
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic overall water splitting on Pt nanocluster-intercalated, restacked KCa2Nb3O10 nanosheets: the promotional effect of co-existing ions†
Takayoshi
Oshima
ab,
Yunan
Wang
c,
Daling
Lu
d,
Toshiyuki
Yokoi
c and
Kazuhiko
Maeda
 *a
*a
aDepartment of Chemistry, School of Science, Tokyo Institute of Technology, 2-12-1-NE-2, Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: maedak@chem.titech.ac.jp
bJapan Society for the Promotion of Science, Kojimachi Business Center Building, 5-3-1, Kojimachi, Chiyoda-ku, Tokyo 102-0083, Japan
cNanospace Catalysis Unit, Institute of Innovative Research, Tokyo Institute of Technology, 4259-S2-5, Nagatsuta, Midori-ku, Yokohama 226-8503, Japan
dCenter for Advanced Materials Analysis, Tokyo Institute of Technology, 4259-R1-34, Nagatsuta-cho, Midori-ku, Yokohama 226-850, Japan
First published on 3rd December 2018
Abstract
Promotional effects of co-existing ions on overall water splitting into H2 and O2 have been studied in bulk-type semiconductor photocatalysts (e.g., TiO2), but such an effect remains unexplored in two-dimensional nanosheet photocatalysts. Here we examined the effect of co-existing ions on the photocatalytic water splitting activity of Pt nanocluster-intercalated KCa2Nb3O10 nanosheets. Interestingly, not only anions, as usually observed in bulk-type photocatalysts, but also cations had a significant influence on the photocatalytic performance. The rates of H2 and O2 evolution over Pt/KCa2Nb3O10 as well as the product stoichiometry were improved in the presence of NaI. I− ions were found to effectively suppress undesirable backward reactions, consistent with the previous work by Abe et al. (Chem. Phys. Lett., 2003, 371, 360–364). On the other hand, Na+ ions in the reaction solution were exchanged for K+ in the interlayer space of KCa2Nb3O10 during the water splitting reaction, which promoted interlayer hydration and consequently improved photocatalytic performance.
Introduction
Clean production of hydrogen by water splitting over a semiconductor photocatalyst has attracted considerable interest in order to meet the social demand for replacing CO2-emitting fossil fuels with clean energy sources.1–3 The efficiency of photocatalytic water splitting, however, still remains unsatisfactory. Therefore, not only enhancing the performance of known photocatalysts but also fundamental investigation, for example understanding the reaction mechanism and exploring new materials, is important in this research field.Certain layered materials undergo exfoliation, producing colloidal suspensions of unilamellar sheets.4–27 The planar size of the exfoliated sheets typically ranges from several hundreds of nanometers to a few micrometers, with a thickness of 1–2 nm. Because of the sheet-like two-dimensional (2D) structure, it is called a nanosheet. The nanosheets containing certain transition metal cations such as Ti4+, Nb5+, Ta5+, and W6+ are known to exhibit photocatalytic activity. They are expected to have several advantages in heterogeneous photocatalysis, compared with conventional bulk-type 3D photocatalysts (e.g., TiO2).
It is known that high crystallinity of a semiconductor has a positive impact on photocatalytic performance for overall water splitting, because of the prolonged lifetime of photogenerated electrons and holes.28 To obtain a highly crystalline material, high calcination temperature is generally required in the synthesis process; however, such harsh calcination conditions facilitate grain growth of photocatalyst particles, resulting in smaller surface area and lower density of reaction sites, and vice versa.29 Moreover, larger size of semiconductor particles can have a negative effect in terms of carrier diffusion to the surface, resulting in recombination between electrons and holes and lowering photocatalytic activity. On the other hand, a nanosheet has both high crystallinity and high surface area owing to its single-crystalline character and anisotropic structure.30 Furthermore, the small thickness of the nanosheet is expected to shorten the migration distance of photogenerated carriers to the surface, thereby reducing the recombination probability.9,12 Despite these fascinating features, there are a limited number of examples of overall water splitting using a nanosheet photocatalyst.
