
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiahelicene-based inherently chiral films for enantioselective electroanalysis†
Serena
Arnaboldi
 *a,
Silvia
Cauteruccio
*a,
Sara
Grecchi
a,
Tiziana
Benincori
b,
Massimo
Marcaccio
c,
Alessio Orbelli
Biroli
d,
Giovanna
Longhi
e,
Emanuela
Licandro
a and
Patrizia Romana
Mussini
*a
*a,
Silvia
Cauteruccio
*a,
Sara
Grecchi
a,
Tiziana
Benincori
b,
Massimo
Marcaccio
c,
Alessio Orbelli
Biroli
d,
Giovanna
Longhi
e,
Emanuela
Licandro
a and
Patrizia Romana
Mussini
*a
aDipartimento di Chimica, Università degli Studi di Milano, Via Golgi, 19, 20133 Milano, Italy. E-mail: serena.arnaboldi@unimi.it; silvia.cauteruccio@unimi.it; patrizia.mussini@unimi.it
bDipartimento di Scienza e Alta Tecnologia, Università degli Studi dell’Insubria, Via Valleggio 11, 22100 Como, Italy
cDipartimento di Chimica “Giacomo Ciamician”, Alma Mater Studiorum Università di Bologna, Via Selmi 2, 40126 Bologna, Italy
dIstituto di Scienze e Tecnologie Molecolari del CNR (CNR-ISTM), SmartMatLab Centre, Via Golgi, 19, 20133 Milano, Italy
eDipartimento di Medicina Molecolare e Traslazionale, Università degli Studi di Brescia, Viale Europa 11, 25123 Brescia, Italy
First published on 29th November 2018
Abstract
Chiral electroanalysis could be regarded as the highest recognition degree in electrochemical sensing, implying the ability to discriminate between specular images of an electroactive molecule, particularly in terms of significant peak potential difference. A groundbreaking strategy was recently proposed, based on the use of “inherently chiral” molecular selectors, with chirality and key functional properties originating from the same structural element. Large differences in peak potentials have been observed for the enantiomers of different chiral molecules, also of applicative interest, using different selectors, all of them based on atropisomeric biheteroaromatic scaffolds of axial stereogenicity. However, helicene systems also provide inherently chiral building blocks with attractive features. In this paper the enantiodiscrimination performances of enantiopure inherently chiral films obtained by electrooxidation of a thiahelicene monomer with helicoidal stereogenicity are presented for the first time. The outstanding potentialities of this novel approach are evaluated towards chiral probes with different chemical nature and bulkiness, in comparison with a representative case of the so far exploited class of inherently chiral selectors with axial stereogenicity. It is also verified that the high enantiodiscrimination ability holds as well for electron spins, as for atropisomeric selectors.
Introduction
In the ongoing search for efficient tools to achieve enantioselective electrochemistry for analytical, preparative and advanced device applications, a major target is to discriminate the enantiomers of chiral probes in terms of significant potential differences, in order to recognize and possibly quantify and/or manage each enantiomer with no preliminary separation steps.1 In this perspective, outstanding results have been recently obtained by different approaches having in common the use of “inherently chiral” selectors, either electrode surfaces2–7 or media.8,9 According to a functional definition,2,3 in inherently chiral molecules and materials both chirality and specific properties must originate from the same structural element, so that they are strictly linked. In the above works, this has been achieved by the insertion of atropisomeric stereogenic elements, introducing a tailored torsion in the main selector backbone. This strategy was implemented on one side in heterocycle-based electroactive monomers, to be converted by electrochemical or chemical oxidation in a mixture of open as well as closed oligomers, exploited as inherently chiral electrode surfaces (employing 3,3′-bibenzothiophene,1–4,6 3,3′-bithiophene,5 or 2,2′-biindole atropisomeric systems,7Fig. 1a).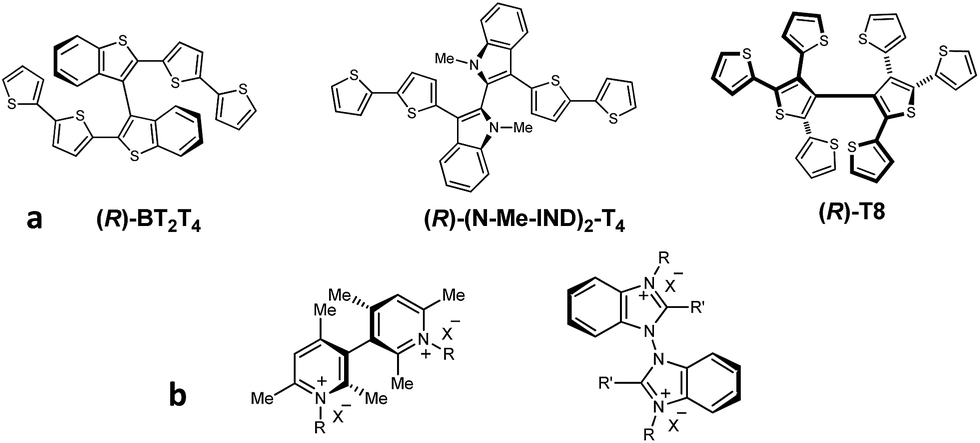 | ||
| Fig. 1 (a): Inherently chiral selectors with stereogenic axis, based on atropisomeric scaffolds for electrodeposition of inherently chiral film surfaces; (b) inherently chiral ionic liquids or additives. | ||
On the other side, the same strategy was implemented in the cations of ionic liquids or supporting electrolytes/additives to be employed as inherently chiral media for electrochemical experiments on achiral electrodes (employing bicollidinium or bibenzimidazolium atropisomeric units,8,9Fig. 1b).
In all cases neat enantiodiscrimination in terms of large potential differences, specular by inversion of selector configuration, was obtained,2–9 confirming the general validity of this strategy. An important feature for the selector performance is C2 symmetry, too. In the ionic liquid case it favours high ordering at the charged electrode/ionic liquid interface, which has been recently shown to resemble a semisolid crystal extending for many layers.10 In the film case, it grants regio- and stereoregularity in oligomerization; in this frame, synergistic progressive addition of atropisomeric scaffolds in the oligomerization process results in very stable macromolecular folding, much more efficient in enantiodiscrimination than chiral polymers with chirality sources external to the conjugated backbone, such as stereocenters localized in attached pendants.11
Attractively, as very recently highlighted by some of us,7 the outstanding enantiodiscrimination ability of inherently chiral films towards chiral electroactive molecular probes holds as well towards the L- and D- circularly polarized light components,2,7,12 and towards spin magnetic moments under application of an external magnetic field (with unprecedented response in terms of potential rather than difference),13 the three contexts being strictly interrelated.7
Focusing on inherently chiral films, so far only atropisomeric systems have been explored, but other architectures could be exploited to achieve the required controlled twisting of the main electroactive (hetero)aromatic backbone.
