
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA versatile strategy for the synthesis of sequence-defined peptoids with side-chain and backbone diversity via amino acid building blocks†
Shixue
Wang
ab,
Yue
Tao
ac,
Jianqun
Wang
ac,
Youhua
Tao
 *ac and
Xianhong
Wang
ac
*ac and
Xianhong
Wang
ac
aKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Renmin Street 5625, Changchun 130022, People's Republic of China. E-mail: youhua.tao@ciac.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100039, People's Republic of China
cUniversity of Science and Technology of China, Hefei 230026, People's Republic of China
First published on 22nd November 2018
Abstract
Designing artificial macromolecules with absolute sequence order is still a long-term challenge in polymer chemistry as opposed to natural biopolymers with perfectly defined sequences like proteins and DNA. Herein, we combined amino acid building blocks and iterative Ugi reactions for the de novo design and synthesis of sequence-defined peptoids. The highly efficient strategy provided excellent yields and enables multigram-scale synthesis of perfectly defined peptoids. This new strategy furnishes the broad structural diversity of side chains, as well as backbones. Importantly, the overall hydrophobicity and lower critical solution temperature (LCST) behaviours of these precisely defined peptoids can be logically altered by variation of the sequence. By following the same Ugi chemistry, these peptoids are also conjugated to DNA in a simple way, facilitating the development of novel therapeutics.
Introduction
The sequence of biopolymers such as DNA, RNA and proteins is elaborately controlled, and functional substituents at well-defined positions cooperatively play an essential role in their special and unique functions.1,2 Recently, sequence control of synthetic polymers has been drawing great interest towards creation of more advanced functions like those of natural polymers,3–6 but the control remains a great challenge in polymer science.Some concepts or methodologies for controlling the monomer sequence in synthetic polymers have been reported, including controlled radical polymerization,7–11 ring-opening metathesis polymerization,12,13 DNA-templated synthesis,14,15 and iterative synthesis.16–35 One-by-one iterative methods, which are generally carried out on a solid support, enable complete regulated monomer sequences for artificial peptides and DNA. Other notable examples include the method for the synthesis of triazene-based polymers,36 poly(alkoxyamine amide)s,37 oligothioetheramides,38–40 and thiolactone aminolysis,41,42 and the multicomponent reaction.43–45 Although solid-phase synthesis enables simple purification, solution-phase synthesis is desired due to its homogenous reaction kinetics and non-necessity for removal of the products from the solid support.46,47
Peptoids (or poly-N-substituted glycines) are a class of biomimetic sequence-defined polymers that have found a wide range of biotechnological applications.48–57 Peptoids are routinely synthesized using the solid-phase submonomer approach developed by Zuckermann et al.58 Up to now, several hundred commercially available amines have been applied to synthesize peptoids with a wide array of side chains.53,54 Although significant progress has been made in peptoids, developing a novel strategy towards the de novo synthesis of sequence-defined peptoids is still imperative. To ensure sufficient molecular diversity and complexity, it is highly desirable to synthesis backbone-variants of traditional α-peptoids, such as β-peptoids, which can expand the structural and functional repertoire of peptoids.52,54,59,60 However, submonomer synthesis similar to that used for α-peptoids was determined to be less efficient for β-peptoids.61 This challenge and the need for increased molecular diversity prompt us to develop a new approach to generate sequence-defined peptoids with broad structural diversity, both of the side chains and the backbones.
Herein, we describe a general route for the assembly of sequence-specific polymers through iterative Ugi reactions of amino acid building blocks (Scheme 1; see the ESI† for the detailed mechanism of the Ugi reaction). We hypothesized that amino acids, which supply the carboxylic acid and amino functions, could sequentially react with an aldehyde and an isocyanide to form a peptoid structure. In order to achieve scalability, we employed the efficient solution-phase synthesis, which theoretically allowed scalable reactions.23–25 The Ugi reaction62–67 was chosen as a coupling strategy due to its ability to introduce broad structural diversity via simply employing different amino acids or isocyanides in each step of the reaction.
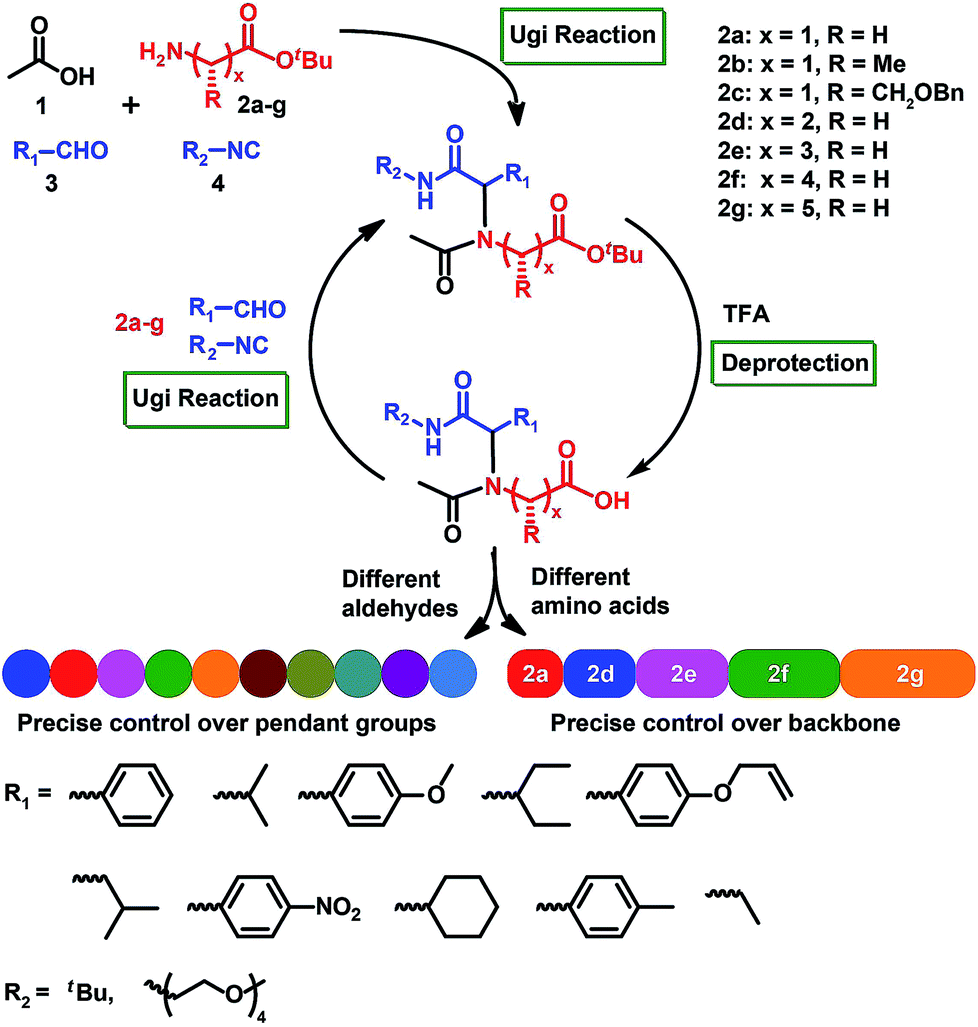 | ||
| Scheme 1 Synthesis strategy towards sequence-defined peptoids via amino acid building blocks and iterative Ugi reactions. | ||
Applying this approach, we performed the synthesis of sequence-defined peptoids with molar masses >4 kDa on a ∼4 g scale. After this effective strategy, we further demonstrated that the variation in the overall hydrophobicity and lower critical solution temperature (LCST) behaviours of these precisely defined peptoids could be logically altered by variation of the sequence. Finally, we showed that these sequence-defined materials could be conjugated to DNA quantitatively, allowing the development of novel therapeutics in a facile way.
