
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal–salen molecular cages as efficient and recyclable heterogeneous catalysts for cycloaddition of CO2 with epoxides under ambient conditions†
Chee Koon
Ng
 ab,
Ren Wei
Toh
a,
Ting Ting
Lin
b,
He-Kuan
Luo
b,
T. S. Andy
Hor
*c and
Jie
Wu
*a
ab,
Ren Wei
Toh
a,
Ting Ting
Lin
b,
He-Kuan
Luo
b,
T. S. Andy
Hor
*c and
Jie
Wu
*a
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore. E-mail: andyhor@hku.hk; chmjie@nus.edu.sg
bInstitute of Materials Research and Engineering, Agency for Science, Technology and Research, #08-03, 2 Fusionopolis Way, Innovis, Singapore 138634, Singapore
cDepartment of Chemistry, The University of Hong Kong, Pokfulam, Hong Kong SAR, China
First published on 29th November 2018
Abstract
A salen based molecular cage, salen@cage, was synthesized and complexed with Co and Al to yield metal–salen molecular cages, Co(II)@cage, Co(III)@cage and Al(III)@cage. These cages were demonstrated to be efficient heterogeneous catalysts for the cycloaddition of CO2 with styrene oxide, achieving full conversion at 25 °C and 1 atm CO2. Good to excellent yields of various cyclic carbonates were also achieved under mild conditions. Al(III)@cage can be reused up to five times without any significant loss of its high catalytic activity. The capability to access a variety of heterogeneous organometallic catalysts with salen@cage offers new prospects for practical CO2 utilization and chemical manufacturing.
Introduction
Molecular cages have been of much interest over the past decade as these materials have a wide range of applications from molecular recognition1 to chemical sensors.2 Moreover, their high surface area and porous structure allow them to find applications in gas separation and storage.3 The reactive sites found within these porous molecular cages are suitable for catalytic applications as they can be used to stabilize metal nanoparticles like Pd4 and Rh,5 increasing their catalytic activities towards organic transformations. Molecular cages can also trap both homogeneous catalysts and substrates via specific binding sites. This preorganization brings the encapsulated substrates and catalyst into a confined cavity which promotes the catalytic process.6Salen ligands and their metal complexes have been well studied over the last two decades in homogeneous catalysis.7 These ligands are versatile as they can coordinate with many transition metals, main group metals, and even lanthanides. Their complexes have been employed as catalysts in a wide variety of organic transformations, e.g. epoxidation of alkenes, Diels–Alder reactions, oxidation, ring opening of epoxides, Michael addition and reduction of ketones.8 One of the most important application of these metal salen complexes is to catalyze the cycloaddition of CO2 with epoxides to form cyclic carbonates. The increasing anthropogenic emissions of CO2 have resulted in excessive global warming, and thus the efficient utilization of CO2 as a C1 source is an appealing subject of investigation.9 Co(III) and Al(III) salen complexes were effective catalysts for the cycloaddition of CO2 to epoxides and the reaction usually proceeds under mild conditions.10 The cyclic carbonate products obtained have a range of different applications, including being used as green solvents,11 electrolytes in lithium-ion batteries12 and precursors in organic synthesis as intermediates to important chemicals like glycols, polyurethanes, dialkyl carbonates, carbamates, purines and pyrimidines. Although homogeneous catalysts like Co(III) and Al(III) salen complexes13 were effective at coupling CO2 with epoxides,14 they suffer from poor catalyst separation and low recyclability. On the other hand, heterogeneous catalysts like metal oxides,15 supported metal complexes,16 metal organic frameworks (MOFs)17 and porous polymers18 have been investigated for the production of cyclic carbonates from CO2, but many of these catalysts require elevated temperatures and pressures or long reaction durations. Therefore, heterogenization of these metal salen complexes to obtain highly efficient catalysts is important not only for the CO2 cycloaddition, but also for shedding light on the diverse metal salen-mediated chemical transformations.8
We herein report that by utilizing dynamic imine condensation,19 molecular cages incorporated with the salen moiety (salen@cage) can be conveniently synthesized in moderate yields, which can undergo complexation with different metal precursors to give Co(II)@cage, Co(III)@cage and Al(III)@cage. These cages show excellent catalytic reactivity for the cycloaddition of epoxide with CO2, giving full conversions of styrene oxide at room temperature and 1 atm CO2, probably because porous molecular cages serve to concentrate CO2 in the pores of the catalyst.
