SiMe2) and a freely rotating Mes group (δH 1.57, 6H, 2,6-C6H2Me2).
The solid-state structure of 5 confirmed the formation of an imido–indium complex (Fig. 6 and Table 3). The compound crystallizes as the non-symmetry related dimer [5]2, with the potassium counter-ions involved in π–aryl interactions with Ar and Mes substituents (C⋯K distances 3.102(3)–3.296(3) Å). The indium centres are distorted trigonal planar (Σangles 358.7°), with In–Nimide bond lengths of 1.986(2) and 1.999(2) Å to N3 and N6, respectively. These represent an average shortening of 3.6% compared with the In–N bonds in the three coordinate amide In(NHMes*)3 (Mes* = 2,4,6-tBu3C6H2),23 although are longer than the neutral imide complex VIb (1.928(3) Å)13d in which the metal is two-coordinate. The imido-nitrogen atoms are bent, with In–N–C angles of 123.9(2)° and 123.8(2)° at N3 and N6, respectively.
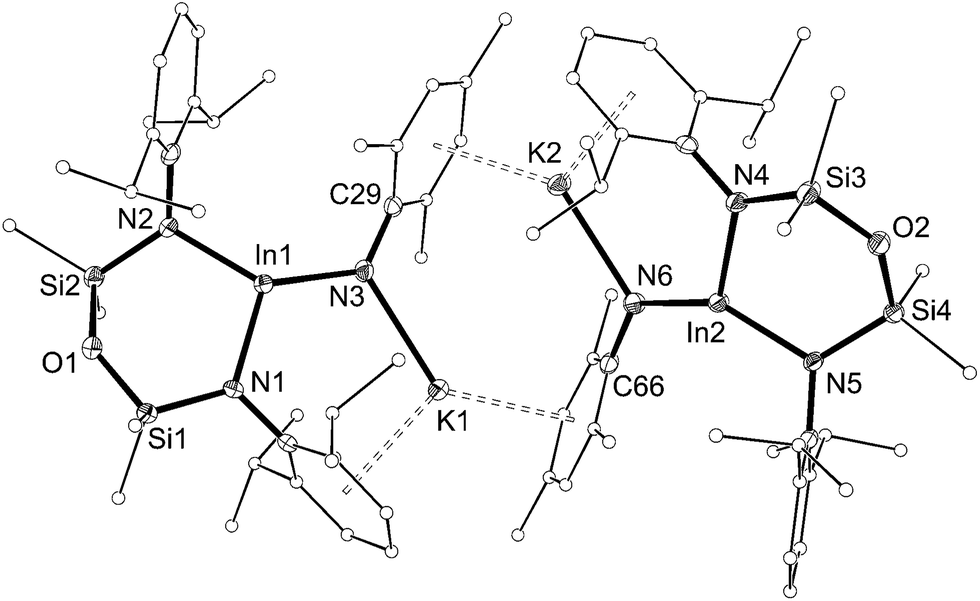 | ||
| Fig. 6 Displacement ellipsoid plot (30% probability, carbon atoms reduced, hydrogen atoms omitted) of [K{In(NONAr)(NMes)}]2 ([5]2). | ||
| Compound | [5]2 | 6 |
|---|---|---|
| a [5]2 = In1, 6 = In. | ||
| Ina–N1 | 2.090(2) | 2.1026(16) |
| Ina–N2 | 2.077(2) | 2.1152(16) |
| Ina–N3 | 1.986(2) | 1.9907(18) |
| In2–N4 | 2.097(2) | — |
| In2–N5 | 2.082(2) | — |
| In2–N6 | 1.999(2) | — |
| N1–Ina–N2 | 104.85(10) | 100.36(6) |
| N1–Ina–N3 | 115.80(10) | 125.54(7) |
| N2–Ina–N3 | 138.01(10) | 130.25(7) |
| N4–In2–N5 | 102.70(9) | — |
| N4–In2–N6 | 115.29(10) | — |
| N5–In2–N6 | 140.66(10) | — |
The length of the In–Nimide bonds in 5 may be influenced by interactions with the potassium cations (N3–K1 2.661(3) Å, N6–K2 2.646(3) Å), which are located closer to the nitrogen atom than in other potassiated imides (range 2.732(3)–3.069(11) Å).24 To isolate the imido-bond from N⋯K interactions, 5 was crystallized in the presence of [2.2.2]-cryptand. The crystal structure of the product confirmed formation of the separated ion-pair [K(crypt-222)][In(NONAr)(NMes)] (6, Fig. 7). Interestingly the In–Nimide bond length (1.9907(18) Å) remains unchanged (within 3σ) and the In–N–C angle is still bent (In–N3–C29 127.43(14)°), suggesting that the N⋯K interactions in 5 have little effect on the structural component of the indium–nitrogen bond.