Results and discussion
Amino acid building blocks
Our approach for achieving synthetic sequence control involves the synthesis of an amino acid-based building block. The building block framework includes a reactive amino group that can undergo Ugi reactions, and a tert-butyl ester protected carboxylic acid function that can undergo efficient deprotection. The amino acid building block can be synthesized from readily available amino acid starting materials (Fig. S1†). A unique feature of the amino acid platform is that a rich variety of amino acids enable the introduction of broad structural diversity into peptoid backbones. For this proof-of-concept study, we synthesized seven different amino acid building blocks 2a–g (Scheme 1, Fig. S1 and S16–S54†).A model short peptoid by iterative Ugi reactions
To verify the feasibility of our approach, we initiated the synthesis of a short peptoid with glycine tert-butyl ester, 2a. The amino functionality of 2a was reacted in an Ugi reaction with, acetic acid 1 as the starting substrate and benzaldehyde, and tert-butyl isocyanide. The use of a 0.1-fold excess of aldehyde and isocyanide is necessary to push the reaction to quantitative transformation (Fig. 1A and B). In addition, purification of the resultant product was realized by flash chromatography on silica using ethyl acetate/dichloromethane/petroleum ether (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) as the eluent. The resultant material of the Ugi reaction bears a tert-butyl ester moiety, which can be used for another Ugi reaction after quantitative hydrolysis with trifluoroacetic acid (TFA) and simple workup by evaporation. The structure of the product after deprotection was confirmed by 1H NMR via the disappearance of the tert-butyl groups at 1.43 ppm (Fig. S2†) and by MALDI-TOF-MS (Fig. S3†). The isolated yield over the two steps was 90%. After this first round of the Ugi reaction and deprotection, we continued peptoid synthesis with another round of reactions. Again, both reactions proceeded smoothly as determined by 1H NMR and MALDI-TOF-MS (Fig. 1C, and S4†). Additionally, it should be noted that the obtained peptoid based on building block 2a lacks chirality, due to the racemic characteristic of the Ugi product.68,69