Other than such structural factors, there are some key factors to achieve high photocatalytic performance. One of them is deposition of metal or metal oxide nanoparticles, which are called co-catalysts, on the photocatalyst surface.1,2 The loaded co-catalyst serves as active reaction sites and as a promoter for charge separation between electrons and holes, leading to drastic enhancement in photocatalytic performance.2
Another one is the reaction conditions, such as co-existing ions and pH.31–37 For example, Pt-loaded anatase TiO2 achieved overall water splitting into H2 and O2 in the presence of highly concentrated HCO3− and/or CO32− under basic conditions (pH = 8–11.5)31 or in a diluted aqueous NaI solution.32 In these reports, the co-existing anions interacted with Pt, resulting in enhancement of water splitting performance. While effects of reaction conditions on activity have been studied mainly in bulk-type 3D photocatalysts, the photocatalytic activity of nanosheets for overall water splitting has not been investigated enough with respect to reaction conditions.
Recently, we have demonstrated overall water splitting using Pt nanocluster-intercalated, restacked KCa2Nb3O10 nanosheets.23,25 In this system, nanosized Pt particles (≤1 nm) were deposited not only on the external surface of KCa2Nb3O10 but also in the interlayer of the restacked nanosheets. The Pt-intercalated photocatalyst exhibited higher performance compared to the previously reported RuO2/KCa2Nb3O10 nanosheets and Pt/KCa2Nb3O10, the latter of which consisted of Pt nanoparticles deposited only on the external surface of restacked KCa2Nb3O10 nanosheets.23 The unprecedented small size of intercalated Pt appears to be responsible for the high performance of this photocatalytic system.
Here we examine, in detail, the photocatalytic water splitting activity of Pt/KCa2Nb3O10 nanosheets with respect to co-existing ions in reaction solution. The unique photocatalytic properties of the 2D nanosheet material are highlighted for the first time.
Experimental
Materials
A Ca2Nb3O10− nanosheet suspension stabilized by tetra(n-butyl)ammonium cations (TBA+) was prepared according to a previous report.9 First, a layered metal oxide, KCa2Nb3O10, was prepared by solid state reaction by heating a mixture of K2CO3 (≥99.5%, Kanto Chemical Co.), CaCO3 (≥99.5%, Kanto Chemical Co.), and Nb2O5 (≥99.95%, Kanto Chemical Co.) in air at 1123 K for 1 h, followed by a second calcination at 1423 K for 10 h in a furnace. The ratio of K, Ca and Nb in the starting mixture was 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3, where 10% excess K was added to compensate for the loss during calcination as reported previously.9 The resulting KCa2Nb3O10 was subjected to stirring in HNO3 (60–61%, Wako Pure Chemicals) solution (1 M) for 3 days to exchange K+ in the interlayer for H+. The resulting solid was collected by centrifugation and washed with water repeatedly until the pH of the supernatant became neutral. After the HCa2Nb3O10 was dried in an oven at 343 K, it was suspended in an aqueous TBA+OH− solution (Aldrich Chemical Co., 40 wt% in H2O), followed by stirring for 1 week, yielding a colloidal TBA+/Ca2Nb3O10− nanosheet suspension. The molar ratio of TBA+OH− to exchangeable cations in the layered solids was 1.0.
:2:3, where 10% excess K was added to compensate for the loss during calcination as reported previously.9 The resulting KCa2Nb3O10 was subjected to stirring in HNO3 (60–61%, Wako Pure Chemicals) solution (1 M) for 3 days to exchange K+ in the interlayer for H+. The resulting solid was collected by centrifugation and washed with water repeatedly until the pH of the supernatant became neutral. After the HCa2Nb3O10 was dried in an oven at 343 K, it was suspended in an aqueous TBA+OH− solution (Aldrich Chemical Co., 40 wt% in H2O), followed by stirring for 1 week, yielding a colloidal TBA+/Ca2Nb3O10− nanosheet suspension. The molar ratio of TBA+OH− to exchangeable cations in the layered solids was 1.0.