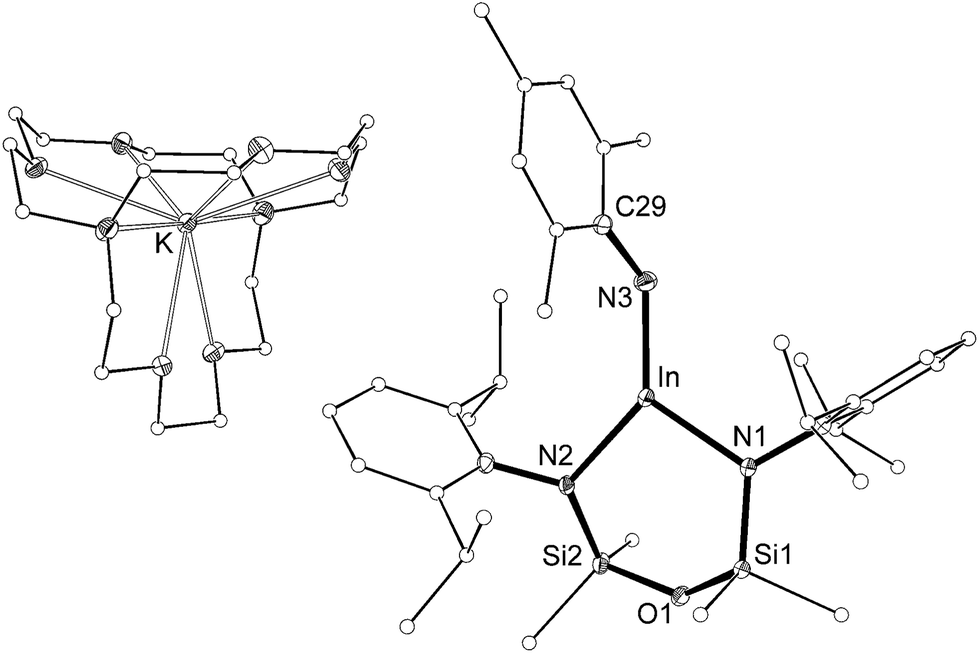 | ||
| Fig. 7 Displacement ellipsoid plot (30% probability, carbon atoms reduced, hydrogen atoms omitted) of [K(crypt-222)][In(NONAr)(NMes)] (6). | ||
Computational analysis of In–Nimide bond
Optimisation and subsequent analysis using density functional theory confirmed the multiple-bond character of the In–Nimide unit in 5 and the isolated anion [In(NONAr)(NMes)]− from compound 6 ([6]−) (see ESI†). This is clearly demonstrated from the increased Wiberg bond orders for this group (5, 0.59; [6]− 0.71) that are substantially higher than the In–N bonds to the NONAr-ligand (5, 0.22/0.29; [6]−, 0.25).To explore the nature of this bond in more detail, plausible resonance structures analogous to those examined for VIb,13d were submitted for NBO calculations (A–D, Scheme 3). The quality criterion used to compare results calculated for the different resonance structures is the percentage of non-Lewis (n-L) components, where a lower non-Lewis percentage indicates a better representation. Resonance form C did not yield a viable solution by this method. However, structures A (triple bond), B (double bond) and D (single bond) all have a low n-L contribution to their overall NBO solution (Table S2†). The best localisations were achieved for multiple-bonded A and B (n-L = 1.965% and 1.990% respectively), while D was only slightly less well localised (n-L = 2.094%). Although it is difficult to extract a precise numerical value for the multiplicity of the In–Nimide bond from these computational data, the results confirm a strong multiple-bond component in accordance with crystallographic results, and observed reactivity (vide infra).