Results and discussion
Synthesis and characterization of salen@cage
The imine-based organic salen@cage was synthesized by the Schiff base reaction as shown in Scheme 1.20 In the ESI-MS spectrum, the salen@cage compound showed only one sharp signal at m/z 1447.83 with the expected isotopic pattern which corresponded to the cation of [2 + 3] salen@cage (Fig. S1 in the ESI†). MALDI-TOF MS in the m/z range of 750–8000 illustrated no further signals other than m/z 1448.0, which excluded the formation of smaller (e.g. [2 + 1] and [2 + 2]) or larger (e.g. [4 + 6] or up to [10 + 15]) condensation cages (Fig. S2 in the ESI†). The formation of salen@cage was further corroborated by high resolution mass spectroscopy, solid state 13C cross polarization magic angle spinning NMR and FT-IR (Fig. S3 and S4a in the ESI†). According to dynamic covalent chemistry, the reversible nature of the imine bonds, high reaction temperature and long reaction duration allowed for the most thermodynamically stable product to be selectively formed in equilibrium.19c,21 The [2 + 3] molecular prism4 was the most enthalpically favoured (least bond angle strain) and entropically favoured (least number of reactants).3d The use of other lower boiling point solvents (e.g. CHCl3 and THF) and shorter reaction durations resulted in the formation of smaller [2 + 2], [2 + 1] and [1 + 2] cages detected by ESI-MS.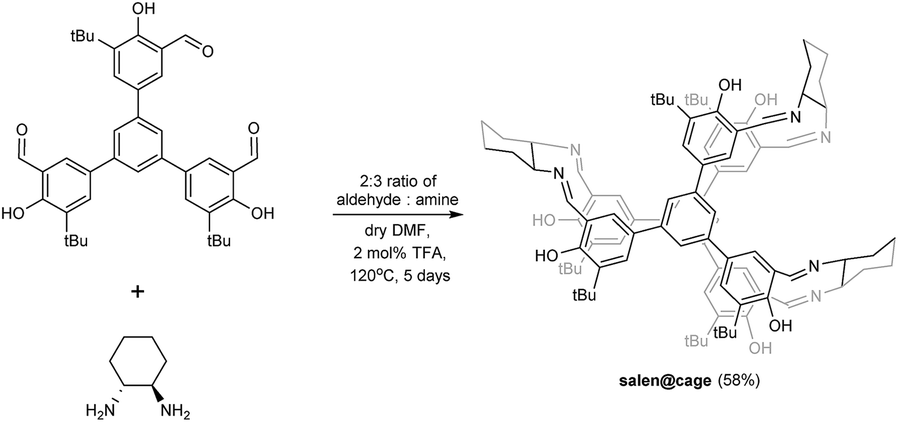 | ||
| Scheme 1 Synthesis of salen@cage. | ||
Synthesis and characterization of Co(II)@cage, Co(III)@cage and Al(III)@cage
Considering the high catalytic activity of Co and Al salen complexes in cycloaddition of CO2 with epoxides,13a,18a,22 Co and Al were subsequently coordinated onto salen@cage via reactions with Co(OAc)2 and Al(OEt)3, respectively (Scheme 2). Further purification of the compounds was done by Soxhlet extraction. Co(II)@cage, Co(III)@cage and Al(III)@cage were all insoluble in common organic solvents, similar to salen@cage. Upon metalation, the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching of the imine bond shifts from 1630 cm−1 in salen@cage to 1607 cm−1 in Co(II)@cage, 1609 cm−1 in Co(III)@cage and 1627 cm−1 in Al(III)@cage (Fig. S4b in the ESI†). The shifts to a lower frequency in FT-IR indicates that Co and Al have been coordinated onto the salen@cage.23 However, a small shoulder at around 1630 cm−1 can still be seen for Co(II)@cage and Co(III)@cage, which may suggest incomplete metalation for these two cage complexes which was confirmed by elemental analysis (Table S1 in the ESI†). In addition, the positive shifts of the N 1s peak in Co(II)@cage, Co(III)@cage and Al(III)@cage as compared to salen@cage in XPS (Fig. S5, ESI†) corroborates the coordination of Co and Al onto the salen@cage.24 The Al 2p peak in the XPS spectrum (Fig. S6a, ESI†) indicates that aluminium is in the +3 oxidation state in Al(III)@cage. The binding energies of the Co 2p peaks in the XPS spectrum (Fig. S6b, ESI†), together with the presence of observable satellite peaks,25 corroborate the +2 oxidation state of cobalt in Co(II)@cage while the slightly higher binding energies of the Co 2p peaks (Fig. S6c, ESI†) and the absence of satellite peaks indicate that the cobalt in Co(III)@cage is in the +3 oxidation state.26