A recent review discusses how helical chirality can be induced by introducing strain into a sufficiently rigid backbone made by fused or linked aromatic rings, classifying the resulting helically twisted ribbons into helicenes, twistacenes and bannisters.14 Helicenes15–18 are quite attractive candidates to be explored to achieve inherently chiral surfaces as an alternative to the above atropisomeric systems. They are curved π-conjugated molecules made of aromatic and/or heteroaromatic fused rings, with a torsional barrier increasing with n ring number, so that from a given n onwards they can be separated into stable enantiomers (M and P). In the last decades the number of available helicene structures has dramatically increased, as well as the range of tested applications,15,18–20 like in stereoselective catalysis,15,18,21 molecular switches, also electrochemically modulated, or molecular machines,15,18,22–24 optoelectronics and non-linear optics,18 supramolecular assemblies,18 liquid crystals,18 biological applications, like nucleic acid interactions and bioimaging,19 sensing devices18etc. However, surprisingly, although electrochemistry has been so far widely employed to characterize the electronic properties of many helicene molecules, in combination with spectroscopy and theoretical computations, only very few electrochemical applications have been so far reported (e.g. the recent example23 concerning electroluminescence25), excluding the significant number of studies focusing on the ion-coordinating ability of racemic helicenes.26 The helicene chirality has been considered in connection with electrochemistry e.g. when dealing with electrochemically activated redox switches,22,24 and very recently, as a tool to achieve spin-selective electrochemistry (although so far in terms of current differences only).27 However, surprisingly, the enantiodiscrimination potential of helicene-based enantiopure electrode surfaces has not been explored yet, although helicene-based polymers and oligomers have been reported, obtained both chemically and electrochemically, with suitable linkers between carbohelicene units,18 or by linking the helicene unit to a thiophene monomer,28 or by direct electrooxidative deposition, of carbohelicene monomers (resulting in enantiopure layers on ITO electrodes starting from enantiopure monomers29) or, in milder conditions, of thiahelicene monomers with free thiophene terminals.30,31
Among the different helicene families, a most convenient one for application as active materials in advanced devices is the thiahelicene series,32,33 alternating condensed thiophene T and benzene B rings, and ending with two thiophene terminals. Angular benzodithiophene TBT is its smaller term, followed by five-membered TBTBT (5-TH), seven-membered TBTBTBT (7-TH, tetrathiahelicene), nine-membered TBTBTBTBT (9-TH, pentathiahelicene) and so on (Fig. 2). The series has huge specific optical rotations (e.g. [α]D ∼ 2990° for 7-TH33) as well as good enantiodiscrimination ability for the circularly polarized light components in chiroptical experiments (e.g.7-TH34); possible correlations have been recently discussed13 between the last feature, typical of inherently chiral electroactive oligoheterocycles,35,36 and their enantiodiscrimination ability for electroactive chiral probes in electrochemical experiments. Moreover, compared to the homologue family of carbohelicenes, consisting of all benzene rings, the thiahelicene family is electron richer, and has higher conjugation efficiency and electrochemical reactivity.30,37
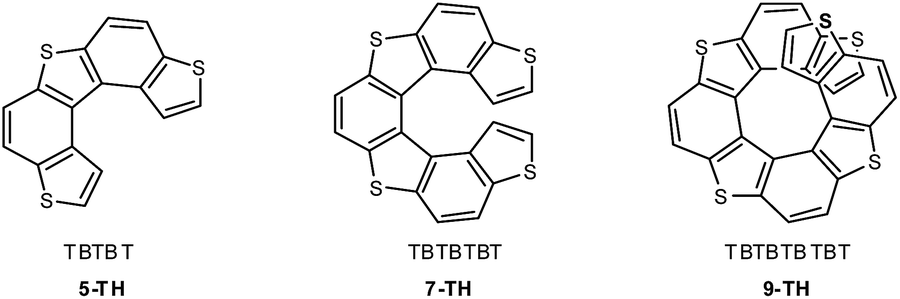 | ||
| Fig. 2 Inherently chiral thiahelicene scaffolds of increasing chain length. | ||
7-TH and some derivatives were electrochemically investigated in detail by some of us several years ago.30,317-TH is the shortest term in the thiahelicene series in which sterical hindrance between thiophene terminals results in a significant torsion with an energy barrier high enough to allow the molecule to exist as stable enantiomers, contrary to 5-TH that undergoes fast racemization at room T, being separable into enantiomers only at low T.33 At the same time, in 7-TH the thiophene terminals are still unhindered enough to enable electrooxidative formation of an electroactive film at a reasonable rate,30 which is achievable even when one of the terminals is unavailable since capped/functionalized.30,31 In longer terms, superimposition of thiophene terminals can result in π-stacking effects and stabilization of the electrochemically formed radical cation intermediates, thus hampering film electrodeposition, as observed for a 11-TH monomer.30
Thus, in the present study we focus on tetrathiahelicene 7-TH, which exhibited a high ability for electrooxidative deposition of electroactive films, and a solubility low enough to allow a good electrodeposition rate.30 The enantiodiscrimination performances are presented for the first time of enantiopure inherently chiral films obtained by 7-TH electrooxidation on chiral electroactive probes in electrochemistry experiments. Moreover, the outstanding potentialities of this novel approach are evaluated in comparison with a representative case of the so far exploited class of inherently chiral selectors with axial stereogenic elements. The electroanalytical enantiodiscrimination tests are also complemented with preliminary tests of interaction with circularly polarized light and with an applied magnetic field, to verify whether the interrelation observed with atropisomeric selectors also hold with helical ones.
Results
1. Redox properties of tetrathiahelicene and electrodeposition of tetrathiahelicene-based inherently chiral films
The electroactivity of 7-TH in dichloromethane (DCM) and acetonitrile (ACN), at constant 0.1 M TBAPF6 supporting electrolyte and 0.2 V s−1 scan rate, is accounted for by the CV patterns reported in Fig. 3.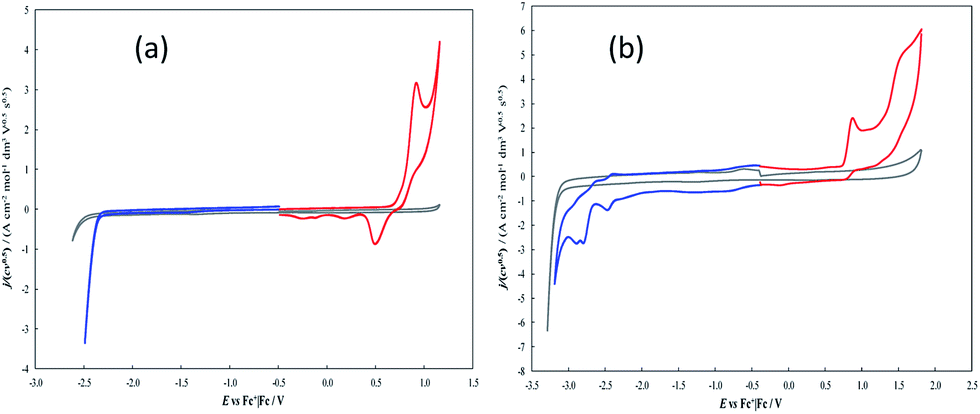 | ||
| Fig. 3 Normalized CV patterns, assembled from first oxidation and first reduction half cycles in order to exclude possible interferences from reciprocal reaction products, obtained for 0.0017 M 7-TH in DCM (a) or ACN (b), with 0.1 M TBAPF6 supporting electrolyte, on GC electrode, at 0.2 V s−1. Background patterns are also reported in grey, for comparison's sake. | ||
In DCM solvent the 7-TH first oxidation peak is located at Ep,Ia ∼0.92 V vs. Fc+|Fc. As usual in thiophene systems with free α positions,38 it corresponds to radical cation formation and subsequent coupling resulting in formation of α,α linked oligomers. In fact, the peak is chemically irreversible, as inferred from the absence of a symmetrical return peak, pointing to a fast chemical follow up, while the non-canonical reduction peak at less positive potentials, regularly increasing upon subsequent oxidative half cycles (Fig. SI.1a†), confirms the progressive growth of an electroactive film on the electrode surface.
On the other hand, first reduction is only perceived as a positive shift of the cathodic background. ACN, having a wider potential window, affords better observation of the reduction pattern, with a first reduction peak at Ep,Ia ∼ −2.47 V vs. Fc+|Fc. In the same solvent the first oxidation peak is located at Ep,Ia ∼0.88 V vs. Fc+|Fc, resulting in a HOMO LUMO gap of ∼3.35 eV, intermediate respect to ∼3.9 eV for linear oligo-T3 and ∼3.1 eV for linear oligo-T4;39 this is consistent with a four-thiophene system in which global conjugation is partially hampered by the helical torsion. While the first reduction peak looks chemically reversible, pointing to a stable radical anion, the first oxidation one appears irreversible as in the DCM case.