:1:1) as the eluent. The resultant material of the Ugi reaction bears a tert-butyl ester moiety, which can be used for another Ugi reaction after quantitative hydrolysis with trifluoroacetic acid (TFA) and simple workup by evaporation. The structure of the product after deprotection was confirmed by 1H NMR via the disappearance of the tert-butyl groups at 1.43 ppm (Fig. S2†) and by MALDI-TOF-MS (Fig. S3†). The isolated yield over the two steps was 90%. After this first round of the Ugi reaction and deprotection, we continued peptoid synthesis with another round of reactions. Again, both reactions proceeded smoothly as determined by 1H NMR and MALDI-TOF-MS (Fig. 1C, and S4†). Additionally, it should be noted that the obtained peptoid based on building block 2a lacks chirality, due to the racemic characteristic of the Ugi product.68,69
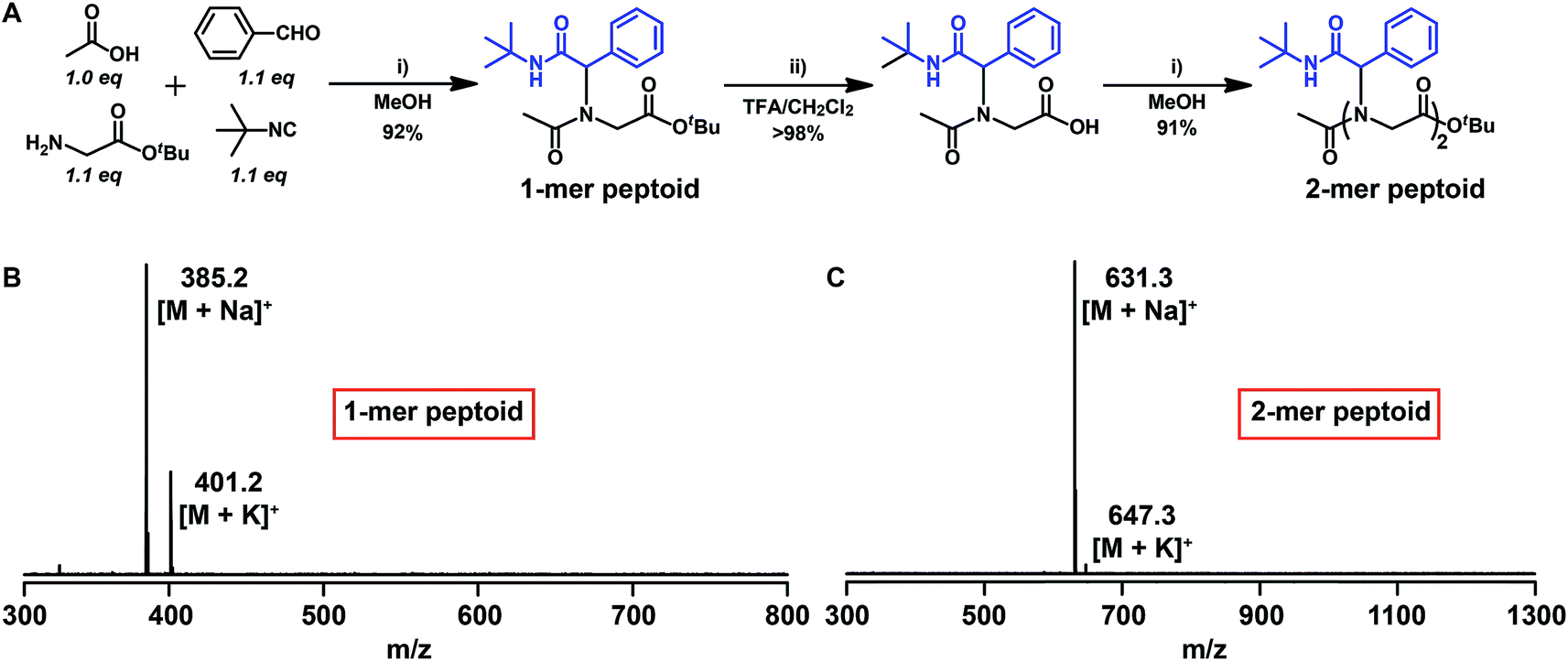 | ||
| Fig. 1 (A) Synthesis of a model short peptoid by iterative Ugi reactions. (B) MALDI-TOF-MS spectrum of a 1-mer peptoid. (C) MALDI-TOF-MS spectrum of a 2-mer peptoid. | ||
Sequence-defined peptoids with side-chain diversity via iterative Ugi reactions
Having demonstrated the efficient synthesis of the model short peptoid, we proceeded with the synthesis of 9-mer and 10-mer sequence polymers, peptoid 1 and peptoid 2 (Fig. 2A and B). Both polymers were synthesized with the same amino acid building block 2a but have different side chain sequences. The synthesis of peptoid 1 and peptoid 2 was followed and confirmed at intermediate steps with MALDI-TOF-MS and SEC (Fig. 2, S5–S6 and S85–S86†). Further structural confirmation of both peptoids was obtained via MALDI-TOF-MS. The observed parent ions of the two peptoids were identical at 2252.4 Da and 2534.1 Da (Fig. 2A and B), and matched with the theoretical value of 2252.0 Da and 2533.9 Da, respectively. Furthermore, by the application of different aldehydes for the Ugi reaction, we could incorporate ten different pendant groups into the sequence-specific peptoid 3, such as aliphatic, aromatic, and olefinic motifs (Fig. 2C, 3, S7 and S71†). We were pleased to note that the strategy for these sequence-defined peptoids is scalable, and multiple grams of linear peptoids were obtained. For instance, peptoid 3 (4.01 g) was produced in a 40% isolated yield over 19 steps as an off-white powder material (Fig. S8†), indicating the high efficiency of this chemistry. Therefore, using the above chemistry can afford sequence-defined polymers for valuable applications, facilitating the exploration of structure–property relationships accurately. Starting from bis–COOH–functionalized PEG, the sequence-defined block polymer (peptoid 4) was synthesized using the above chemistry. The SEC curve (Fig. 2D) of the obtained peptoid 4 shifted to higher molecular weights (Mn ∼ 4.1 kDa), which was consistent with the successful formation of the desired symmetrical sequence-specific products.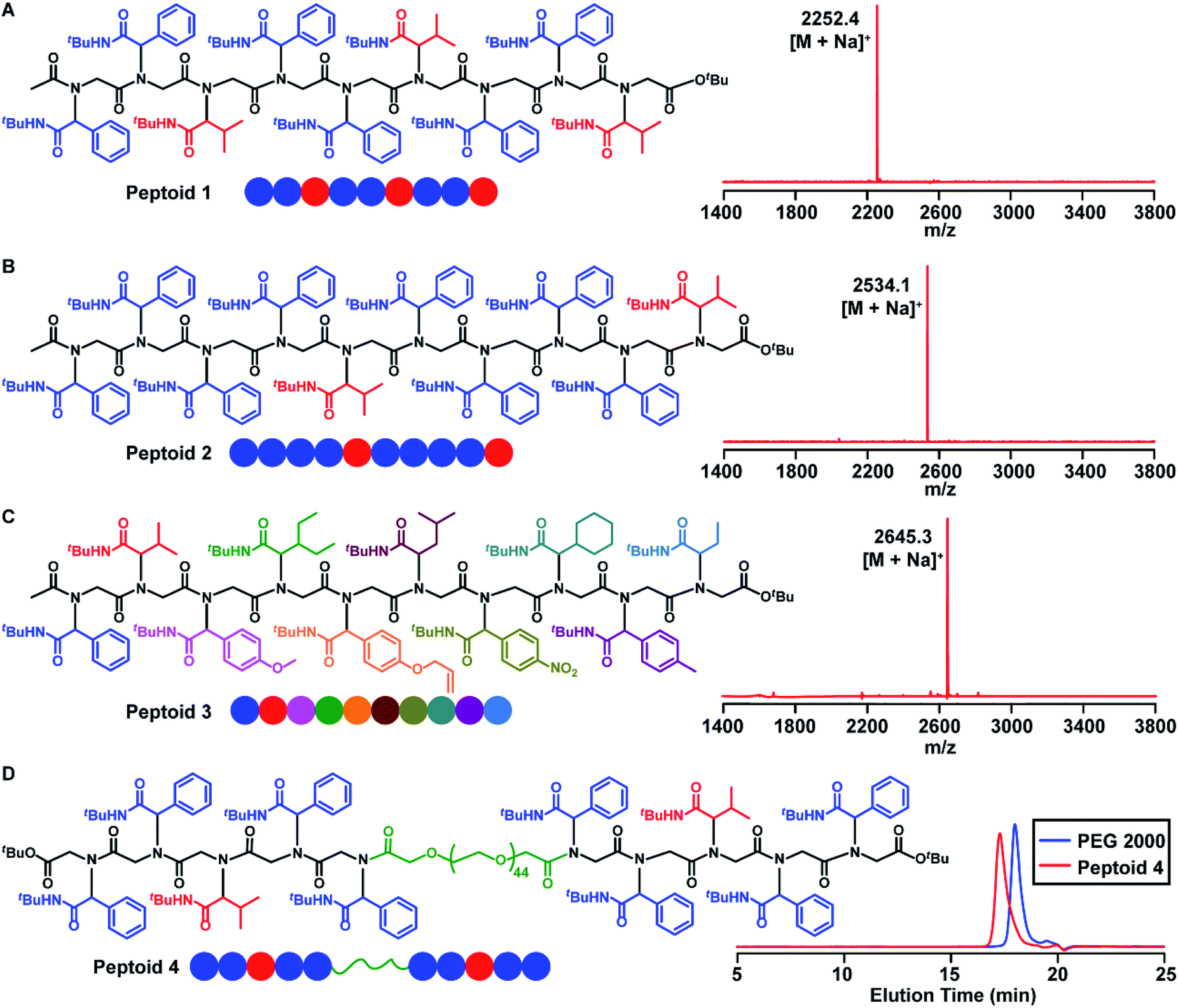 | ||