N stretching of the imine bond shifts from 1630 cm−1 in salen@cage to 1607 cm−1 in Co(II)@cage, 1609 cm−1 in Co(III)@cage and 1627 cm−1 in Al(III)@cage (Fig. S4b in the ESI†). The shifts to a lower frequency in FT-IR indicates that Co and Al have been coordinated onto the salen@cage.23 However, a small shoulder at around 1630 cm−1 can still be seen for Co(II)@cage and Co(III)@cage, which may suggest incomplete metalation for these two cage complexes which was confirmed by elemental analysis (Table S1 in the ESI†). In addition, the positive shifts of the N 1s peak in Co(II)@cage, Co(III)@cage and Al(III)@cage as compared to salen@cage in XPS (Fig. S5, ESI†) corroborates the coordination of Co and Al onto the salen@cage.24 The Al 2p peak in the XPS spectrum (Fig. S6a, ESI†) indicates that aluminium is in the +3 oxidation state in Al(III)@cage. The binding energies of the Co 2p peaks in the XPS spectrum (Fig. S6b, ESI†), together with the presence of observable satellite peaks,25 corroborate the +2 oxidation state of cobalt in Co(II)@cage while the slightly higher binding energies of the Co 2p peaks (Fig. S6c, ESI†) and the absence of satellite peaks indicate that the cobalt in Co(III)@cage is in the +3 oxidation state.26
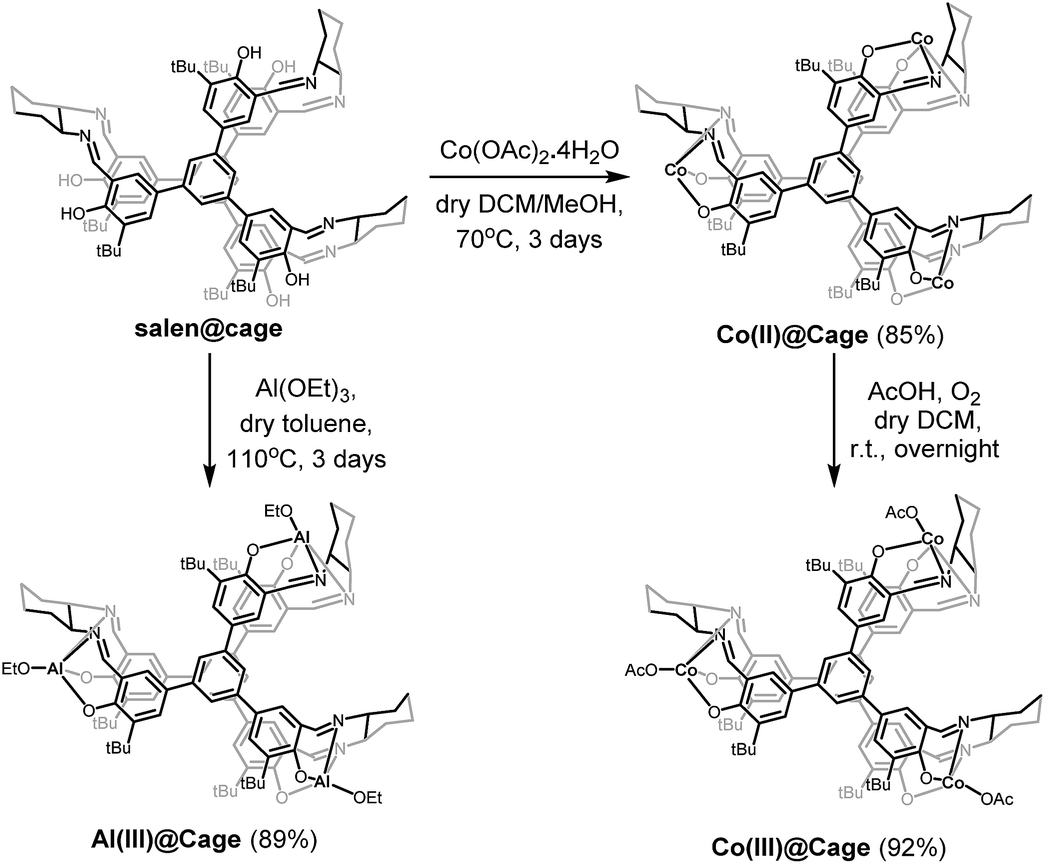 | ||
| Scheme 2 Synthesis of Co(II)@cage, Co(III)@cage and Al(III)@cage. | ||
We then studied the porosity and CO2 adsorption of these materials as these properties would affect the CO2 uptake and conversion. The porosity of these materials was studied by gas sorption experiments with N2 at 77 K and CO2 at 298 K (Fig. S9 and S10 in the ESI†). All the cages showed type I nitrogen gas adsorption isotherms according to the IUPAC classifications,27 indicating that these cage complexes consisted of both micropores and mesopores. DFT calculations of the molecular structure of Al(III)@cage suggested that the micropores (<2 nm) likely originated from the intramolecular space within the cage compound (Fig. S11 in the ESI†), while the mesopores (2–50 nm) might have originated from the intermolecular packing between each cage molecule (Fig. S12 in the ESI†). All the synthesized cage complexes were considered to be moderately porous, with Al(III)@cage displaying a higher BET surface area (771 m2 g−1) as compared to the other cage complexes (610–635 m2 g−1) (Table S2 in the ESI†). The CO2 adsorption capability of salen@cage (35.8 mg g−1) increased once it was coordinated to a metal (Co or Al). This was likely due to the Lewis acidic metal sites which polarized CO2 and led to greater adsorption.28 Due to the larger surface area, Al(III)@cage displayed a larger CO2 adsorption capability (70.4 mg g−1) as compared to the rest of the cage complexes (35.8–49.2 mg g−1), which was comparable to that of the conjugated microporous polymers synthesized by Deng et al.18a
Cycloaddition of CO2 with epoxides
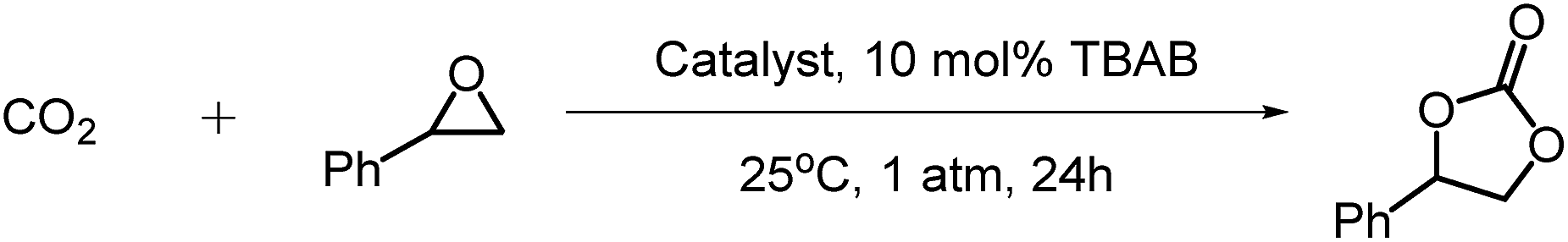
We then investigated the cycloaddition of styrene oxide (SO) with CO2 to give styrene carbonate (SC) using our cage complexes at 25 °C and 1 atm CO2. Only 9% conversion was achieved after 24 hours in the absence of a catalyst (entry 1, Table 1). Using salen@cage as a catalyst did not improve the conversion (8%, entry 2). This proved that the metal centres in the metal cages were the active catalytic sites and the salen@cage served as the framework to support the metal salen complexes and to increase the surface area of the catalyst for the efficient diffusion of substrates (CO2 and SO). The heterogeneous Co(III)@cage proved to be more active than Co(II)@cage (100% vs. 55%, entries 4 and 3 respectively), because the more electrophilic Co(III) could coordinate strongly with the epoxide and activated it for ring opening.13a Co(III)@cage was also more efficient than Al(III)@cage (100% vs. 75%, entries 4 and 7 respectively), although Al(III)@cage could also achieve full conversion after 48 h (entry 8). DFT calculations by Deng and co-workers suggested that the Co–salen catalysts gave better catalytic activity as compared to the Al–salen catalysts due to the lower activation barrier of the Co–salen catalysts.29 When the catalyst loading of Co(III)@cage was reduced to 0.17 mol%, the conversion of SO to SC decreased to 76% (entry 5). A longer reaction duration (48 instead of 24 h) at this reduced catalyst loading could achieve 98% conversion (entry 6). The