Performing preparative potentiodynamic electrodepositions cycling around the first oxidation peak(s), very little formation of electroactive product is observed on the electrode surface when working in ACN (Fig. SI.1b†). Instead, in DCM satisfactory electrodeposition can be achieved (Fig. 4a top), proceeding slowly but steadily as well as reproducibly (in experiments with different total cycle numbers, CV patterns are similar at constant cycle number). Moreover, the electroactive layers appear stable upon repeated “stability cycles”, performed around the first oxidation peak in monomer free solution in order to eliminate monomer residuals and check the film stability in repeated doping/undoping cycles (Fig. 4a bottom). The film thickness appears to increase with cycle number as expected from the electrodeposition CV pattern and confirmed by profilometry data (SI.2†); however, the correlation between number of deposition cycles and exchanged charge in the corresponding stability cycles appears irregular; this could be a consequence of overoxidation and/or some other process after the first radical cation coupling, partially hampering the film electroactivity but with little effect on the film morphological features.
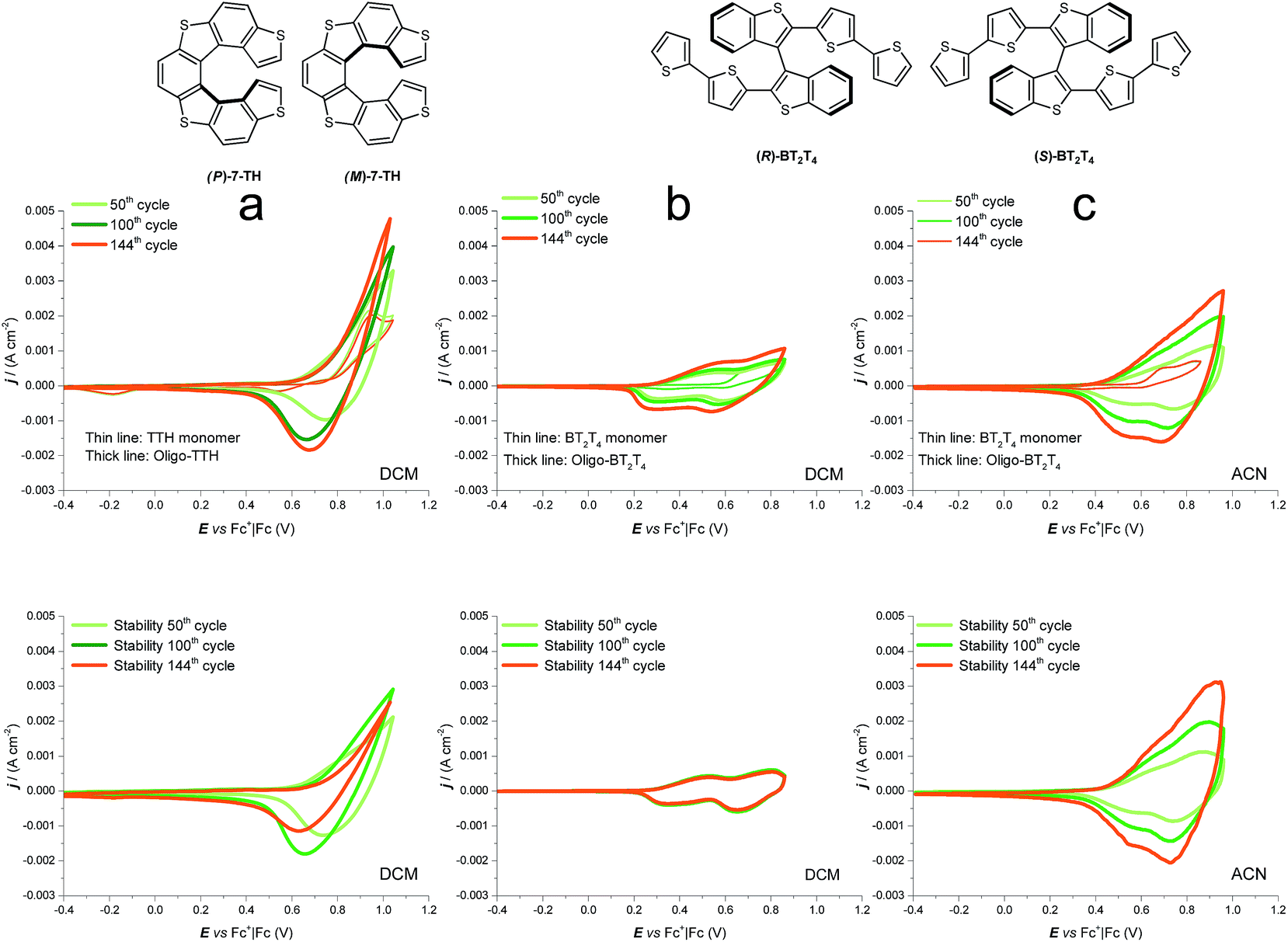 | ||
| Fig. 4 Top: electrodeposition experiments from (a) monomer 7-TH in DCM, (b) monomer BT2T4 in DCM and (c) monomer BT2T4 in ACN, all of them carried out by potential cycling on glassy carbon electrode, at constant 0.0025 M monomer concentration, with 0.1 M TBAPF6 supporting electrolyte and at 0.2 V s−1 potential scan rate, as a function of the total number of potential cycles (in particular, 50, 100, and 144 cycles). Bottom: last of five stability cycles in monomer free solution for each deposited film. | ||
MALDI analysis of the material electrodeposited by electrooligomerization of a racemic mixture of 7-TH monomer, proved hard to perform and the recorded spectra are complex; however, they confirm the presence of dimeric species in the oligomer films, particularly cyclic ones (SI.3†). Very interestingly, all assigned MALDI signals (SI.3†) point to dimer structures in which, besides the expected “interhelicene” links between α positions of thiophene terminals resulting from radical cation coupling (according to the usual thiophene electrooligomerization scheme38,39) and besides the S-atom oxidation (Fig. SI.3.3†), the unexpected formation of one or two “intrahelicene” single bonds, linking the β positions of the thiophene terminals of the same tetrathiahelicene unit, is observed (Fig. SI. 3.3–5†). This feature leads to attractive unprecedented structures, like a closed dimer having a cyclooctatetraene “antiaromatic” enclave (Fig. 5a) framed within two tetrathiahelicene half crowns. Preliminary quantum chemical calculations at B3LYP/6-31G* DFT level40 point to the HOMO being mostly localized on the central cyclooctatetraene, while the LUMO is mostly delocalized on the tetrathiahelicene systems (Fig. SI. 4.2†). Calculations also highlight the effect of the configuration of the monomer units on the possible dimer structures. While contorted structures implying open dimers should originate from the coupling of a M monomer with a P one (Fig. SI. 4.1c†), the coupling of two M- or two P- enantiomers should result in more planar, although twisted, geometries, favouring ring closure (Fig. SI. 4.1a and b†). This feature is similar to that recently observed by some of us for BT2T4 inherently chiral dimers, with cyclic ones originating from coupling of monomers having the same R- or S- configuration.41 However in the present case the sequence of C–C bond formation should also be evaluated, considering both α,α “interhelicene” and β,β “intrahelicene” ones. In the MALDI spectrum of (P,M)-7-TH a group of signals has been assigned to an open dimer with a single α,α interhelicene bond and two β,β intrahelicene ones, which could be either homochiral, or heterochiral, or both of them; concerning the homochiral one, calculations show (SI.4†) that ring closure by formation of the second α,α bond is highly favoured.