Pt was deposited on the KCa2Nb3O10 restacked nanosheets in the same manner as our previous reports.23,25 An aqueous [Pt(NH3)4]Cl2 (Wako Pure Chemicals) solution (1 mM) was added dropwise into the nanosheet suspension containing 5 g of Ca2Nb3O10− nanosheets per L, and the resulting suspension was stirred for one day to adsorb the Pt precursor on the nanosheets. Then, an aqueous KOH (≥86.0%, Kanto Chemical Co.) solution was added to the nanosheet suspension to restack nanosheets, followed by thorough washing with H2O and drying in an oven. The dried solid was ground into powder with a mortar and a pestle, and heated at 473 K for 1 h under H2 flow (20 mL min−1) to obtain Pt/KCa2Nb3O10 restacked nanosheets.
Adsorption isotherms of NaI on bare KCa2Nb3O10 or NaCa2Nb3O10 restacked nanosheets were measured as follows. Typically, 20 mg of sample was dispersed in an aqueous NaI solution with different concentrations (10 mL) and kept at least for one day to achieve adsorption and desorption equilibrium. The resulting suspension was filtered and the filtrate was analyzed by UV-visible absorption spectroscopy (V-565, Jasco). The adsorbed amount of NaI was quantified by a reduction of absorbance at 226.5 nm.
Characterization
X-ray diffraction (XRD) patterns were recorded on a Rigaku MiniFlex600 powder diffractometer employing monochromatic Cu Kα radiation and operating at 15 mA and 40 kV. Transmission electron microscopy (TEM) images were acquired using a JEM-2010F electron microscope (Jeol). X-ray photoelectron spectra (XPS) were acquired using an ESCA-3400 X-ray photoelectron spectrometer (Shimadzu). The binding energies determined using XPS were corrected with reference to the C 1s peak (284.6 eV) for each sample. N2 adsorption isotherms were measured using a BELSORP-mini instrument (MicrotracBEL) at liquid nitrogen temperature. H2O adsorption isotherms were acquired at 298 K using a BELSORP-max (MicrotracBEL) instrument. Samples were heated at 473 K for 1 h under vacuum prior to the measurements for both N2 and H2O adsorption isotherms.Photocatalytic reaction
Photocatalytic reactions were performed in an air-free, glass, closed circulation system. A top-irradiation type reaction cell made of Pyrex glass was used, combined with a cooling water bath to maintain the temperature of the reactor at approximately 293 K throughout the reaction. In each reaction, a 50 mg quantity of the restacked nanosheets was suspended in an aqueous solution (100 mL) containing various salts [NaI (≥99.5%, Kanto Chemical Co.), KI (≥99.5%, Kanto Chemical Co.), Na2SO4 (≥99.0%, Kanto Chemical Co.), or K2SO4 (≥99.0%, Kanto Chemical Co.)]. After degassing the reaction cell, a small amount (ca. 3 kPa) of argon gas was introduced and the suspension was irradiated using a 300 W xenon lamp (λ ≥ 300 nm) to photoexcite KCa2Nb3O10 that has a band gap of ca. 3.6 eV. The light intensity at 350 nm was approximately 1.1 × 10−3 einstein per h. The evolved gases were analyzed by gas chromatography (Shimadzu, GC-8A with a TCD detector and an MS-5A column, argon carrier gas).Results and discussion