| Fig. 2 (A) Chemical structure and MALDI-TOF-MS spectrum of a 9-mer sequence polymer (peptoid 1). (B) Chemical structure and MALDI-TOF-MS spectrum of a 10-mer sequence polymer (peptoid 2). (C) Chemical structure and MALDI-TOF-MS spectrum of a 10-mer sequence polymer with ten different pendant groups (peptoid 3). (D) Chemical structure and SEC traces of a sequence-defined block polymer (peptoid 4). | ||
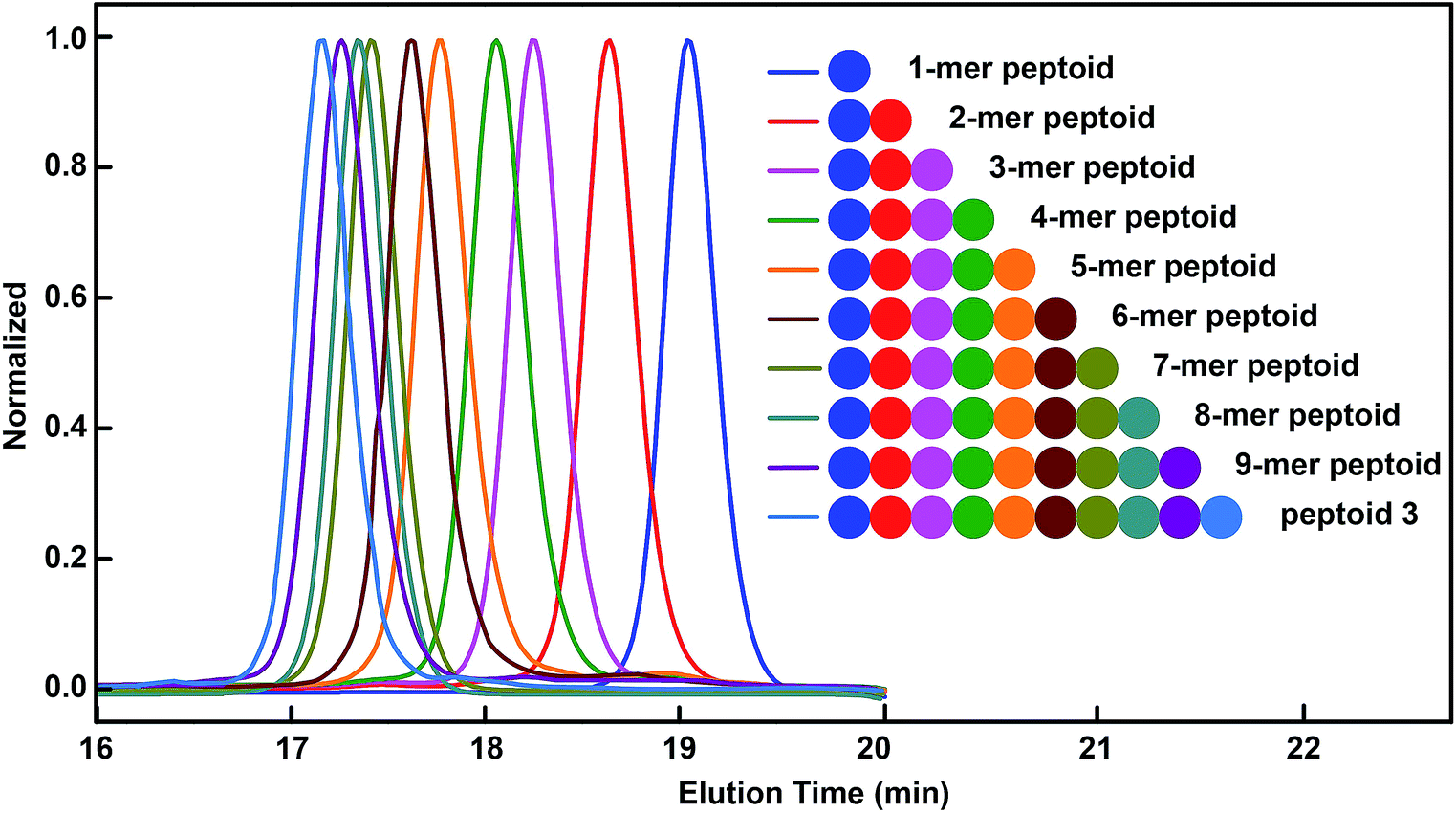 | ||
| Fig. 3 SEC traces obtained after Ugi reactions in the synthesis of the sequence-defined peptoid 3 (in THF, 35 °C, polystyrene as a standard). | ||
To illustrate the versatility of this strategy, we synthesized a 5-mer α-peptoid 5 (Fig. S9†) consisting of L-alanine (2b) and benzyl-protected L-serine (2c) building blocks. SEC and MALDI-TOF-MS spectra revealed its structural integrity. The corresponding peptoid 6 (Fig. S9 and S75†) with pendant hydroxyl groups was recovered by hydrolysis of tert-butyl ester followed by removal of the benzyl protecting group. The presence of hydroxyl groups in its side chains would enhance the hydrophilicity of the resultant peptoids, facilitating its biomedical applications.
Sequence-defined peptoids with backbone diversity via iterative Ugi reactions
Faced with the ever-present challenge of β-peptoid preparation using the submonomer method,54 we further synthesized a 10-mer β-peptoid 7 with β-alanine tert-butyl ester 2d as the building block, according to the aforementioned procedure (Fig. 4A, S76 and S87†). Importantly, the chemistry for β-peptoids is high efficient, and the isolated yield in each cycle over two successive steps (that is the Ugi reaction and deprotection) was ∼85%.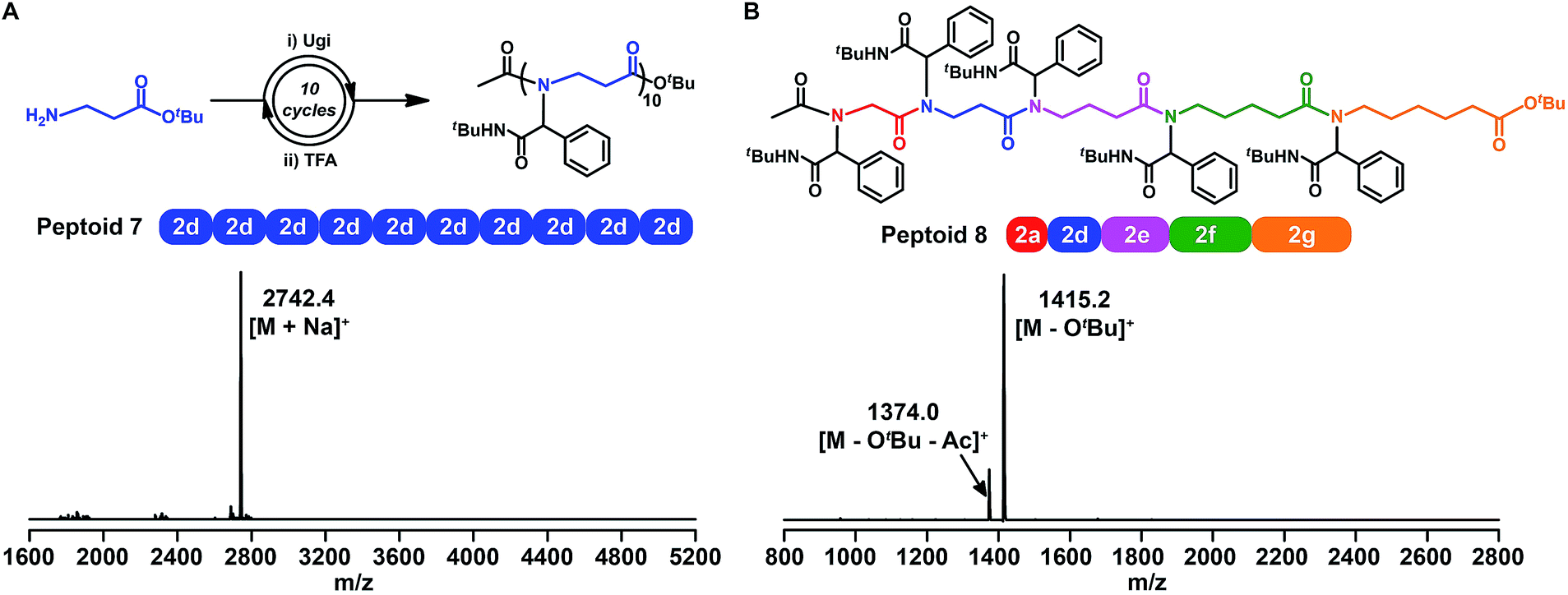 | ||
| Fig. 4 (A) Synthetic route and MALDI-TOF-MS spectrum of 10-mer β-peptoid 7 obtained by iterative Ugi reactions using β-alanine tert-butyl ester as the building block. (B) Chemical structure and MALDI-TOF-MS spectrum of a 5-mer peptoid consisting of all five amino acid building blocks (peptoid 8). | ||
The versatility and robustness of the above chemistry for building sequence-defined polymers were further demonstrated by constructing backbone-variants of traditional α-peptoids. We synthesized a 5-mer peptoid (Fig. 4B, S77 and S88†) consisting of all five amino acid building blocks. The resulting peptoid 8 was characterized by the MALDI-TOF-MS spectrum, and the doubly charged [M − OtBu]+, and [M − OtBu − Ac]+ ions were clearly observed. Thus, the iterative Ugi reaction combined with amino acid building blocks is indeed a very powerful new approach towards sequence-defined peptoids with backbone diversity.