 | ||
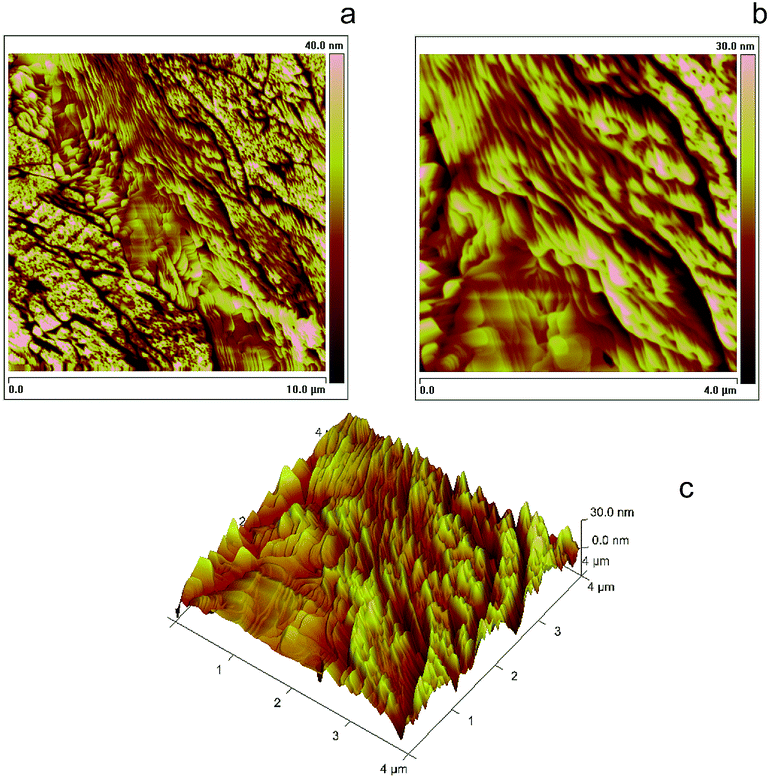
Fig. 5 (a and a′): Assumed general formula as well as structure optimized at DFT B3LYP/6-31G* level for (M, M) or (P, P) cyclic dimers included in the 7-TH oligomer film, with indication of possible additional oxidation sites by orange/red lines (additional intramolecular C–C links between terminal beta positions) or pink circle (S→S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). (b and b′): Tapping Mode AFM images of 10 × 10 μm2 and 4 × 4 μm2 areas of a thin layer electrodeposited from 1.7 mM monomer racemate onto uflat-ITO electrodes (2 cycles, DCM solution, 0.2 V s−1); (c and c′): same protocol applied to films electrodeposited from enantiopure monomers. (d) Profilometric analysis for the c′ film sample, along the white lines with red and blue markers in Fig. 5c′. O). (b and b′): Tapping Mode AFM images of 10 × 10 μm2 and 4 × 4 μm2 areas of a thin layer electrodeposited from 1.7 mM monomer racemate onto uflat-ITO electrodes (2 cycles, DCM solution, 0.2 V s−1); (c and c′): same protocol applied to films electrodeposited from enantiopure monomers. (d) Profilometric analysis for the c′ film sample, along the white lines with red and blue markers in Fig. 5c′. | ||
Homochiral dimers, particularly the discoid cyclic ones, are also more suitable for self-assembling by π,π-stacking, as for many polyaromatic hydrocarbons with extended structures.42
When the oligomerization is carried out starting from an enantiopure monomer, rather than on the monomer racemate, less freedom degrees should be implied affording more selectivity in the resulting dimer structures, promoting the above cyclic, easily stackable ones. An atomic force microscopy (AFM) investigation was carried out on thin films electrodeposited in the first two potential cycles on uflat-ITO substrate from racemate monomers (Fig. 5b/b′) vs. enantiopure ones (Fig. 5c/c′) in DCM. In the enantiopure film case nanorods are observed, with dimensions that are roughly one order of magnitude larger than the size of oligomers themselves (about 1.5 nm as obtained by molecular modeling). This is consistent with the above discussed cyclic dimer structures resulting from homochiral monomers, which can allow self-assembling into nanostructured rods similarly e.g. to the hexabenzocoronene case.43 Scratching the film with the AFM tip allowed to estimate the layer thickness about 15 nm, corresponding to the height of the nanometric rods. This is also shown by AFM section analysis (Fig. 5d), along the white lines in the AFM image of Fig. 5c′, where it is possible to see that the average heights of the rods are about 15 nm with a length of 250 nm. Instead, applying the same protocol to the film obtained from the racemate results in a dispersion of closer, bigger and less uniform nanostructured aggregates, clearly constituted by spherical deposits (average size 15–35 nm) agglomerating along lines. Such a pattern, albeit showing some degree of ordering, reminds the first stages of deposition of achiral traditional conducting polymers.
2. Enantioselectivity performance of tetrathiahelicene-based inherently chiral films
Enantiopure films gave outstanding enantiodiscrimination performances in a series of tests with a collection of different chiral probes. Concerning the preparation protocol of the enantiopure electrode surfaces, we selected 100 as a convenient cycle number as a compromise to obtain a film of reasonable thickness (∼1.5 μm according to profilometry tests, SI.2†), while maintaining a good charging/discharging ability.Fig. 6a shows the enantiopure film performance in our standard enantiodiscrimination test, carried out with the commercially available (S)-(−)- and (R)-(+)-N,N-dimethyl-1-ferrocenylethylamine, (S)-Fc and (R)-Fc. We usually employ such chiral probe in first tests of new selectors, on account of its undergoing facile and reversible electron transfer at a mild oxidation potential, with Ep,Ia ∼ 0.05 V on bare GC electrode. At such potential, 7-TH-based films should be still uncharged, considering the film stability CV patterns in Fig. 4a. However, it must be taken into account that probe/film interactions can modify film activation conditions.
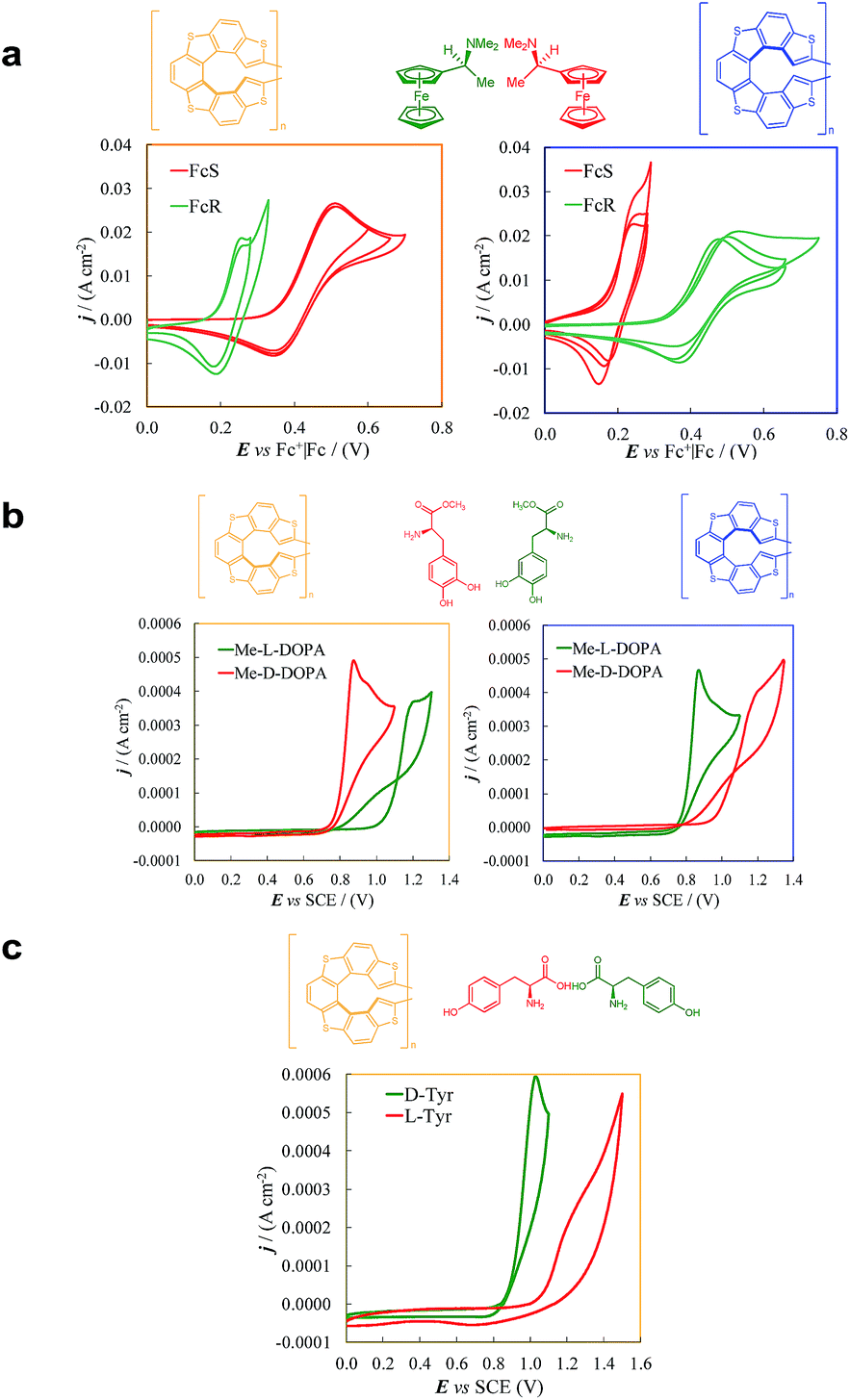 | ||
| Fig. 6 Enantioselection performance of enantiopure (P)-7-TH (yellow box) and (M)-7-TH (blue box) electrode surfaces towards chiral electroactive (a) ferrocenyl probes, named (S)-Fc and (R)-Fc, (b) methyl L- and D-DOPA and (c) L- and D-tyrosine. CVs were recorded at 0.05 V s−1 potential scan rate. | ||
As evident in Fig. 6a, outstanding enantiodiscrimination in terms of peak potential values (E°′, calculated as the average between forward and backward peak potentials), is obtained. In fact, two quite different CV peaks, both well defined and partially reversible, are observed for (i) (R)-Fc on (P)-film (Fig. 6a, left) or (S)-Fc on (M)-film (Fig. 6a, right) specular combinations, at a formal potential 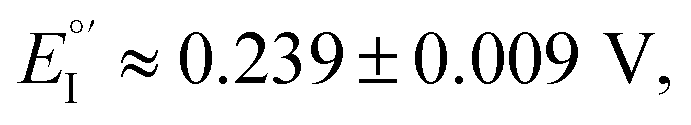 (ii) (R)-Fc on (M)-film (Fig. 6a, right), or (S)-Fc on (P)-film (Fig. 6a, left), specular combinations, in a diastereomeric relationship with respect to the two former ones at
(ii) (R)-Fc on (M)-film (Fig. 6a, right), or (S)-Fc on (P)-film (Fig. 6a, left), specular combinations, in a diastereomeric relationship with respect to the two former ones at 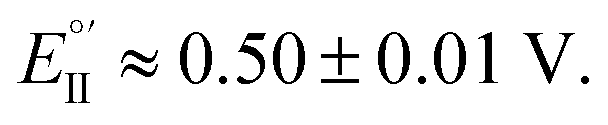 Thus, a huge formal potential difference of