Photocatalytic performance of Pt/KCa2Nb3O10 for overall water splitting in the presence of various salts
Fig. 1 shows the TEM images of KCa2Nb3O10 restacked nanosheets with deposited Pt. Very small Pt nanoparticles with homogeneous dispersion were observed on the restacked nanosheets as dark spots due to a difference in electron density between Pt and KCa2Nb3O10. Our previous study revealed that the deposited Pt was intercalated in the KCa2Nb3O10 restacked nanosheets with an average size of smaller than 1 nm.23,24 The details of the structural characterization of the Pt-intercalated KCa2Nb3O10 nanosheets can be found elsewhere.23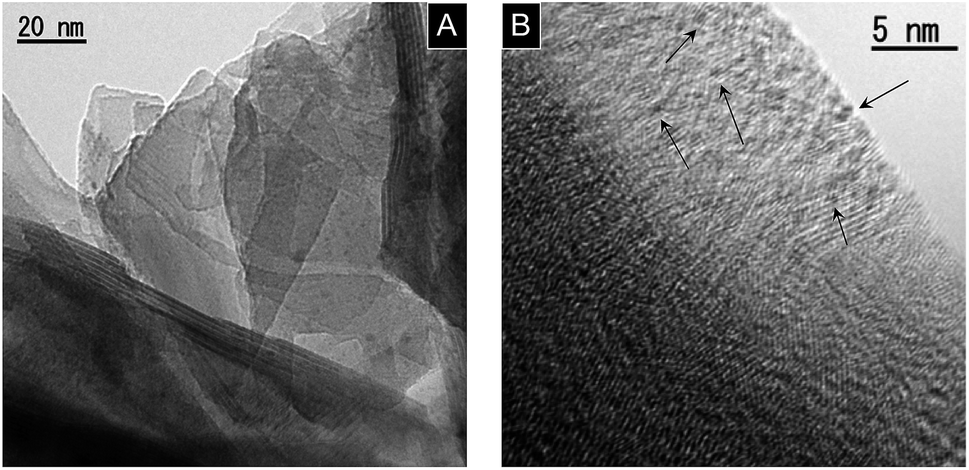 | ||
| Fig. 1 TEM images of 1.0 wt% Pt/KCa2Nb3O10 restacked nanosheets. (A) Lower and (B) higher magnification images. Arrows in (B) indicate some deposited Pt nanoclusters. | ||
Photocatalytic reactions were performed using the Pt/KCa2Nb3O10 restacked nanosheets in the presence of various salts. Table 1 summarizes the amounts of H2 and O2 evolved in the reaction after 10 h irradiation. When the reaction was carried out in the absence of salts (i.e. pure water), a certain amount of H2 was evolved, with a low level of O2 that was far below the stoichiometric value (H2/O2 < 2). On the other hand, addition of NaI to pure water dramatically improved the photocatalytic performance in terms of both gas evolution amount and H2/O2 stoichiometry. The experimental error in gas evolution rate was approximately 15%. Neither deactivation nor decomposition of Pt/KCa2Nb3O10 has been confirmed by XRD and TEM after 30 h irradiation.23
| Dissolved salt | Amount of evolved gases/μmol | H2/O2 ratio | |
|---|---|---|---|
| H2 | O2 | ||
| a Reaction conditions: catalyst, 50 mg; reaction solution, 100 mL; light source, a 300 W Xe lamp (λ ≥ 300 nm). Reaction time: 10 h. | |||
| Pure water | 21.6 | 3.6 | 6.0 |
| NaI, 10 mM | 85.5 | 37.4 | 2.3 |
| KI, 10 mM | 33.3 | 12.0 | 2.8 |
| K2SO4, 5 mM | 20.5 | 2.2 | 9.3 |
| Na2SO4, 5 mM | 56.4 | 20.0 | 2.8 |
In order to investigate in detail the roles of Na+ and I− in promoting the overall water splitting, KI, Na2SO4 or K2SO4 was dissolved in the reaction solution, and photocatalytic activity was examined. The addition of KI to the reaction solution improved the H2/O2 evolution rates and the stoichiometry, compared to the pure water case, but not as much as that with the addition of NaI. The photocatalytic activity in the presence of K2SO4 was almost the same as that in pure water, suggesting that both K+ and SO42− had little impact on photocatalytic performance. On the other hand, the co-existence of Na2SO4 in the reaction solution enhanced the performance, although the extent was lower than that observed in the presence of NaI. These results indicate that both Na+ and I− have a positive influence on the photocatalytic performance of Pt/KCa2Nb3O10.