homogeneous metal salts Co(OAc)2·4H2O and Al(OEt)3 exhibited much lower reactivity (59% and 24%, entries 9 and 10) as compared to the heterogeneous Co(III)@cage and Al(III)@cage. Previous reports on utilizing molecular cage complexes for CO2 cycloaddition are rare, although some molecular cages showed selective uptake of CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20,30 and were able to trap CO2 as carbonate anions within the cage framework.31 In 2014, Martinez and Dufaud demonstrated that an azaphosphatrane–hemicryptophane cage complex was able to catalyze the CO2 cycloaddition to give styrene carbonate in 82% yield at 100 °C and 1 atm CO2.32
20,30 and were able to trap CO2 as carbonate anions within the cage framework.31 In 2014, Martinez and Dufaud demonstrated that an azaphosphatrane–hemicryptophane cage complex was able to catalyze the CO2 cycloaddition to give styrene carbonate in 82% yield at 100 °C and 1 atm CO2.32
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Catalyst loading/mol% | Conversionb/% |
| a Typical reaction conditions: 5.0 mmol styrene oxide, 0.5 mmol TBAB, and catalyst under 1 atm CO2 pressure at 25 °C for 24 h. b Conversions calculated from the crude 1H NMR spectra. c Reaction time = 48 h. d Corresponding to approximately 1 mol% of Co. e Corresponding to approximately 0.5 mol% of Co. f Corresponding to approximately 1 mol% of Al. | |||
| 1 | NIL | NIL | 9 |
| 2 | salen@cage | 0.33 | 8 |
| 3 | Co(II)@cage | 0.33d | 55 |
| 4 | Co(III)@cage | 0.33d | >99 |
| 5 | Co(III)@cage | 0.17e | 76 |
| 6 | Co(III)@cage | 0.17e | 98c |
| 7 | Al(III)@cage | 0.33f | 75 |
| 8 | Al(III)@cage | 0.33f | >99c |
| 9 | Co(OAc)2·4H2O | 1 | 59 |
| 10 | Al(OEt)3 | 1 | 24 |
We also investigated the enantioselectivity of the CO2 cycloaddition as the metal cage complexes were synthesized with chiral (S,S)-trans-1,2-diaminocyclohexane, which might impart a chiral environment to the heterogeneous catalysts. A low selectivity (13.6% ee) was obtained during the cycloaddition of CO2 with propylene oxide. However, the ee decreased to 2.3% when styrene oxide was used as the substrate (Fig. S15 and S16, ESI†).22b,33
The recyclability of the metal cage complexes was subsequently evaluated. Al(III)@cage retained most of its catalytic activity even after five runs, with the conversions dropping only slightly from 100% to 94% (Fig. 1). ICP-OES analysis of the reaction mixture after filtering shows only 52 ppm of Al, indicating negligible leaching of Al into the reaction mixture. However, Co(III)@cage performed less convincingly in its recyclability testing, with conversions dropping from 100% to 67% (Fig. S17 in the ESI†). Finally, we investigated the epoxide scope with Al(III)@cage under optimized conditions (Fig. 2). Good to excellent yields of the cyclic carbonates were achieved under mild conditions, illustrating that this catalytic system was effective with both alkyl and aryl epoxides, tolerating functionalities including halides, ethers, alkenes and alkynes. The modest yield for propylene carbonate was due to the high volatility of propylene oxide even at room temperature. In comparison with other high performing heterogeneous catalysts for this transformation, the catalytic activity of metal salen cages outperforms that of many supported metal complexes,16b–d,34, porous polymers35 and MOFs,36 but there have been recent reports of some MOFs37 and metalated porous organic polymers (POPs)18a,d–f,38 with similar or higher catalytic activities. The advantages of these cages include high stability, ease of synthesis, use of readily available materials, ability to adapt to different metals, and good recyclability with high productivity. Further optimisation can be focused on the control of porosity through molecular manipulation of the salen framework and its metal compatibility.