Thus, a huge formal potential difference of 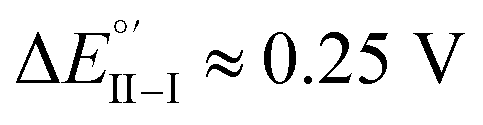 is observed between potentials of the two (R)-Fc and (S)-Fc enantiomers using both (P)- or (M)-films, as a consequence of a diastereomeric interaction between probe and selector, which produces energetically different conditions. Remarkable specular differences also concern peak morphology; this points to significant differences in the electron transfer mechanism and follow-up, which could involve film activation, too. In particular, the CV pattern for the (R,P) or (S,M) combinations, resembling a former case studied by some of us of an electroactive film constituted by tetrathiahelicene units functionalized with a ferrocene terminal via an ethynyl bridge,31 suggests the Fc|Fc+ couple acting as redox mediator for film oxidation activation at lower potential.
is observed between potentials of the two (R)-Fc and (S)-Fc enantiomers using both (P)- or (M)-films, as a consequence of a diastereomeric interaction between probe and selector, which produces energetically different conditions. Remarkable specular differences also concern peak morphology; this points to significant differences in the electron transfer mechanism and follow-up, which could involve film activation, too. In particular, the CV pattern for the (R,P) or (S,M) combinations, resembling a former case studied by some of us of an electroactive film constituted by tetrathiahelicene units functionalized with a ferrocene terminal via an ethynyl bridge,31 suggests the Fc|Fc+ couple acting as redox mediator for film oxidation activation at lower potential.
Reproducibility is confirmed by experiment repetitions and, even more reliably, particularly to rule out systematic errors, by the nice specularity of the signals obtained inverting probe or film configuration (Fig. 6a).
The thiahelicene-based enantiopure films have also been submitted to enantioselection tests of more applicative character, in aqueous buffered solution, with chemically different chiral probes of pharmaceutical interest, i.e.L- and D-DOPA methyl ester (Me-DOPA) and L- and D-tyrosine, both of them at pH 4 (Fig. 6b and c), in which condition they give on bare electrode a chemically irreversible first oxidation peak at 0.51 V vs. SCE for Me-DOPA and at 0.94 V vs. SCE for tyrosine.6 Again, a huge difference is obtained in both CV peak potentials and morphology for the Me-DOPA enantiomers (Fig. 6b). In particular a chemical irreversible first oxidation is observed (in this case we consider peak potentials rather than formal potentials): (i) at Ep,Ia,I ≈ 0.80 V for specular combinations D-Me-DOPA on (P)-film and L-Me-DOPA on (M)-film; (ii) at Ep,Ia,II ≈ 1.15 V for specular combinations D-Me-DOPA and (M)-film + or L-Me-DOPA on (P)-film, that are in a diastereomeric relationship with respect to the two former ones. Thus, an outstanding peak potential difference, ΔEp,Ia,II–I ≈ 0.35 V, even larger than that obtained with the ferrocenyl probes, is observed for probe + selector combinations obtained by inverting either the probe or the film configuration. This feature can allow to clearly distinguish the enantiomer configuration with no preliminary separation steps; at the same time the peak current, with suitable standardization, can provide quantitative information. A neat peak difference is also observed with tyrosine (Fig. 6c), resulting in Ep,Ia,I ≈ 0.95 V and Ep,Ia,II ≈ 1.15 V, with a potential difference of Ep,Ia,II–I ≈ 0.2 V.
Such results soundly confirm the general character of the huge discrimination ability of the new selector surfaces, holding for molecules of different structures and reactivities, including probes of practical interest.
Such enantiodiscrimination performances could be justified considering the electrochemical process to take place in diasteromeric and thus energetically different situations, resulting from intermolecular interactions between enantiopure selector and enantiopure probe, particularly hinging on the many available aromatic rings and heteroatoms. Moreover, diastereomeric conditions could also originate from modulation of the internal magnetic field of the chiral film in the presence of the (R)- or (S)- probe, as we recently pointed out in a very recent study highlighting the spin-filter properties of inherently chiral films with atropisomeric scaffold.13
In this context, the new selector films based on helical elements were also preliminarily tested concerning interactions with electron spin moments under applied magnetic field, following the approach that very recently enabled us to unveil outstanding spin filter properties in films obtained from atropisomeric monomers.7,13 CV patterns were recorded for the achiral, reversible ferrocyanide/ferricyanide couple in aqueous solution, both on ITO and on screen-printed single-walled carbon nanotube (SWNT SPE) working electrode, modified with a very thin enantiopure oligo-P-7-TH film electrodeposited in a single CV cycle. Application of an external magnetic field resulted in both cases in remarkable formal potential shift according to the N/S or S/N magnet orientation (Fig. 7), an impressive effect recently observed on inherently chiral films based on atropisomeric units (oligo-BT2T4 films13 and on 2,2′-biindole-based films7), besides a chiral C2-symmetry PEDOT. The phenomenon has been tentatively justified considering the combined effects of the internal magnetic field intrinsic of the chiral layer (particularly effective for an inherently chiral one) and of the external applied magnetic field on the electron energy levels, as a function of the α or β spin magnetic moment.13 Two couples of equivalent α,β energy level splitting situations can be considered, one corresponding to both magnetic fields favouring the same spin orientation and one corresponding to them having opposite effects.