Effect of the sequence structure of peptoids on materials properties
The above chemistry can afford an array of sequence-defined polymers for accurately exploring the structure–property relationship. In particular, the study of the properties of polymers with similar chemical structures but different sequence structures is a fundamental topic in the polymer science area. For this purpose, we employed the tert-butyl isocyanide and oligo-ethylene-glycol isocyanide to make 12-mer peptoids 9–12 containing hydrophobic and hydrophilic side chains (Fig. 5A and B, S10–S12, and S78–S81†). SEC measurements demonstrate the successful synthesis of the desired peptoids by the shifting towards higher molecular weights after each step (Fig. S89–S92†). Each of these peptoids differs in the placement fashion of the hydrophobic and hydrophilic units, which were grouped into a block size of one (peptoid 12), two (peptoid 11), three (peptoid 10), and six (peptoid 9). The resultant peptoids were soluble in cold water. The lower critical solution temperature (LCST) behaviours of these peptoids were investigated. As shown in Fig. 5C, a gradual increase in LCST was detected as the block size decreased from six in peptoid 9 to one in peptoid 12, demonstrating a gradual increase in the overall hydrophilicity. This may be due to the fact that the more water-soluble peptoid 12 may adopt a structure in which the hydrophilic OEG moieties provide better shielding of the hydrophobic portion from water.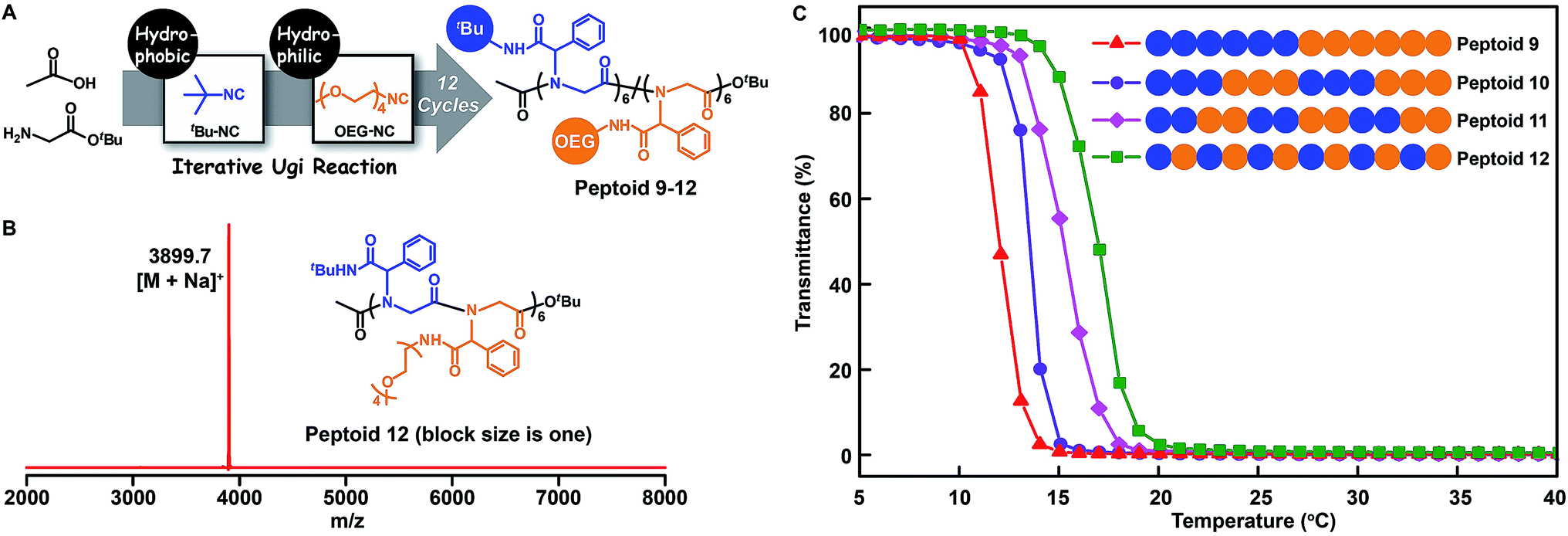 | ||
| Fig. 5 (A) Illustration of the side-chain sequence-regulated peptoids 9–12 synthesized by iterative Ugi reactions. (B) MALDI-TOF-MS spectrum of 12-mer peptoid 12. (C) Temperature dependence of transmittance for the aqueous solutions (2 mg mL−1) of sequence-regulated peptoids 9–12 (500 nm, heating at a rate of 1 °C min−1). | ||
Furthermore, to elucidate the backbone monomer sequence on the polymer properties, we applied the glycine tert-butyl ester 2a and oligo-ethylene-glycol based amino acid tert-butyl ester 2h (Fig. S1 and S55–S61†) as building blocks to make a series of main-chain sequence-regulated peptoids 13–15 (Fig. 6). Peptoids with ABB and AABBB sequence-regulated backbone structures, and peptoids with a block sequence in the backbone were verified by MALDI-TOF-MS and SEC measurements (Fig. 6B, S13–S14, and S93–S95†). The solubility of these peptoids was then assessed in the mixed solvents of EtOH and H2O at different temperatures via determining the light transmittance of the peptoid solutions. As shown in Fig. 6C, the peptoids with the same composition but different monomer sequences in the backbones, i.e., peptoid 13, peptoid 14 and peptoid 15, showed different solubilities; the peptoid with an ABB alternating sequence in the backbone (peptoid 15) was more soluble than the others: it became turbid at Tc = 45 °C (Tc: cloud point), whereas its block counterpart (peptoid 13) did so at a higher temperature, Tc = 53 °C. These experiments again suggested that not only the side-chain sequences but also the backbone sequences were important for determining the solubility of the peptoids and that the solubility could be gradually adjusted by both backbone and side-chain monomer sequences. Actually, the interaction preference between the polymer and solvent might be changed depending on the placement fashion of monomer units.