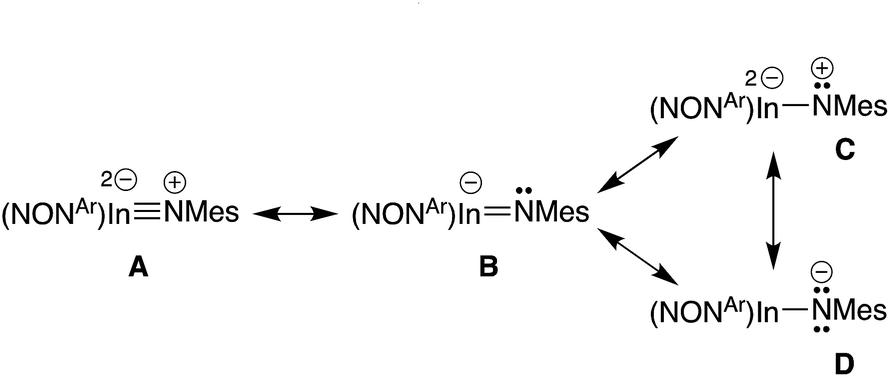 | ||
| Scheme 3 Possible resonance structures for indium imide anions. | ||
Quantum Theory of Atoms In Molecules (QTAIM) analysis of 5 and [6]− has been performed. The bond critical point between the In and Nimide bonds have a low ellipiticity (ε) of 0.079 and 0.072 for 5 and [6]−, respectively, inconsistent with a conventional InN double where a larger value (>0.25) is predicted. These data suggest a non-elliptical cross-section of electron density in the In–N bond vector. This is consistent with a model proposed by Power and co-workers in related gallium imides related to VIa,13b in which an organogallium(I) species interacts with a singlet nitrene, with incomplete donation of electron pairs (represented by dashed lines in Fig. 8).
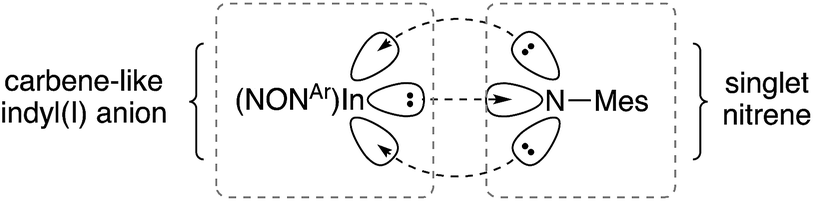 | ||
| Fig. 8 Proposed bonding model for In–Nimide interaction in [6]−, based on a model proposed Power and by co-workers (see ref. 13b). | ||
Reactivity of indium imides with organic azides
As the imido-mesityl substituents are considerably less bulky than the terphenyl groups in IV–VI, we wished to determine whether the In–Nimide bond in 5 was available for controlled reactivity studies. Inspired by the proposed formation of metallotetrazenes from group 13 metal imides (Scheme 1), we investigated the reactivity of isolated samples of 5 with organic azides RN3 (R = Mes, SiMe3).Addition of a solution of RN3 to an orange solution of 5 at room temperature resulted in decolorization over an approximate 5 minute period (Scheme 4). NMR spectra show changes corresponding to the addition of mesityl (7) or SiMe3 (8) groups, consistent with their incorporation in the product. In agreement with these data, the composition of the products as the first examples of indium tetrazenido compounds was confirmed by X-ray crystallography (Fig. 9 and 10, Table 4). These results demonstrate that in this instance, the potassium atoms in 5 do not adversely affect the reactivity of the In–Nimide bond. We therefore propose that 5 behaves chemically as ‘InNMes’, and that the formation of the unsymmetrical tetrazene 8 strongly endorses the previously assumed (2 + 3)-cycloaddition pathway of transient imides.