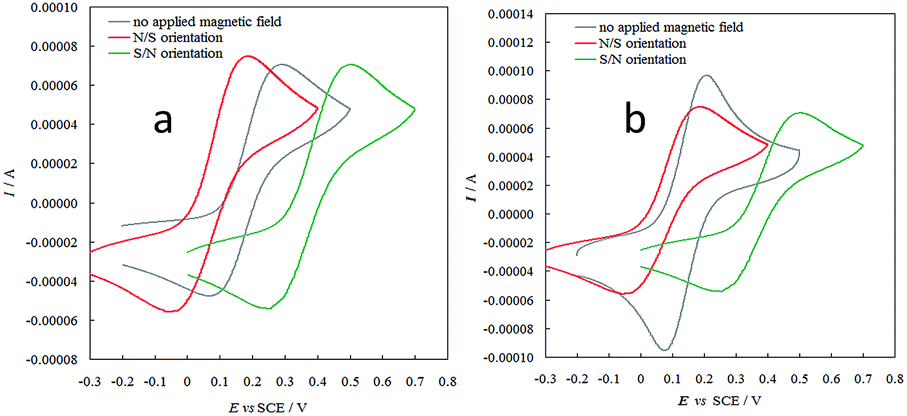 | ||
| Fig. 7 CV patterns for the achiral Fe(CN)63−|Fe(CN)64− redox couple, recorded at 0.05 V s−1 in aqueous solution on ITO electrodes (a) or SWCN SPEs (b) modified by a thin layer of (P)-enantiopure oligo-7-TH thin film, as such and under application of an external magnet with N/S or S/N orientation (red or green lines respectively). | ||
Since the two cases are in diastereomeric relationship, they are energetically different, which could explain the shift in formal potentials upon application of opposite external magnetic fields.
Such “spin modulated electrochemical potentials” are of great potential interest from both fundamental and applicative perspectives.13
3. Comparison of 7-TH electrochemical performance with those of BT2T4, a representative case of axial stereogenicity
We consider interesting and useful to compare the outstanding features and performance of the helicene selectors with the C2-symmetric bibenzothiophene BT2T4 and its electrodeposited films.1 In fact (i) both monomers 7-TH and BT2T4 are endowed with inherent chirality, but on account of different stereogenic elements, i.e. helicity vs. stereogenic axis.; (ii) both monomers have electroactivity based on thiophene conjugated systems: in the case of 7-TH, a system of four thiophene rings alternated to condensed benzene ones with an helicoidal torsion; in the case of BT2T4, two weakly interacting linear terthiophene systems; (iii) the torsional barrier to be overcome for racemization is similar (i.e. ∼45 kcal mol−1 for BT2T42 and ∼39 kcal mol−1 for 7-TH30); (iv) both monomers have two free homotopic thiophene terminals for stereo- and regio regular electrodeposition of oligomer films.For comparison's sake, in this paper we have prepared and tested BT2T4 films with the same protocol used for the 7-TH helical selector, in both DCM and ACN solvents. Unlike the 7-TH case, BT2T4 oligomerization is very slow in DCM, with the resulting films appearing very thin (Fig. 4b and SI.2†), while it proceeds fast, regularly and with no apparent limitation in ACN (Fig. 4c) as formerly accounted, too.4,6
The electrodeposited layers appear quite different from the thiahelicene-based ones, as revealed by an AFM morphological investigation carried out on very thin films electrodeposited by the first two CV cycles, with the same protocol above applied to the oligo-7-TH case. In particular, the layers display a lamellar structure (Fig. 8a and b) and are significantly thicker (in the range 70–140 nm, Fig. 8c), as it results from the scratch of the film by the AFM tip.
 | ||
| Fig. 8 Tapping Mode AFM images (a) 10 × 10 μm2, (b) and (c) 4 × 4 μm2 of BT2T4 oligomers electrochemically deposited onto uflat-ITO electrode by 2 voltammetric cycles at 0.2 V s−1 from a DCM solution of 1.7 mM racemic monomer BT2T4. | ||
Enantiopure BT2T4 films electrodeposited in DCM with the same 100-cycle protocol adopted for 7-TH gave poor enantiodiscrimination in our model test with ferrocenyl chiral probes, resulting in 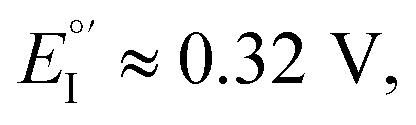
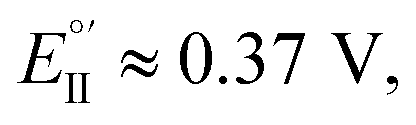
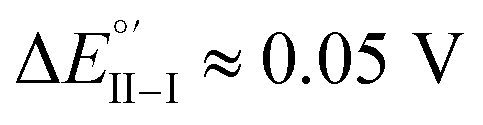 (Fig. 9, top). This is consistent with the thinness and fragility of the films obtained with this protocol. Instead good discrimination was obtained on the thick, although quite rough, BT2T4 films electrodeposited in the same conditions but in ACN solvent (Fig. 9, bottom). In this case two signals significantly different in potential and shape (again, possibly involving film activation, too) are observed at
(Fig. 9, top). This is consistent with the thinness and fragility of the films obtained with this protocol. Instead good discrimination was obtained on the thick, although quite rough, BT2T4 films electrodeposited in the same conditions but in ACN solvent (Fig. 9, bottom). In this case two signals significantly different in potential and shape (again, possibly involving film activation, too) are observed at 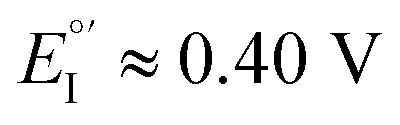 and
and 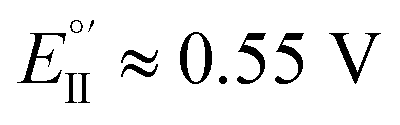 with a peak potential difference of
with a peak potential difference of 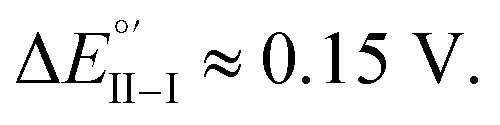
 | ||
| Fig. 9 Enantioselection performance of enantiopure (S)-BT2T4 (yellow) and (R)-BT2T4 (blue) films towards chiral electroactive ferrocenyl probes, named (S)-Fc and (R)-Fc, in DCM (top) and ACN (bottom) as solvents. | ||
Such observations confirm first of all the outstanding enantiodiscrimination potentialities of the helical selector, even compared to the atropisomeric one. We have to consider that the comparison between the two systems unfortunately is not fully achieved because we could not electrodeposit 7-TH monomer in ACN and vice versaBT2T4 monomer in DCM.
Conclusions
Enantiopure thiahelicenes can be regarded as valuable inherently chiral building blocks for the preparation of the corresponding enantiopure inherently chiral electroactive surfaces, exhibiting very high enantiodiscrimination ability, hinging on a helical stereogenic element rather than an axial one as in former cases. Furthermore, the availability of a large potential window on the oxidation side allows to use the thiahelicene-based electrodes for a wide range of chiral probes in enantioselection tests.Our convenient preparation protocol affords electrodeposition of compact films of adequate thickness and stability. The inherently chiral electroactive surfaces display outstanding enantiodiscrimination performance for different chiral probes, in terms of peak potential and morphology differences, competitive with those obtained by an inherently chiral selector with axial stereogenicity. A new, important helicene application is thus unveiled, adding a novel class to the growing palette of inherently chiral selectors for enantioselective electroanalysis.