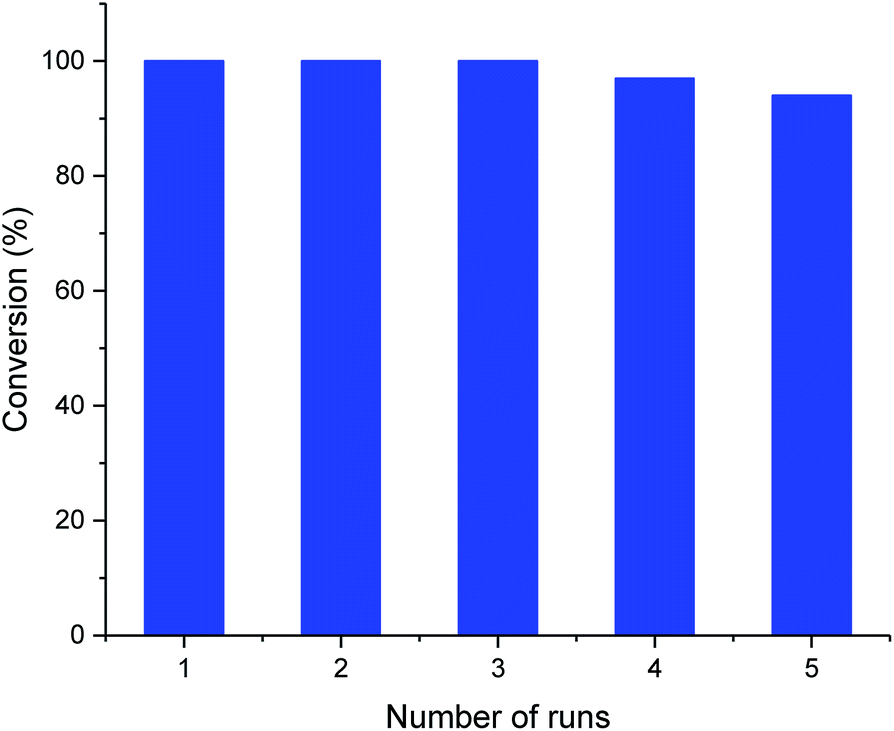 | ||
| Fig. 1 Recyclability of Al(III)@cage. Typical reaction conditions: 5.0 mmol styrene oxide, 10 mol% TBAB and 0.33 mol% Al(III)@cage under 1 atm CO2 pressure at 25 °C for 48 h. | ||
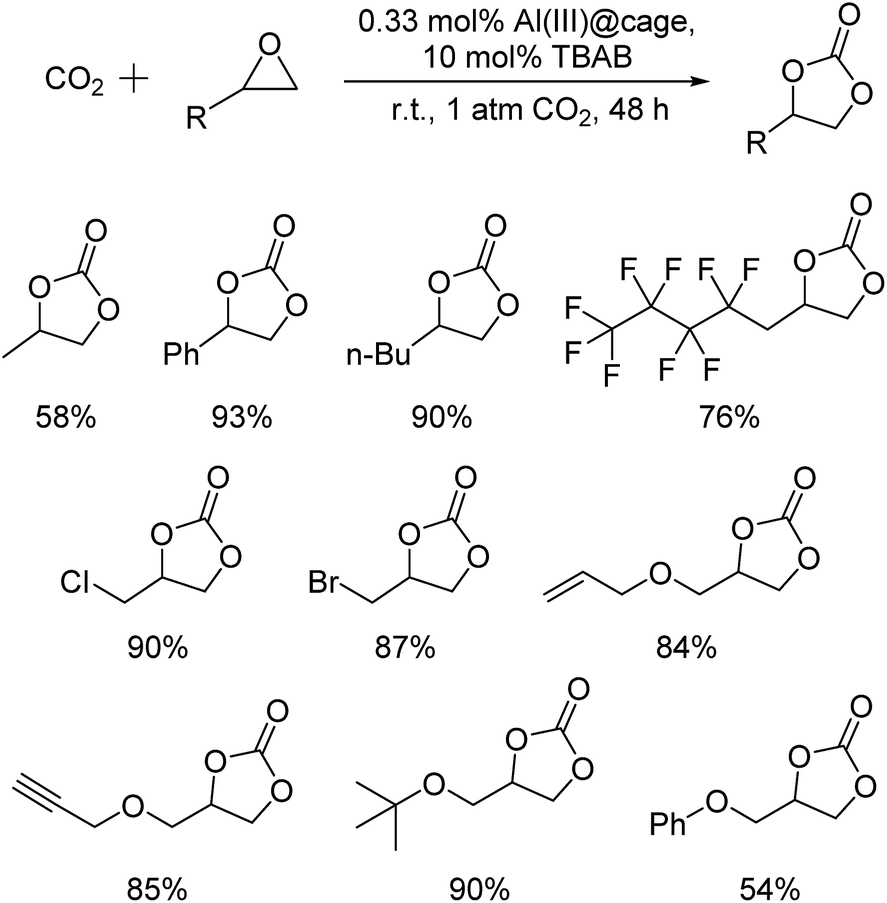 | ||
| Fig. 2 Isolated yields of cyclic carbonates formed by the cycloaddition of CO2 with epoxides catalyzed by Al(III)@cage; reaction conditions: 1 mmol epoxide, 0.33 mol% Al(III)@cage, 10 mol% TBAB, r.t., 1 atm CO2, and 48 h. | ||
Conclusions
We have successfully synthesized and characterized a salen-based [2 + 3] molecular cage, salen@cage. As a proof of concept for the heterogenization of metal salen complexes, Co and Al were coordinated onto the salen@cage to give Co(II)@cage, Co(III)@cage and Al(III)@cage in excellent yields. Co(III)@cage and Al(III)@cage proved to be excellent heterogeneous catalysts for the cycloaddition of CO2 with styrene oxide, giving full conversions of styrene oxide to styrene carbonate at 25 °C and 1 atm CO2 with only 0.33 mol% catalyst loading. Al(III)@cage could be reused up to five times without any significant decrease in its catalytic activity. The catalytic performances of molecular cage catalysts exceed that of many other heterogeneous CO2 cycloaddition catalysts under mild conditions which makes them suitable heterogeneous catalysts for CO2 conversion under ambient conditions. Moreover, our study opens the possibility of utilizing other metals with the salen@cage framework to yield a variety of efficient heterogeneous metal salen catalysts without the use of external heterogeneous supports like silica. These heterogeneous metal salen catalysts can potentially be applied in a variety of other transformations and also can be fitted for continuous-flow synthesis as a packed-bed catalyst. These studies are currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support provided by GSK-EDB (R-143-000-687-592), A*STAR RIE2020 AME (A1783c0013). We would like to acknowledge the A*STAR Computational Resource Centre for their support of the computation work through the use of its high performance computing facilities.Notes and references