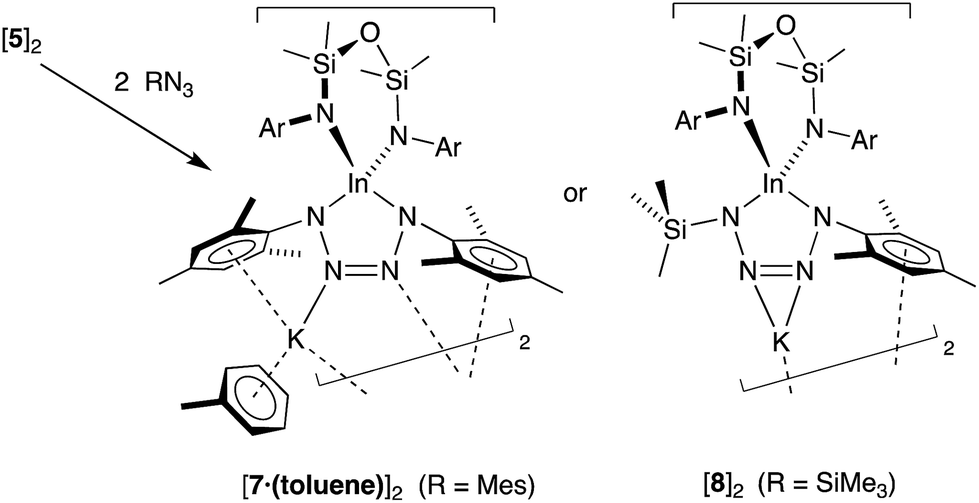 | ||
| Scheme 4 Reaction of K[In(NONAr)(NMes)] (5) with RN3 (R = Mes, SiMe3). | ||
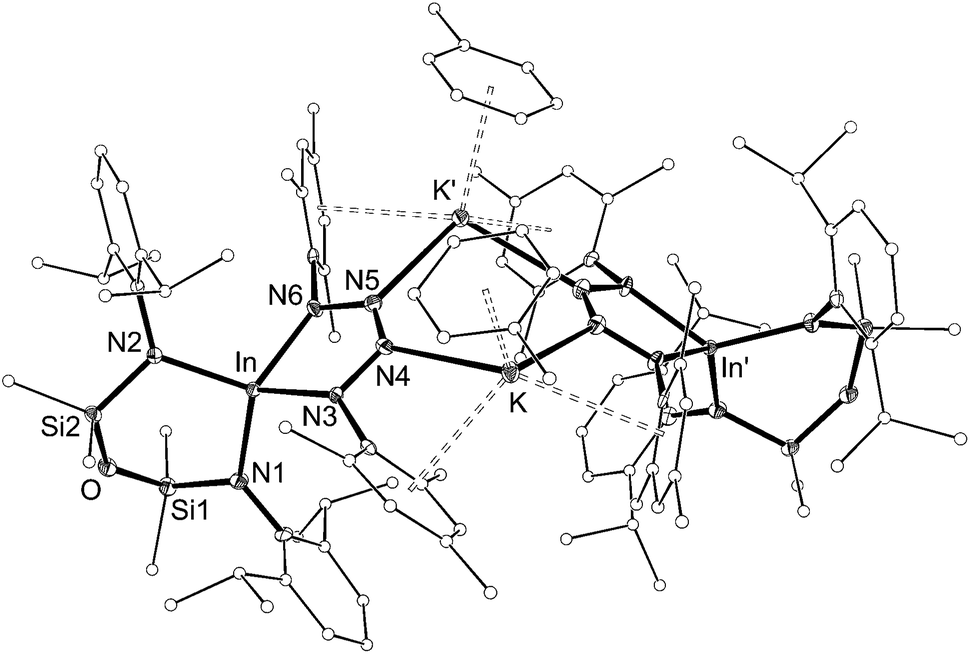 | ||
| Fig. 9 Displacement ellipsoid plot (30% probability, carbon atoms reduced, hydrogen atoms omitted) of [K{In(NONAr)(κ-N,N′-N4{Mes}2–1,4)(C7H8)}]2 ([7·(toluene)]2). | ||
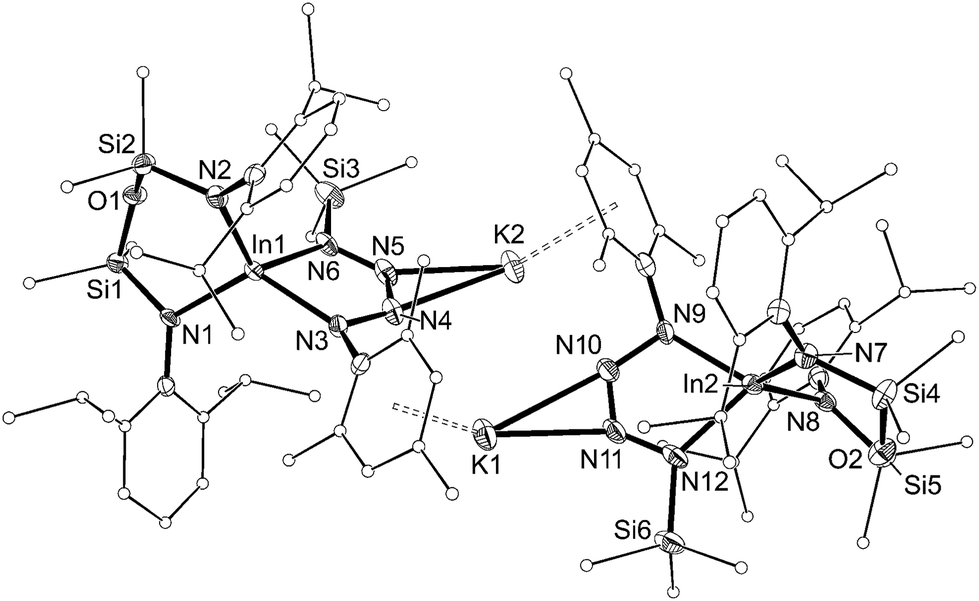 | ||
| Fig. 10 Displacement ellipsoid plot (30% probability, carbon atoms reduced, hydrogen atoms omitted) of [K{In(NONAr)(κ-N,N′-N4{Mes}{SiMe3}-1,4)}]2 ([8]2). | ||
| Compound | [7·(toluene)]2 | [8]2 |
|---|---|---|
| a [7·(toluene)]2 = In1, 6 = In. | ||
| Ina–N1 | 2.143(4) | 2.137(5) |
| Ina–N2 | 2.134(4) | 2.117(5) |
| Ina–N3 | 2.127(4) | 2.116(5) |
| Ina–N6 | 2.140(4) | 2.147(5) |
| In2–N7 | — | 2.091(6) |
| In2–N8 | — | 2.132(5) |
| In2–N9 | — | 2.117(6) |
| In2–N12 | — | 2.146(6) |
| N3–N4 | 1.391(6) | 1.398(8) |
| N4–N5 | 1.273(6) | 1.277(9) |
| N5–N6 | 1.376(6) | 1.373(9) |
| N9–N10 | — | 1.402(8) |
| N10–N11 | — | 1.272(9) |
| N11–N12 | — | 1.347(10) |
| N1–Ina–N2 | 103.12(16) | 106.1(2) |
| N3–Ina–N6 | 74.67(16) | 76.0(2) |
| N7–In2–N8 | 105.8(2) | |
| N9–In2–N12 | 74.9(2) | |