Moreover, the wide-scope effectiveness of the inherent chirality strategy is further confirmed by the observation that inherently chiral helical electroactive molecules share with atropisomeric ones the same all-around discrimination ability, encompassing besides circularly polarized light components electroactive chiral probes as well as electron spin magnetic moments.
Developments could involve not only the exploration of a wider casebook of probe/selector/protocol combinations, particularly of applicative interest, but also possible interrelations as well as combinations with the optoelectronic and spintronic peculiarities.
Methods
1. Synthesis and separation of enantiopure antipodes of 7-TH
Tetrathia[7]helicene (±)-7-TH was synthesized as previously reported.44 The two enantiomers of (±)-7-TH were separated using an Agilent 1100 series HPLC, equipped with DAD analyzer and the chiral semipreparative column Chiralpak IA (10 × 250 mm), in a hexane/dichloromethane mixture (92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8, v/v) as the mobile phase.45 The earlier eluting fraction consisted of the enantiomer exhibiting a positive optical rotation (+)-7-TH, while the later eluting fraction contained the enantiomer with negative optical rotation (−)-7-TH. On the basis of their X-ray structures,46 (+)-7-TH corresponds to the absolute configuration of a right-handed helix (P), and, vice versa, (−)-7-TH corresponds to the absolute configuration of a left-handed helix (M).
:8, v/v) as the mobile phase.45 The earlier eluting fraction consisted of the enantiomer exhibiting a positive optical rotation (+)-7-TH, while the later eluting fraction contained the enantiomer with negative optical rotation (−)-7-TH. On the basis of their X-ray structures,46 (+)-7-TH corresponds to the absolute configuration of a right-handed helix (P), and, vice versa, (−)-7-TH corresponds to the absolute configuration of a left-handed helix (M).
2. Electrochemistry
All electrodepositions and stability tests (cycling 5 times the oligomer in a monomer free solution) were performed in traditional solvents, acetonitrile (ACN, Sigma-Aldrich, anhydrous, 99.8%) and dichloromethane (DCM, Sigma-Aldrich anhydrous, 99.8%).
Electrodepositions of enantiopure (R)- and (S)-BT2T4 films and enantiopure (P)-(+)-7-TH and (M)-(−)-7-TH ones were performed on the GC disk electrode with 2.5 mM (R)- or (S)- corresponding monomer solutions in DCM and ACN with 0.1 M tetrabutylammonium hexafluorophosphate as supporting electrolyte (TBAPF6, Sigma-Aldrich, electrochemical analysis). 50, 100, 144 consecutive oxidative potential cycles at 0.2 V s−1 around the first oxidation peaks for both starting monomers were performed, followed by repeated potential cycling in a monomer free solution, until the CV curves became stable. These films were then analysed by means of profilometry experiments.
Other tests on the enantiorecognition capability were performed at 0.05 V s−1 scan rate, only on the (P)-(+)-7-TH and (M)-(−)-7-TH electrode surfaces in 0.003 M solutions of two different probes of pharmaceutical interest (i) L- and D-tyrosine (Alfa-Aesar) and (ii) L- and D-methyl-DOPA hydrochloride prepared according to a literature protocol48 in a buffer pH 4 solution (Fluka, prepared with citric acid, NaOH, and NaCl).
All experiments were carried out in a three-electrode minicell (with 3 cm3 of solution), with a glass-embedded glassy carbon disk (GC, Metrohm, S = 0.033 cm2) as the working electrode, a Pt disk as the counter electrode, and an aqueous saturated calomel electrode (SCE) as the reference electrode operating in a double bridge filled with the working medium to avoid water and KCl leakage into the working solutions. N2 purging before measurement was not necessary since all experiments were included in a potential range more positive than O2 reduction. The optimised preliminary polishing procedure for the GC disk electrode consisted of a sonication for 5 minutes of the GC tip in acetone and after a treatment with a diamond powder of 1 mm diameter (Aldrich) on a wet DP-Nap cloth (Struers®).
The enantiopure (P)- and (M)-oligo-thiahelicenes films were prepared by electrooligomerization of the monomer enantiomers on SPEs from commercial ionic liquid 1-butyl-3-methylimidazolium bis(trifluoro-methylsulfonyl)imide (BMIMTFSI).
The magnetic field was applied by an external magnet (nickel coated NdFeB B88X0 Grade N42 K&J Magnet, Inc.; magnetic field strength at the surface = 6353 Gauss) placed under the SPEs during the cyclic voltammetric experiments.
Cyclic voltammograms of the achiral redox couple Fe(CN)63−|Fe(CN)64− (with each component at 2.5 mM concentration and 400 mM KCl as supporting electrolyte) were recorded at 0.05 V s−1 potential scan rate by depositing a drop of the aqueous solution on SPEs and by flipping the external magnet north vs. south orientations.
3. Profilometry measurements
Thickness measurements were directly carried out on the working electrode tip, scratching the film deposited with a wooden toothpick wet of acetone. The step obtained were analysed by a Bruker DektakXT contact profilometer using a 12.5 μm radius stylus in Low-Force mode. The working electrode tip was fixed to the vacuum chuck by a home-made holder. The values of thickness and roughness were determinate by Vision64 software.4. AFM investigations
AFM imaging and analysis were performed in air with a Digital NanoScope 3D Multimode microscope (Veeco, USA) using phosphorus n-doped Silicon probes (spring constant, 20–80 N m−1; resonance frequency 280–320 kHz; nominal tip radius <10 nm) and operating in tapping mode. Samples for the AFM experiments were prepared by depositing the oligomeric film through electrochemical polymerization, of either a ACN or DCM monomer solution, on ultra-flat ITO (uflat-ITO), with a sheet resistance of 7 Ω sq−1, RMS roughness of about 0.6 nm (1 × 1 μm2), provided by Kuramoto Co. (Kurihara, Japan). The images were obtained by optimizing the setpoint tapping amplitude with respect to the free amplitude of the probe, near the resonance frequency of the probe itself, as reported elsewhere.47 The thickness of the deposit was evaluated by scratching an area of the deposit with the AFM tip by applying a minimum constant force (130 nN) in contact mode and subsequently performing the imaging of the selected surface part in tapping mode. NanoScope 6.14r1 software was used for data analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Authors are grateful to Dr Marco Pappini (UNIMI CIGA) for support in obtaining and interpreting the MALDI spectrum. The authors' inherent chirality work is financially supported by Fondazione Cariplo and Regione Lombardia (2016-0923 RST—Avviso congiunto FC-RL Sottomisura B) rafforzamento (Enhancing VINCE (Versatile INherently Chiral Electrochemistry)). Moreover, the support of SmartMatLab Centre (cofunded by Regione Lombardia, Fondazione Cariplo and Università degli Studi di Milano, grant No. 2013-1766) for profilometry experiments and by the Università di Bologna are gratefully acknowledged.Notes and references
- S. Arnaboldi, M. Magni and P. R. Mussini, Curr. Opin. Electrochem., 2018, 8, 60 CrossRef CAS.
- F. Sannicolò, S. Arnaboldi, T. Benincori, V. Bonometti, R. Cirilli, L. Dunsch, W. Kutner, G. Longhi, P. R. Mussini, M. Panigati, M. Pierini and S. Rizzo, Angew. Chem., Int. Ed., 2014, 53, 2623 CrossRef PubMed.
- F. Sannicolò, P. R. Mussini, T. Benincori, R. Cirilli, S. Abbate, S. Arnaboldi, S. Casolo, E. Castiglioni, G. Longhi, R. Martinazzo, M. Panigati, M. Pappini, E. Quartapelle Procopio and S. Rizzo, Chem.–Eur. J., 2014, 20, 15261 CrossRef.