- (a) F. Hof, S. L. Craig, C. Nuckolls and J. J. Rebek, Angew. Chem., Int. Ed., 2002, 41, 1488–1508 CrossRef CAS; (b) A. V. Leontiev and D. M. Rudkevich, Chem. Commun., 2004, 1468–1469 RSC; (c) J. Rebek, Angew. Chem., Int. Ed., 2005, 44, 2068–2078 CrossRef CAS PubMed; (d) S. M. Biros and J. J. Rebek, Chem. Soc. Rev., 2007, 36, 93–104 RSC.
- (a) A. P. Bisson, V. M. Lynch, M.-K. C. Monahan and E. V. Anslyn, Angew. Chem., Int. Ed., 1997, 36, 2340–2342 CrossRef CAS; (b) Y. Ferrand, M. P. Crump and A. P. Davis, Science, 2007, 318, 619–622 CrossRef CAS PubMed.
- (a) J. L. Atwood, L. J. Barbour and A. Jerga, Science, 2002, 296, 2367–2369 CrossRef CAS PubMed; (b) K. Swaminathan Iyer, M. Norret, S. J. Dalgarno, J. L. Atwood and C. L. Raston, Angew. Chem., Int. Ed., 2008, 47, 6362–6366 CrossRef PubMed; (c) T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed; (d) Y. Jin, B. A. Voss, A. Jin, H. Long, R. D. Noble and W. Zhang, J. Am. Chem. Soc., 2011, 133, 6650–6658 CrossRef CAS; (e) M. Petryk, J. Szymkowiak, B. Gierczyk, G. Spólnik, Ł. Popenda, A. Janiak and M. Kwit, Org. Biomol. Chem., 2016, 14, 7495–7499 RSC.
- B. Mondal, K. Acharyya, P. Howlader and P. S. Mukherjee, J. Am. Chem. Soc., 2016, 138, 1709–1716 CrossRef CAS PubMed.
- J.-K. Sun, W.-W. Zhan, T. Akita and Q. Xu, J. Am. Chem. Soc., 2015, 137, 7063–7066 CrossRef CAS PubMed.
- (a) W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Nat. Chem., 2016, 8, 231 CrossRef CAS; (b) Q.-Q. Wang, S. Gonell, S. H. A. M. Leenders, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Nat. Chem., 2016, 8, 225 CrossRef CAS PubMed; (c) C. Tan, J. Jiao, Z. Li, Y. Liu, X. Han and Y. Cui, Angew. Chem., Int. Ed., 2017, 57, 2085–2090 CrossRef PubMed; (d) J. Jiao, C. Tan, Z. Li, Y. Liu, X. Han and Y. Cui, J. Am. Chem. Soc., 2018, 140, 2251–2259 CrossRef CAS PubMed.