Effect of I−
A clear difference was also observed in the time course of overall water splitting in the presence of I− anions. Fig. 2 shows the time courses of H2 and O2 evolution in an aqueous Na2SO4 or NaI solution. In the case of Na2SO4, rates of H2 and O2 evolution gradually decelerated over reaction time, although the initial evolution rates were almost the same as those recorded in the case of NaI, suggesting that backward reaction proceeded as the evolved H2 and O2 were accumulated in the reaction system. On the other hand, the gas evolution rates remained unchanged in the presence of NaI. Moreover, the stoichiometry of evolved H2 and O2 was also improved by NaI addition (H2/O2 ≈ 2.3 for NaI, and 2.8 for Na2SO4). We observed a similar trend when photocatalytic reactions were performed in aqueous K2SO4 and KI solution (Fig. S1†). These results suggest that the undesirable reaction (e.g., H2/O2 recombination and O2 photoreduction) was suppressed in the presence of I−.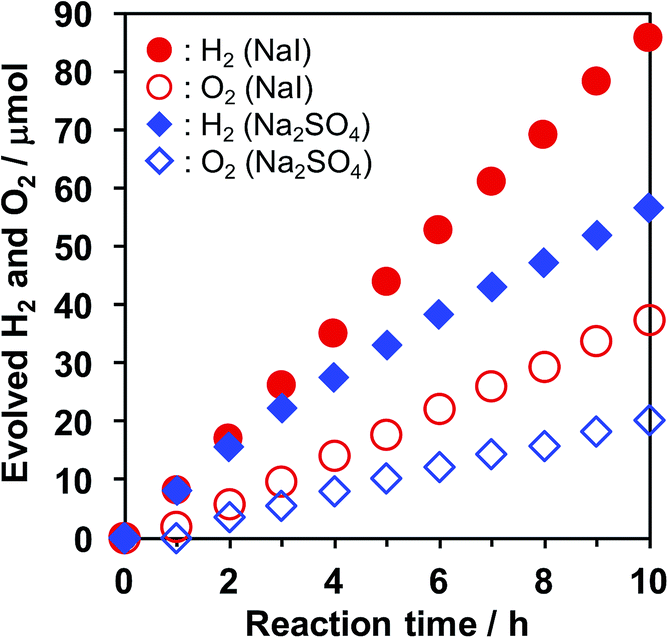 | ||
| Fig. 2 Time courses of H2 and O2 evolution over Pt/KCa2Nb3O10 in aqueous solution containing NaI (10 mM, red marks) or Na2SO4 (5 mM, blue marks). Closed marks: H2; open marks: O2. Reaction conditions: catalyst, 50 mg; reaction solution, 100 mL; light source, a 300 W Xe lamp (λ ≥ 300 nm). | ||
Abe et al. reported that Pt-loaded TiO2 exhibited much higher water splitting performance in the presence of I−, because the iodine layer formed on Pt nanoparticles could suppress the backward reaction.32 We also confirmed the effect of I− on backward reaction in this study. A certain amount of H2 and O2 was introduced in a closed reaction system and the amount was monitored under dark conditions, as shown in Fig. 3. Although the backward reaction over bare KCa2Nb3O10 was negligible in pure water, a decrease of H2 and O2 was observed over Pt/KCa2Nb3O10, indicating that the backward reaction took place on Pt. The water formation rate over Pt/KCa2Nb3O10 became slower in the presence of NaI than in pure H2O. On the other hand, the backward reaction rate in aqueous Na2SO4 solution was almost the same as that in pure water. These results suggest that the I−, not Na+, interacted with Pt and suppressed the backward reactions, which are most likely to occur on the externally deposited Pt, contributing to higher photocatalytic activity. Note here that SO42− is in principle unreactive during the photocatalytic reaction.35
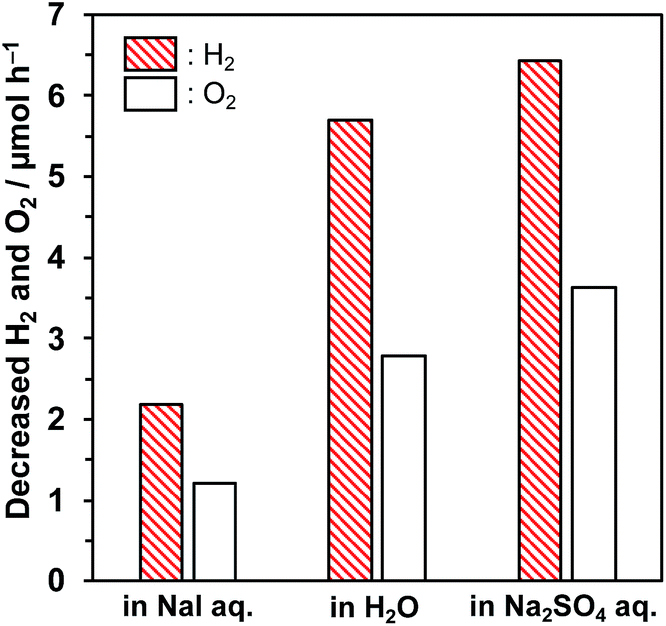 | ||