The dimesityl derivative [7·(toluene)]2 lies on a two-fold rotation axis that forms a dimeric unit, with K⋯π–aryl interactions between Mes-groups and an incorporated toluene molecule that encapsulates the cation (C⋯K distances 3.006(5)–3.993(5) Å). Crystallization of [7·(toluene)]2 in the presence of 18-crown-6 (18-c-6) disrupts dimer formation to afford [K(18-c-6)][In(NONAr)(N4{Mes}2–1,4)] (7 (18-c-6)). This salt forms a contact ion-pair linked by K⋯N interactions to the two central nitrogen atoms of the InN4-tetrazene ring (2.820(2) Å and 2.939(2) Å, Fig. S27†). The trimethylsilyl derivative K[In(NONAr)(N4{Mes}{SiMe3}-1,4)] also crystallizes as the dimer [8]2, with the potassium cations linking non-symmetry related units through a combination of K⋯N (2.641(6)–2.913(6) Å) and K⋯π–aryl (3.024(9)–3.264(8) Å) interactions.
In all cases the anion comprises two approximately orthogonal rings fused at a four-coordinate indium centre. The metallotetrazene rings are essentially planar, with nitrogen–nitrogen bond lengths indicating double-bond character between atoms in the 3- and 4-positions of the heterocycle (see VII, Scheme 1). These parameters are consistent with neutral aluminum-17,18 and gallium-18b derivatives, although we note that compounds 7 and 8 represent the first structurally characterized indium compounds containing the tetrazenide ligand, and are unique examples where the MN4-heterocycle is a component of an anionic species.