- S. Arnaboldi, T. Benincori, R. Cirilli, W. Kutner, M. Magni, P. R. Mussini, K. Noworyta and F. Sannicolò, Chem. Sci., 2015, 6, 1706 RSC.
- F. Sannicolò, P. R. Mussini, T. Benincori, R. Martinazzo, S. Arnaboldi, G. Appoloni, M. Panigati, E. Quartapelle Procopio, V. Marino, R. Cirilli, S. Casolo, W. Kutner, K. Noworyta, A. Pietrzyk-Le, Z. Iskierko and K. Bartold, Chem.–Eur. J., 2016, 22, 10839 CrossRef.
- S. Arnaboldi, T. Benincori, R. Cirilli, S. Grecchi, L. Santagostini, F. Sannicolò and P. R. Mussini, Anal. Bioanal. Chem., 2016, 408, 7243 CrossRef CAS PubMed.
- S. Arnaboldi, T. Benincori, A. Penoni, L. Vaghi, R. Cirilli, S. Abbate, G. Longhi, G. Mazzeo, M. Panigati and P. R. Mussini, Chem. Sci., 2018 Search PubMed , under revision.
- S. Rizzo, S. Arnaboldi, V. Mihali, R. Cirilli, A. Forni, A. Gennaro, A. A. Isse, M. Pierini, P. R. Mussini and F. Sannicolò, Angew. Chem., Int. Ed., 2017, 56, 2079 CrossRef CAS PubMed.
- S. Rizzo, S. Arnaboldi, R. Cirilli, A. Gennaro, A. A. Isse, F. Sannicolò and P. R. Mussini, Electrochem. Commun., 2018, 89, 57 CrossRef CAS.
- K. Ma, R. Jarosova, G. M. Swain and G. J. Blanchard, Langmuir, 2016, 32, 9507 CrossRef CAS PubMed.
- L. A. P. Kane-Maguire and G. G. Wallace, Chem. Soc. Rev., 2010, 39, 2545 RSC.
- G. Longhi, S. Abbate, G. Mazzeo, E. Castiglioni, P. Mussini, T. Benincori, R. Martinazzo and F. Sannicolò, J. Phys. Chem. C, 2014, 118, 16019–16027 CrossRef CAS.
- T. Benincori, S. Arnaboldi, M. Magni, S. Grecchi, C. Fontanesi and P. R. Mussini, Chem. Sci., 2018 Search PubMed , under revision.
- M. Rickhaus, M. Mayor and M. Jurícek, Chem. Soc. Rev., 2016, 45, 1542 RSC.
- Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463 CrossRef CAS.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 968 RSC.
- M. Gingras, G. Felix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007 RSC.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 1051 RSC.
- J. Bosson, J. Gouin and J. Lacour, Chem. Soc. Rev., 2014, 43, 2824 RSC.
- N. Saleh, C. Shen and J. Crassous, Chem. Sci., 2014, 5, 3680 RSC.
- P. Aillard, A. Voituriez and A. Marinetti, Dalton Trans., 2014, 43, 15263 RSC.
- C. Nuckolls, R. Shao, W.-G. Jang, N. A. Clark, D. M. Walba and T. J. Katz, Chem. Mater., 2002, 14, 773 CrossRef CAS.
- H. Isla and J. Crassous, C. R. Chim., 2016, 19, 39 CrossRef CAS.
- L. Pospíšil, L. Bednárová, P. Štepánek, P. Slavíček, J. Vávra, M. Hromadová, H. Dlouhá, J. Taráábek and F. Teplý, J. Am. Chem. Soc., 2014, 136, 10826 CrossRef PubMed.
- J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743 CrossRef CAS PubMed.
- M. Tounsi, M. Ben Braiek, A. Baraket, M. Lee, N. Zine, M. Zabala, J. Bausells, F. Aloui, B. Ben Hassine, A. Maaref and A. Errachid, Electroanalysis, 2016, 28, 2892 CrossRef CAS.
- V. Kiran, S. P. Mathew, S. R. Cohen, I. Hernández Delgado, J. Lacour and R. Naaman, Adv. Mater., 2016, 28, 1957 CrossRef CAS PubMed.
- J. Hrbac, J. Storch, V. Halouzka, V. Cirkva, P. Matejka and J. Vacek, RSC Adv., 2014, 4, 46102 RSC.
- J. Vacek, J. Hrbáč, T. Strašák, V. Církva, J. Sýkora, L. Fekete, J. Pokorný, J. Bulíř, M. Hromadová, J. Crassous and J. Storch, ChemElectroChem, 2018, 5, 2080 CrossRef CAS.
- A. Bossi, L. Falciola, C. Graiff, S. Maiorana, C. Rigamonti, A. Tiripicchio, E. Licandro and P. R. Mussini, Electrochim. Acta, 2009, 54, 5083 CrossRef CAS.
- F. Rose-Munch, M. Li, E. Rose, J. C. Daran, A. Bossi, E. Licandro and P. R. Mussini, Organometallics, 2012, 31, 92 CrossRef CAS.
- E. Licandro, S. Cauteruccio and D. Dova, Adv. Heterocycl. Chem., 2016, 118, 1 CrossRef CAS.
- K. Yamada, H. Nakagawa and H. Kawazura, Bull. Chem. Soc. Jpn., 1986, 59, 2429 CrossRef CAS.
- H. Nakagawa, K. Gomi and K. Yamada, Enantiomer, 1998, 3, 175–179 CAS.
- N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914 RSC.
- J. T. Vázquez, Tetrahedron: Asymmetry, 2017, 28, 1199 CrossRef.
- H. Sakai, T. Kubota, J. Yuasa, Y. Araki, T. Sakanoue, T. Takenobu, T. Wada, T. Kawai and T. Hasobe, J. Phys. Chem. C, 2016, 120, 7860 CrossRef CAS.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724 CrossRef CAS PubMed.
- K. Meerholz and J. Heinze, Electrochim. Acta, 1996, 41, 1839 CrossRef CAS.
- (a) All the geometry optimization and single-point DFT calculations, were carried out at the B3LYP/6-31G* level of theory in the gas phase implemented in Gaussian 09.; (b) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- E. Quartapelle Procopio, T. Benincori, G. Appoloni, P. R. Mussini, S. Arnaboldi, C. Carbonera, R. Cirilli, A. Cominetti, L. Longo, R. Martinazzo, M. Panigati and R. Po', New J. Chem., 2017, 41, 10009 RSC.
- C. Bruno, M. Marcaccio, D. Paolucci, C. Castellarin-Cudia, A. Goldoni, A. V. Streletskii, T. Drewello, S. Barison, A. Venturini, F. Zerbetto and F. Paolucci, J. Am. Chem. Soc., 2008, 130, 3788–3796 CrossRef CAS PubMed.
- L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119–1122 CrossRef CAS PubMed.
- S. Maiorana, A. Papagni, E. Licandro, R. Annunziata, P. Paravidino, D. Perdicchia, C. Giannini, M. Bencini, K. Clays and A. Persoons, Tetrahedron, 2003, 59, 6481 CrossRef CAS.
- T. Kawasaki, K. Suzuki, E. Licandro, A. Bossi, S. Maiorana and K. Soai, Tetrahedron: Asymmetry, 2006, 17, 2050 CrossRef CAS.
- H. Nakagawa, A. Obata, K. Yamada and H. Kawazura, J. Chem. Soc., Perkin Trans. 2, 1985, 1899 RSC.
- L. Petrizza, D. Genovese, G. Valenti, M. Iurlo, A. Fiorani, F. Paolucci, S. Rapino and M. Marcaccio, Electroanalysis, 2016, 28, 2777 CrossRef CAS.
- S. H. Park, H. S. Oh, M. A. Kang, H. Cho, J. B. Prasad, J. Won and K. H. Lee, Bioorg. Med. Chem., 2007, 15, 3938 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03337d |
| This journal is © The Royal Society of Chemistry 2019 |