| Fig. 3 Decreasing rates of H2 and O2 evolution over Pt/KCa2Nb3O10 in aqueous NaI solution (10 mM), pure water, and Na2SO4 solution (5 mM) under dark conditions. Reaction conditions: photocatalyst, 50 mg; reactant volume, 100 mL; introduced amount of H2, ca. 200 μmol and O2, ca. 100 μmol. | ||
We attempted to detect the iodine species adsorbed on Pt/KCa2Nb3O10 after the water splitting reaction. However, no iodine signal was observed in this sample (Fig. S2†), most likely because I− cannot interact with the intercalated Pt due to electrostatic repulsion between negatively charged nanosheets and I−. On the other hand, a small, but distinct photoelectron signal attributable to I 3d was detected in the restacked KCa2Nb3O10 nanosheets that consisted of externally deposited Pt nanoparticles. It in turn indicates that the number of externally deposited Pt nanoparticles in the present Pt/KCa2Nb3O10 was not large enough to produce an observable quantity of iodine layers by XPS. Nevertheless, backward reactions that occurred on such lower density Pt islands are significant, and the suppression of the backward reactions is essential. In the photocatalytic reaction, I− also improved the H2/O2 stoichiometry. It is known that photo-reduction of O2 proceeds very efficiently on Pt nanoparticles, reducing the O2 evolution performance.38 Therefore, the improved stoichiometry would originate from the suppression of O2 photo-reduction on Pt as well.
I− is often used as an electron mediator between H2 and O2 evolution photocatalysts in Z-scheme water splitting.33,38–41 One may suspect that I− has a negative impact on photocatalytic performance because the oxidation of I− on the semiconductor surface hinders water oxidation, which has been observed in Pt/TiO2 at high concentration of NaI.32 Thus, we investigated the dependence of water splitting performance of Pt/KCa2Nb3O10 on the NaI concentration. Fig. 4 displays a relationship between H2/O2 evolution rates in the water splitting reaction and NaI concentration. H2 and O2 evolution rates monotonically increased up to 10 mM NaI and reached a plateau at higher concentration of NaI. UV-visible absorption spectroscopy indicated that before and after the photocatalytic reaction in 10 mM NaI solution, the concentration of I− remained almost unchanged. These facts indicate that the oxidation of H2O, not I−, dominated on KCa2Nb3O10. Adsorption isotherm measurement of NaI revealed that no adsorption of I− took place on the surface of bare KCa2Nb3O10. The surface properties inhibited the oxidation of I− on KCa2Nb3O10, leading to O2 evolution even in the presence of higher concentration of NaI.