Conclusions
This work describes the first detailed reactivity study of an indyl-anion. We confirm that the negative charge associated with the indium centre does not adversely affect their ability to act as a reducing agent towards organic azides. The reactions proceed cleanly with elimination of dinitrogen and oxidation of the indium(I) to In(III). The isolated compounds have been structurally verified as a new class of anionic indium imide, shown computationally to contain In–Nimide multiple bonds. Furthermore, we demonstrate that the reduced size of the imide-substituent in this work compared with previous examples allows access to the In–Nimide bond, demonstrated by the reaction with additional equivalents of azide. The products from this (2 + 3)-cycloaddition are the first time that this reaction has been extended to indium, and crystallographic analysis confirms a planar InN4-heterocycle as a component of the anion.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed; (b) S. González-Gallardo, T. Bollermann, R. A. Fischer and R. Murugavel, Chem. Rev., 2012, 112, 3136–3170 CrossRef PubMed; (c) H. Schnöckel, Chem. Rev., 2010, 110, 4125–4163 CrossRef PubMed.
- (a) Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113–115 CrossRef CAS PubMed; (b) T. B. Marder, Science, 2006, 314, 69–70 CrossRef CAS PubMed.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
- (a) C. Jones, D. P. Mills, R. P. Rose and A. Stasch, Dalton Trans., 2008, 4395–4408 RSC; (b) G. Jin, C. Jones, P. C. Junk, A. Stasch and W. D. Woodul, New J. Chem., 2008, 32, 835–842 RSC; (c) C. Jones, P. C. Junk, J. A. Platts and A. Stasch, J. Am. Chem. Soc., 2006, 128, 2206–2207 CrossRef CAS PubMed.
- (a) M. S. Hill and P. B. Hitchcock, Chem. Commun., 2004, 1818–1819 RSC; (b) M. S. Hill, P. B. Hitchcock and R. Pongtavornpinyo, Dalton Trans., 2005, 273–277 RSC; (c) M. S. Hill, P. B. Hitchcock and R. Pongtavornpinyo, Angew. Chem., Int. Ed., 2005, 44, 4231–4235 CrossRef CAS PubMed; (d) N. J. Hardman, B. E. Eichler and P. P. Power, Chem. Commun., 2000, 1991–1992 RSC; (e) M. Stender and P. P. Power, Polyhedron, 2002, 21, 525–529 CrossRef CAS; (f) C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS.
- (a) M. S. Hill, P. B. Hitchcock and R. Pongtavornpinyo, Science, 2006, 311, 1904–1907 CrossRef CAS PubMed; (b) A. V. Protchenko, D. Dange, J. R. Harmer, C. Y. Tang, A. D. Schwarz, M. J. Kelly, N. Phillips, R. Tirfoin, K. H. Birjkumar, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Nat. Chem., 2014, 6, 315 CrossRef CAS PubMed.
- (a) R. J. Baker, R. D. Farley, C. Jones, M. Kloth and D. M. Murphy, J. Chem. Soc., Dalton Trans., 2002, 3844–3850 RSC; (b) R. J. Baker, C. Jones and J. A. Platts, J. Am. Chem. Soc., 2003, 125, 10534–10535 CrossRef CAS PubMed; (c) R. J. Baker, C. Jones and M. Kloth, Dalton Trans., 2005, 2106–2110 RSC; (d) S. Aldridge, R. J. Baker, N. D. Coombs, C. Jones, R. P. Rose, A. Rossin and D. J. Willock, Dalton Trans., 2006, 3313–3320 RSC; (e) R. J. Baker, C. Jones, D. P. Mills, D. M. Murphy, E. Hey-Hawkins and R. Wolf, Dalton Trans., 2006, 64–72 RSC; (f) O. Bonello, C. Jones, A. Stasch and W. D. Woodul, Organometallics, 2010, 29, 4914–4922 CrossRef CAS; (g) E. S. Schmidt, A. Jockisch and H. Schmidbaur, J. Am. Chem. Soc., 1999, 121, 9758–9759 CrossRef CAS.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed.
- R. J. Schwamm, M. D. Anker, M. Lein, M. P. Coles and C. M. Fitchett, Angew. Chem., Int. Ed., 2018, 57, 5885–5887 CrossRef CAS PubMed.
- A. Hinz and F. Breher, Angew. Chem., Int. Ed., 2018, 57, 8818–8820 CrossRef CAS PubMed.
- (a) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed; (b) M. F. Lappert, P. P. Power, A. V. Protchenko and A. L. Seeber, Metal Amide Chemistry, Wiley, New York, 2009 Search PubMed.
- M. A. Malik, M. Afzaal and P. O'Brien, Chem. Rev., 2010, 110, 4417–4446 CrossRef CAS PubMed.