- S. J. Wezenberg and A. W. Kleij, Angew. Chem., Int. Ed., 2008, 47, 2354–2364 CrossRef CAS PubMed.
- (a) P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC; (b) K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS.
- (a) D. Yu, S. P. Teong and Y. Zhang, Coord. Chem. Rev., 2015, 293–294, 279–291 CrossRef CAS; (b) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (c) J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS PubMed; (d) Q. W. Song, Z. H. Zhou and L. N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- (a) A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS; (b) M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kuhn, ChemSusChem, 2015, 8, 2436–2454 CrossRef CAS PubMed; (c) F. D. Bobbink, D. Vasilyev, M. Hulla, S. Chamam, F. Menoud, G. Laurenczy, S. Katsyuba and P. J. Dyson, ACS Catal., 2018, 8, 2589–2594 CrossRef CAS; (d) R. R. Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2018, 8, 419–450 CrossRef CAS.
- (a) M. North, F. Pizzato and P. Villuendas, ChemSusChem, 2009, 2, 862–865 CrossRef CAS PubMed; (b) B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- S. S. Zhang, J. Power Sources, 2006, 162, 1379–1394 CrossRef CAS.
- (a) A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS PubMed; (b) X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- W.-L. Dai, S.-L. Luo, S.-F. Yin and C.-T. Au, Appl. Catal., A, 2009, 366, 2–12 CrossRef CAS.
- (a) N. A. M. Razali, K. T. Lee, S. Bhatia and A. R. Mohamed, Renewable Sustainable Energy Rev., 2012, 16, 4951–4964 CrossRef CAS; (b) T. Ema, Y. Miyazaki, T. Taniguchi and J. Takada, Green Chem., 2013, 15, 2485–2492 RSC; (c) A. A. L. Gonçalves, A. C. Fonseca, J. F. J. Coelho and A. C. Serra, Curr. Green Chem., 2015, 2, 43–65 CrossRef; (d) V. D'Elia, H. Dong, A. J. Rossini, C. M. Widdifield, S. V. C. Vummaleti, Y. Minenkov, A. Poater, E. Abou-Hamad, J. D. A. Pelletier, L. Cavallo, L. Emsley and J.-M. Basset, J. Am. Chem. Soc., 2015, 137, 7728–7739 CrossRef.
- (a) A. C. Kathalikkattil, R. Babu, J. Tharun, R. Roshan and D.-W. Park, Catal. Surv. Asia, 2015, 19, 223–235 CrossRef CAS; (b) H. He, J. A. Perman, G. Zhu and S. Ma, Small, 2016, 12, 6309–6324 CrossRef CAS PubMed; (c) J. W. Maina, C. Pozo-Gonzalo, L. Kong, J. Schütz, M. Hill and L. F. Dumée, Mater. Horiz., 2017, 4, 345–361 RSC.
- (a) Y. Xie, T.-T. Wang, X.-H. Liu, K. Zou and W.-Q. Deng, Nat. Commun., 2013, 4, 1960 CrossRef PubMed; (b) T.-T. Liu, J. Liang, Y.-B. Huang and R. Cao, Chem. Commun., 2016, 52, 13288–13291 RSC; (c) G. Ji, Z. Yang, H. Zhang, Y. Zhao, B. Yu, Z. Ma and Z. Liu, Angew. Chem., Int. Ed., 2016, 55, 9685–9689 CrossRef CAS PubMed; (d) Z. Dai, Q. Sun, X. Liu, C. Bian, Q. Wu, S. Pan, L. Wang, X. Meng, F. Deng and F.-S. Xiao, J. Catal., 2016, 338, 202–209 CrossRef CAS; (e) Y. Chen, R. Luo, Q. Xu, J. Jiang, X. Zhou and H. Ji, ChemSusChem, 2017, 10, 2534–2541 CrossRef CAS PubMed; (f) S. Wang, K. Song, C. Zhang, Y. Shu, T. Li and B. Tan, J. Mater. Chem. A, 2017, 5, 1509–1515 RSC.
- (a) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef; (b) M. Mastalerz, Angew. Chem., Int. Ed., 2010, 49, 5042–5053 CrossRef CAS PubMed; (c) Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS PubMed.
- S. M. Elbert, F. Rominger and M. Mastalerz, Chem.–Eur. J., 2014, 20, 16707–16720 CrossRef CAS PubMed.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- (a) J. Meléndez, M. North and R. Pasquale, Eur. J. Inorg. Chem., 2007, 2007, 3323–3326 CrossRef; (b) T. Roy, R. I. Kureshy, N.-u. H. Khan, S. H. R. Abdi and H. C. Bajaj, Catal. Sci. Technol., 2013, 3, 2661–2667 RSC; (c) J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- (a) D. Gong, B. Wang, X. Jia and X. Zhang, Dalton Trans., 2014, 43, 4169–4178 RSC; (b) H. Keypour, S. G. Saremi, H. Veisi and R. Azadbakht, RSC Adv., 2016, 6, 77020–77029 RSC.