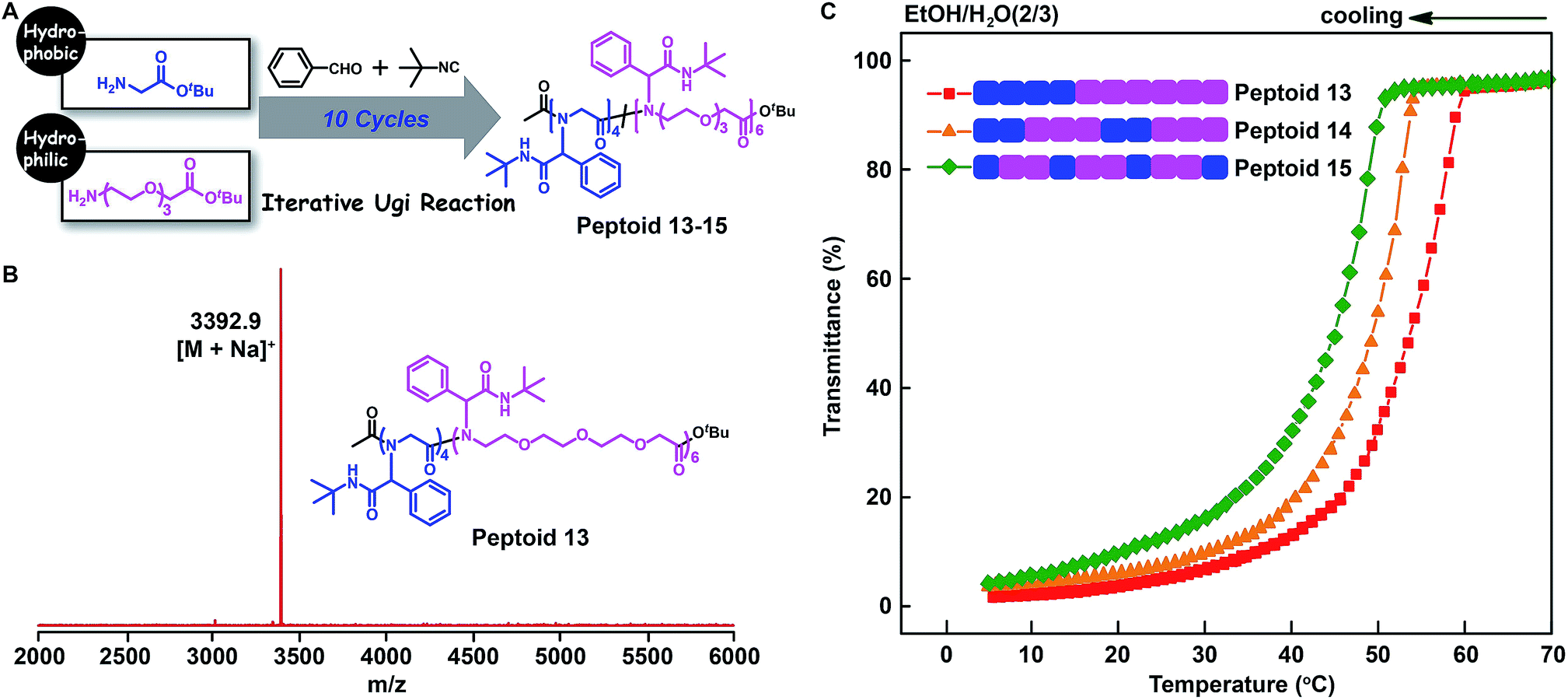 | ||
| Fig. 6 (A) Illustration of the backbone sequence-regulated peptoids 13–15 obtained by iterative Ugi reactions using glycine tert-butyl ester and oligo-ethylene-glycol based amino acid tert-butyl ester as building blocks. (B) MALDI-TOF-MS spectrum of 10-mer peptoid 13. (C) Transmittance of the backbone sequence-regulated peptoids 13–15 at varying temperatures in EtOH/H2O (2/3) (2 mg mL−1, 500 nm, cooling at a rate of 2 °C min−1). | ||
Bioconjugation of peptoids
One possible application of the resultant sequence-specific peptoids may be in nucleic acid delivery. In addition, research on the combination of synthetic materials and biologics through bioconjugation has been the subject of increased study because this integration enables the development of novel therapeutics.19,70 Therefore, we evaluated whether our peptoids could be coupled to DNA for biomedical applications. By applying the same Ugi reaction and 3-azidopropan-1-amine as a component, we attached an azide functionality to a peptoid (Fig. 7A and S15†). The azide-modified peptoid 16 was coupled to DNA with a dibenzocyclooctyne (DBCO) group under ambient conditions without catalysts with acetonitrile and PBS as solvents. DBCO-DNA specifically conjugated to peptoid 16 based on gel electrophoresis shifts, and a 24 h reaction with DNA: peptoid molar ratio = 1 was found to be efficient based on the disappearance of the unconjugated DNA gel band (Fig. 7B). Further verification of conjugation was obtained via MALDI-TOF-MS (Fig. 7C).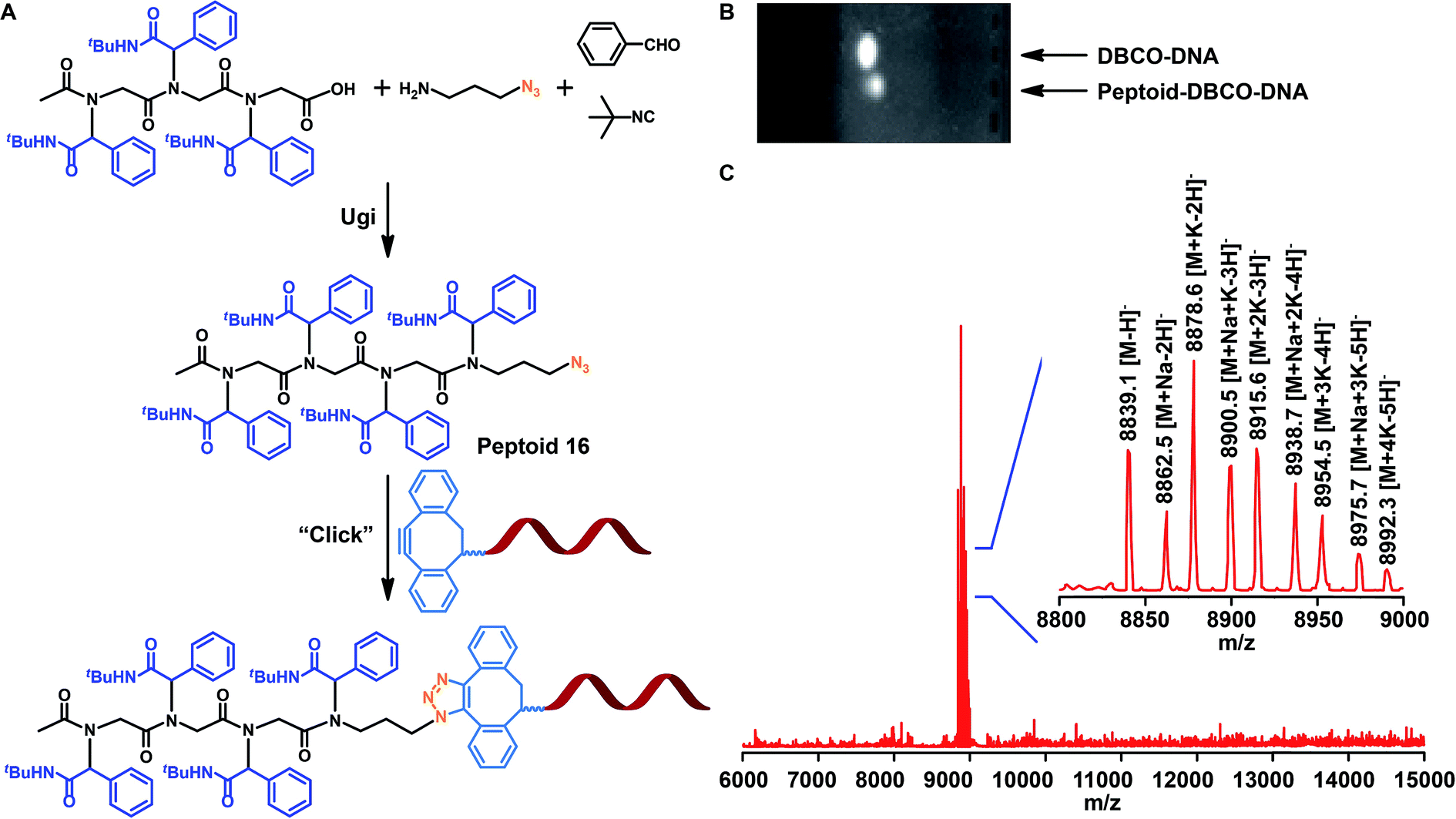 | ||
| Fig. 7 (A) Conjugation of sequence-defined peptoid 16 to DNA (DNA: peptoid molar ratio = 1, 24 h reaction in CH3CN/PBS). (B) Gel-electrophoresis result for the peptoid-DNA conjugation reaction. (C) MALDI-TOF-MS analysis of DNA conjugates. | ||
Conclusions
In summary, we have described herein the development of amino acid building blocks coupled with iterative Ugi reactions for the efficient and multigram-scale assembly of sequence-defined peptoids. This efficient chemistry provides much feasibility for structural diversity, synthetically varying and sequencing both the side chains and the backbones. Activating agents were unnecessary, rendering this strategy advantageous over other stepwise methods, such as the traditional peptide or peptoid synthesis. We further demonstrated that the alteration in the overall hydrophobicity and LCST behaviours of these precisely defined peptoids could be accordingly changed by variation of the sequence. Regulation of sequence specific hydrophobic aggregation within a polymer is a significant result. We also showed that these materials could be conjugated to biomacromolecules, allowing the development of novel therapeutics. We anticipate that this versatile strategy may afford new peptoids for application in therapeutics and as supramolecular foldamers or simple protein mimics for the investigation of advanced self-assembly driven by hydrophobic or other supramolecular interactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 21474101, 21704097 and 51873211), Jilin Science and Technology Bureau (Grant No. 20160414001GH and 20180201070GX), the Opening Foundations of Key Laboratory of Synthetic and Biological Colloids, Ministry of Education, Jiangnan University (Grant No. JDSJ2017-03) and State Key Laboratory of Supramolecular Structure and Materials, Jilin University (Grant No. sklssm201817).Notes and references
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS
.
- A. E. Stross, G. Iadevaia, D. Núñez-Villanueva and C. A. Hunter, J. Am. Chem. Soc., 2017, 139, 12655 CrossRef CAS PubMed
- N. Badi and J.-F. Lutz, Chem. Soc. Rev., 2009, 38, 3383 RSC