- (a) J. Li, X. Li, W. Huang, H. Hu, J. Zhang and C. Cui, Chem. - Eur. J., 2012, 18, 15263–15266 CrossRef CAS PubMed; (b) R. J. Wright, M. Brynda, J. C. Fettinger, A. R. Betzer and P. P. Power, J. Am. Chem. Soc., 2006, 128, 12498–12509 CrossRef CAS PubMed; (c) N. J. Hardman, C. Cui, H. W. Roesky, W. H. Fink and P. P. Power, Angew. Chem., Int. Ed., 2001, 40, 2172–2174 CrossRef CAS; (d) R. J. Wright, A. D. Phillips, T. L. Allen, W. H. Fink and P. P. Power, J. Am. Chem. Soc., 2003, 125, 1694–1695 CrossRef CAS PubMed.
- A. Y. Timoshkin, Coord. Chem. Rev., 2005, 249, 2094–2131 CrossRef CAS.
- (a) T. Chu, S. F. Vyboishchikov, B. M. Gabidullin and G. I. Nikonov, J. Am. Chem. Soc., 2017, 139, 8804–8807 CrossRef CAS PubMed; (b) H. Zhu, J. Chai, V. Chandrasekhar, H. W. Roesky, J. Magull, D. Vidovic, H.-G. Schmidt, M. Noltemeyer, P. P. Power and W. A. Merrill, J. Am. Chem. Soc., 2004, 126, 9472–9473 CrossRef CAS PubMed; (c) S. Schulz, L. Häming, R. Herbst-Irmer, H. W. Roesky and G. M. Sheldrick, Angew. Chem., Int. Ed. Engl., 1994, 33, 969–970 CrossRef.
- C. Cui, S. Köpke, R. Herbst-Irmer, H. W. Roesky, M. Noltemeyer, H.-G. Schmidt and B. Wrackmeyer, J. Am. Chem. Soc., 2001, 123, 9091–9098 CrossRef CAS PubMed.
- H. Zhu, Z. Yang, J. Magull, H. W. Roesky, H.-G. Schmidt and M. Noltemeyer, Organometallics, 2005, 24, 6420–6425 CrossRef CAS.
- (a) C. Cui, H. W. Roesky, H.-G. Schmidt and M. Noltemeyer, Angew. Chem., Int. Ed., 2000, 39, 4531–4533 CrossRef CAS; (b) N. J. Hardman and P. P. Power, Chem. Commun., 2001, 1184–1185 RSC.
- D. B. Leznoff, G. Mund, K. C. Jantunen, P. H. Bhatia, A. J. Gabert and R. J. Batchelor, J. Nucl. Sci. Technol., 2002, 39, 406–409 CrossRef.
- (a) R. J. Baker, C. Jones, M. Kloth and J. A. Platts, Angew. Chem., Int. Ed., 2003, 42, 2660–2663 CrossRef CAS PubMed; (b) O. T. Beachley, S. H. L. Chao, M. R. Churchill and R. F. See, Organometallics, 1992, 11, 1486–1491 CrossRef CAS.
- E. W. Y. Wong, D. Dange, L. Fohlmeister, T. J. Hadlington and C. Jones, Aust. J. Chem., 2013, 66, 1144–1154 CAS.
- T. J. Hadlington and C. Jones, Chem. Commun., 2014, 50, 2321–2323 RSC.
- J. S. Silverman, C. J. Carmalt, A. H. Cowley, R. D. Culp, R. A. Jones and B. G. McBurnett, Inorg. Chem., 1999, 38, 296–300 CrossRef CAS.
- (a) N. H. Anderson, J. Xie, D. Ray, M. Zeller, L. Gagliardi and S. C. Bart, Nat. Chem., 2017, 9, 850 CrossRef CAS PubMed; (b) L. A. Solola, A. V. Zabula, W. L. Dorfner, B. C. Manor, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 6928–6931 CrossRef CAS PubMed; (c) K. C. Mullane, A. J. Lewis, H. Yin, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2014, 53, 9129–9139 CrossRef CAS PubMed; (d) G. Bai, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Organometallics, 2002, 21, 2789–2792 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, copies of NMR spectra, details of X-ray experiments and additional figures, xyz-coordinates from DFT calculations. CCDC 1863650–1863661. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04078h |
| This journal is © The Royal Society of Chemistry 2019 |