- (a) J. H. Burness, J. G. Dillard and L. T. Taylor, J. Am. Chem. Soc., 1975, 97, 6080–6088 CrossRef CAS; (b) H. Liu, X. Xue, T. Li, J. Wang, W. Xu, M. Liu, P. Chen and Y. Wu, RSC Adv., 2016, 6, 84815–84824 RSC.
- (a) S. Laokroekkiat, M. Hara, S. Nagano and Y. Nagao, Langmuir, 2016, 32, 6648–6655 CrossRef CAS PubMed; (b) J. Li, Y. Fan, Y. Ren, J. Liao, C. Qi and H. Jiang, Inorg. Chem., 2018, 57, 1203–1212 CrossRef CAS PubMed.
- D. Patel, G. R. Kurrey, S. S. Shinde, P. Kumar, G.-J. Kim and S. S. Thakur, RSC Adv., 2015, 5, 82699–82703 RSC.
- S. Brunauer, L. S. Deming, W. E. Deming and E. Teller, J. Am. Chem. Soc., 1940, 62, 1723–1732 CrossRef CAS.
- Z. Zhang, Z.-Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868–2899 RSC.
- T.-T. Wang, Y. Xie and W.-Q. Deng, J. Phys. Chem. A, 2014, 118, 9239–9243 CrossRef CAS PubMed.
- (a) S.-T. Zheng, J. T. Bu, Y. Li, T. Wu, F. Zuo, P. Feng and X. Bu, J. Am. Chem. Soc., 2010, 132, 17062–17064 CrossRef CAS PubMed; (b) B. X. Dong, S. Y. Zhang, W. L. Liu, Y. C. Wu, J. Ge, L. Song and Y. L. Teng, Chem. Commun., 2015, 51, 5691–5694 RSC; (c) W.-P. Chen, P.-Q. Liao, Y. Yu, Z. Zheng, X.-M. Chen and Y.-Z. Zheng, Angew. Chem., Int. Ed., 2016, 55, 9375–9379 CrossRef CAS PubMed.
- (a) D. Sun, G.-G. Luo, N. Zhang, R.-B. Huang and L.-S. Zheng, Chem. Commun., 2011, 47, 1461–1463 RSC; (b) S. K. Dey, R. Chutia and G. Das, Inorg. Chem., 2012, 51, 1727–1738 CrossRef CAS PubMed; (c) A. M. Garcia-Deibe, C. Portela-Garcia, M. Fondo, A. J. Mota and J. Sanmartin-Matalobos, Chem. Commun., 2012, 48, 9915–9917 RSC.
- B. Chatelet, L. Joucla, J.-P. Dutasta, A. Martinez and V. Dufaud, Chem.–Eur. J., 2014, 20, 8571–8574 CrossRef CAS PubMed.
- (a) P. Yan and H. Jing, Adv. Synth. Catal., 2009, 351, 1325–1332 CrossRef CAS; (b) S. Zhang, Y. Huang, H. Jing, W. Yao and P. Yan, Green Chem., 2009, 11, 935–938 RSC; (c) D. Chen, R. Luo, M. Li, M. Wen, Y. Li, C. Chen and N. Zhang, Chem. Commun., 2017, 53, 10930–10933 RSC.
- S. Kumar, S. L. Jain and B. Sain, Catal. Today, 2012, 198, 204–208 CrossRef CAS.
- (a) Y. Xie, R.-X. Yang, N.-Y. Huang, H.-J. Luo and W.-Q. Deng, J. Energy Chem., 2014, 23, 22–28 CrossRef CAS; (b) Y. Xie, T.-T. Wang, R.-X. Yang, N.-Y. Huang, K. Zou and W.-Q. Deng, ChemSusChem, 2014, 7, 2110–2114 CrossRef CAS PubMed.
- (a) M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861–15864 CrossRef CAS PubMed; (b) Z.-R. Jiang, H. Wang, Y. Hu, J. Lu and H.-L. Jiang, ChemSusChem, 2015, 8, 878–885 CrossRef CAS PubMed.
- (a) W.-Y. Gao, L. Wojtas and S. Ma, Chem. Commun., 2014, 50, 5316–5318 RSC; (b) W.-Y. Gao, Y. Chen, Y. Niu, K. Williams, L. Cash, P. J. Perez, L. Wojtas, J. Cai, Y.-S. Chen and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 2615–2619 CrossRef CAS PubMed; (c) W.-Y. Gao, C.-Y. Tsai, L. Wojtas, T. Thiounn, C.-C. Lin and S. Ma, Inorg. Chem., 2016, 55, 7291–7294 CrossRef CAS PubMed; (d) H. He, Q. Sun, W. Gao, J. A. Perman, F. Sun, G. Zhu, B. Aguila, K. Forrest, B. Space and S. Ma, Angew. Chem., Int. Ed., 2018, 57, 4657–4662 CrossRef CAS PubMed.
- Y. Chen, R. Luo, Q. Xu, W. Zhang, X. Zhou and H. Ji, ChemCatChem, 2017, 9, 767–773 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization details, computational methods and supplementary catalytic data. See DOI: 10.1039/c8sc05019h |
| This journal is © The Royal Society of Chemistry 2019 |