- M. Ouchi, N. Badi, J.-F. Lutz and M. Sawamoto, Nat. Chem., 2011, 3, 917 CrossRef CAS PubMed
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 628 CrossRef CAS PubMed
- S. C. Solleder, R. V. Schneider, K. S. Wetzel, A. C. Boukis and M. A. R. Meier, Macromol. Rapid Commun., 2017, 38, 1600711 CrossRef PubMed
- N. G. Engelis, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Nat. Chem., 2017, 9, 171 CrossRef CAS PubMed
- S. Pfeifer and J.-F. Lutz, J. Am. Chem. Soc., 2007, 129, 9542 CrossRef CAS PubMed
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef PubMed
- M. Ouchi, M. Nakano, T. Nakanishi and M. Sawamoto, Angew. Chem., Int. Ed., 2016, 55, 14584 CrossRef CAS PubMed
- M. Ojika, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2017, 56, 1789 CrossRef CAS PubMed
- W. R. Gutekunst and C. J. Hawker, J. Am. Chem. Soc., 2015, 137, 8038 CrossRef CAS PubMed
- J. Zhang, M. E. Matta and M. A. Hillmyer, ACS Macro Lett., 2012, 1, 1383 CrossRef CAS
- R. E. Kleiner, Y. Brudno, M. E. Birnbaum and D. R. Liu, J. Am. Chem. Soc., 2008, 130, 4646 CrossRef CAS PubMed
- D. M. Rosenbaum and D. R. Liu, J. Am. Chem. Soc., 2003, 125, 13924 CrossRef CAS PubMed
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149 CrossRef CAS
- K. Takizawa, C. Tang and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 1718 CrossRef CAS PubMed
- S. Binauld, D. Damiron, L. A. Connal, C. J. Hawker and E. Drockenmuller, Macromol. Rapid Commun., 2011, 32, 147 CrossRef CAS PubMed
- T. G. W. Edwardson, K. M. M. Carneiro, C. J. Serpell and H. F. Sleiman, Angew. Chem., Int. Ed., 2014, 53, 4567 CrossRef CAS PubMed
- Y.-H. Wu, J. Zhang, F.-S. Du and Z.-C. Li, ACS Macro Lett., 2017, 6, 1398 CrossRef CAS
- M. R. Golder, Y. Jiang, P. E. Teichen, H. V. T. Nguyen, W. Wang, N. Milos, S. A. Freedman, A. P. Willard and J. A. Johnson, J. Am. Chem. Soc., 2018, 140, 1596 CrossRef CAS PubMed
- Y. Jiang, M. R. Golder, H. V. T. Nguyen, Y. Wang, M. Zhong, J. C. Barnes, D. J. C. Ehrlich and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 9369 CrossRef CAS PubMed
- F. A. Leibfarth, J. A. Johnson and T. F. Jamison, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10617 CrossRef CAS PubMed
- J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810 CrossRef CAS PubMed
- Z. Huang, J. Zhao, Z. Wang, F. Meng, K. Ding, X. Pan, N. Zhou, X. Li, Z. Zhang and X. Zhu, Angew. Chem., Int. Ed., 2017, 56, 13612 CrossRef CAS PubMed
- G. Cavallo, A. Al Ouahabi, L. Oswald, L. Charles and J.-F. Lutz, J. Am. Chem. Soc., 2016, 138, 9417 CrossRef CAS PubMed
- N. Zydziak, W. Konrad, F. Feist, S. Afonin, S. Weidner and C. Barner-Kowollik, Nat. Commun., 2016, 7, 13672 CrossRef PubMed
- S. Binauld, C. J. Hawker, E. Fleury and E. Drockenmuller, Angew. Chem., Int. Ed., 2009, 48, 6654 CrossRef CAS PubMed
- B. Van Genabeek, B. F. M. de Waal, M. M. J. Gosens, L. M. Pitet, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2016, 138, 4210 CrossRef CAS PubMed
- F. Amir, Z. Jia and M. J. Monteiro, J. Am. Chem. Soc., 2016, 138, 16600 CrossRef CAS PubMed
- J. P. Sadighi, R. A. Singer and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 4960 CrossRef CAS
- T. Soejima, K. Satoh and M. Kamigaito, J. Am. Chem. Soc., 2016, 138, 944 CrossRef CAS PubMed
- H. J. Olivos, P. G. Alluri, M. M. Reddy, D. Salony and T. Kodadek, Org. Lett., 2002, 4, 4057 CrossRef CAS PubMed
- O. I. Paynter, D. J. Simmonds and M. C. Whiting, J. Chem. Soc., Chem. Commun., 1982, 1165 RSC
- J. S. Schumm, D. L. Pearson and J. M. Tour, Angew. Chem., Int. Ed., 1994, 33, 1360 CrossRef
- J. W. Grate, K.-F. Mo and M. D. Daily, Angew. Chem., Int. Ed., 2016, 55, 3925 CrossRef CAS PubMed
- R. K. Roy, A. Meszynska, C. Laure, L. Charles, C. Verchin and J.-F. Lutz, Nat. Commun., 2015, 6, 7237 CrossRef PubMed
- M. Porel, D. N. Thornlow, N. N. Phan and C. A. Alabi, Nat. Chem., 2016, 8, 590 CrossRef CAS PubMed
- M. Porel, D. N. Thornlow, C. M. Artim and C. A. Alabi, ACS Chem. Biol., 2017, 12, 715 CrossRef CAS PubMed
- M. Porel and C. A. Alabi, J. Am. Chem. Soc., 2014, 136, 13162 CrossRef CAS PubMed
- P. Espeel, L. L. G. Carrette, K. Bury, S. Capenberghs, J. C. Martins, F. E. Du Prez and A. Madder, Angew. Chem., Int. Ed., 2013, 52, 13261 CrossRef CAS PubMed
- S. Martens, J. Van den Begin, A. Madder, F. E. Du Prez and P. Espeel, J. Am. Chem. Soc., 2016, 138, 14182 CrossRef CAS PubMed
- S. C. Solleder and M. A. R. Meier, Angew. Chem., Int. Ed., 2014, 53, 711 CrossRef CAS PubMed
- S. C. Solleder, D. Zengel, K. S. Wetzel and M. A. R. Meier, Angew. Chem., Int. Ed., 2016, 55, 1204 CrossRef CAS PubMed
- S. C. Solleder, K. S. Wetzel and M. A. R. Meier, Polym. Chem., 2015, 6, 3201 RSC
- S. Pfeifer, Z. Zarafshani, N. Badi and J.-F. Lutz, J. Am. Chem. Soc., 2009, 131, 9195 CrossRef CAS PubMed
- R. L. Kanasty, A. J. Vegas, L. M. Ceo, M. Maier, K. Charisse, J. K. Nair, R. Langer and D. G. Anderson, Angew. Chem., Int. Ed., 2016, 55, 9529 CrossRef CAS PubMed
- N. Gangloff, J. Ulbricht, T. Lorson, H. Schlaad and R. Luxenhofer, Chem. Rev., 2016, 116, 1753 CrossRef CAS PubMed
- R. Luxenhofer, C. Fetsch and A. Grossmann, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2731 CrossRef CAS
- C. Hörtz, A. Birke, L. Kaps, S. Decker, E. Wächtersbach, K. Fischer, D. Schuppan, M. Barz and M. Schmidt, Macromolecules, 2015, 48, 2074 CrossRef
- G. L. Sternhagen, S. Gupta, Y. Zhang, V. John, G. J. Schneider and D. Zhang, J. Am. Chem. Soc., 2018, 140, 4100 CrossRef CAS PubMed
- D. Zhang, S. H. Lahasky, L. Guo, C.-U. Lee and M. Lavan, Macromolecules, 2012, 45, 5833 CrossRef CAS
- E. J. Robertson, A. Battigelli, C. Proulx, R. V. Mannige, T. K. Haxton, L. Yun, S. Whitelam and R. N. Zuckermann, Acc. Chem. Res., 2016, 49, 379 CrossRef CAS PubMed
- A. S. Knight, E. Y. Zhou, M. B. Francis and R. N. Zuckermann, Adv. Mater., 2015, 27, 5665 CrossRef CAS PubMed
- S. A. Fowler and H. E. Blackwell, Org. Biomol. Chem., 2009, 7, 1508 RSC
- T. D. Michelle, K. Rinki and E. B. Annelise, Curr. Pharm. Des., 2011, 17, 2732 CrossRef
- B. Yoo and K. Kirshenbaum, Curr. Opin. Chem. Biol., 2008, 12, 714 CrossRef CAS PubMed
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646 CrossRef CAS
- J. S. Laursen, J. Engel-Andreasen and C. A. Olsen, Acc. Chem. Res., 2015, 48, 2696 CrossRef CAS PubMed
- J. S. Laursen, P. Harris, P. Fristrup and C. A. Olsen, Nat. Commun., 2015, 6, 7013 CrossRef CAS PubMed
- C. A. Olsen, M. Lambert, M. Witt, H. Franzyk and J. W. Jaroszewski, Amino Acids, 2008, 34, 465 CrossRef CAS PubMed
- I. Ugi, Angew. Chem., Int. Ed., 1982, 21, 810 CrossRef
- A. Llevot, A. C. Boukis, S. Oelmann, K. Wetzel and M. A. R. Meier, Top. Curr. Chem., 2017, 375, 66 CrossRef PubMed
- B. Yang, Y. Zhao, Y. Wei, C. Fu and L. Tao, Polym. Chem., 2015, 6, 8233 RSC
- B. Yang, Y. Zhao, S. Wang, Y. Zhang, C. Fu, Y. Wei and L. Tao, Macromolecules, 2014, 47, 5607 CrossRef CAS
- X. Zhang, S. Wang, J. Liu, Z. Xie, S. Luan, C. Xiao, Y. Tao and X. Wang, ACS Macro Lett., 2016, 5, 1049 CrossRef CAS
- Y. Tao, S. Wang, X. Zhang, Z. Wang, Y. Tao and X. Wang, Biomacromolecules, 2018, 19, 936 CrossRef CAS PubMed
- J. Zhang, P. Yu, S.-Y. Li, H. Sun, S.-H. Xiang, J. Wang, K. N. Houk and B. Tan, Science, 2018, 361, 1087 CrossRef CAS PubMed
- Q. Wang, D.-X. Wang, M.-X. Wang and J. Zhu, Acc. Chem. Res., 2018, 51, 1290 CrossRef CAS PubMed
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater, 2013, 12, 967 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03415j |
| This journal is © The Royal Society of Chemistry 2019 |