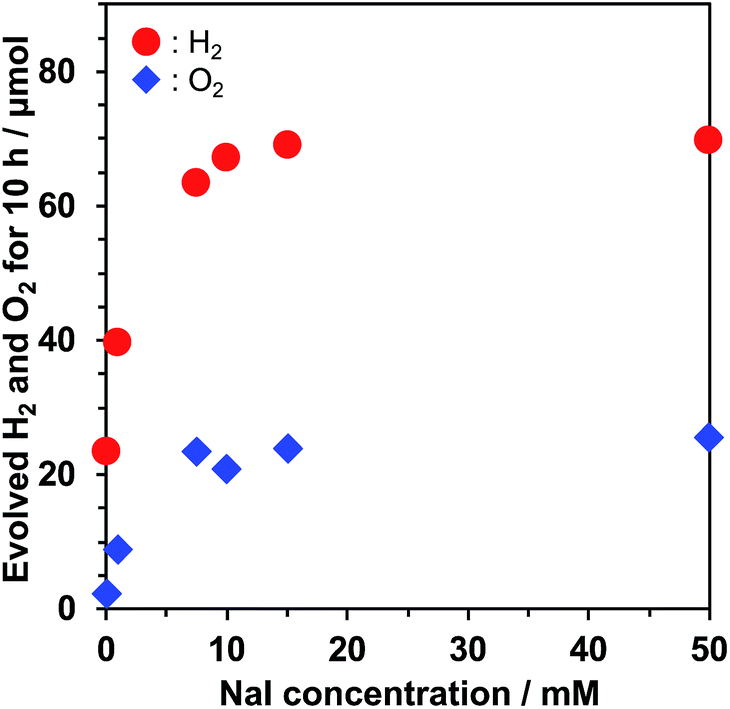 | ||
| Fig. 4 Dependence of the photocatalytic water splitting activity of Pt/KCa2Nb3O10 on the NaI concentration. Reaction conditions: catalyst, 50 mg; reaction solution, 100 mL; light source, a 300 W Xe lamp (λ ≥ 300 nm). | ||
Effect of Na+
As demonstrated above, the photocatalytic activity of Pt/KCa2Nb3O10 restacked nanosheets for overall water splitting was promoted in the presence of Na+ as well. In order to elucidate the reason, the reacted Pt/KCa2Nb3O10 samples in the presence of various salts were characterized by XRD. Fig. 5 displays the X-ray diffraction peaks of Pt/KCa2Nb3O10 attributed to the (002) plane, which is the stacking direction of restacked nanosheets. The diffraction peaks were shifted to lower two-theta directions after photocatalytic reactions in all cases, indicating that the (002) plane distance, namely the interlayer distance of restacked nanosheets, was expanded most likely due to an intercalation of water in the interlayer space, as reported previously.6 It should be noted that the peak positions were altered depending on the cations in the reaction solution. Pt/KCa2Nb3O10 after photocatalytic reaction in the presence of Na+ exhibited 002 diffraction peaks at lower two-theta positions compared to the peaks in the presence of K+, suggesting that a structural change occurred during the water splitting reaction.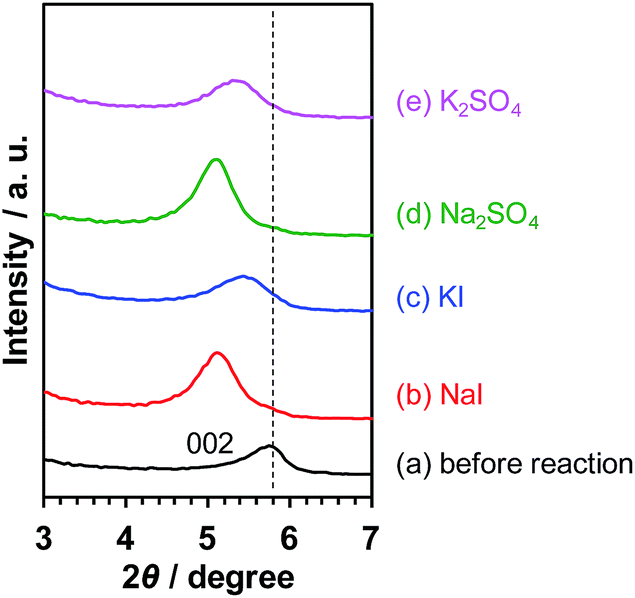 | ||
| Fig. 5 002 diffraction peaks of Pt/KCa2Nb3O10 in XRD (a) before reaction and after reaction in aqueous (b) NaI (10 mM), (c) KI (10 mM), (d) Na2SO4 (5 mM), and (e) K2SO4 (5 mM) solution. | ||
It is known that interlayer cations of certain layered materials can be exchanged for H+ and/or other kinds of alkali cations.42 Therefore, one possibility is that the K+ in the interlayer space of Pt/KCa2Nb3O10 was exchanged for Na+, which was present in the reaction solution. As shown in Fig. 6, XPS analysis revealed that photoelectron signals attributable to the K 2p orbital almost disappeared, while new peaks corresponding to the Na 1s orbital were clearly detected after reactions in aqueous NaI and Na2SO4 solution. In contrast, photoelectron singles of not Na 1s, but K 2p were observed before and after reaction in aqueous KI and K2SO4 solution. The results indicate that the ion-exchange of K+ in the interlayer for Na+ occurred, and that the structure of Pt/KCa2Nb3O10 restacked nanosheets after reaction turned into Pt/NaCa2Nb3O10.
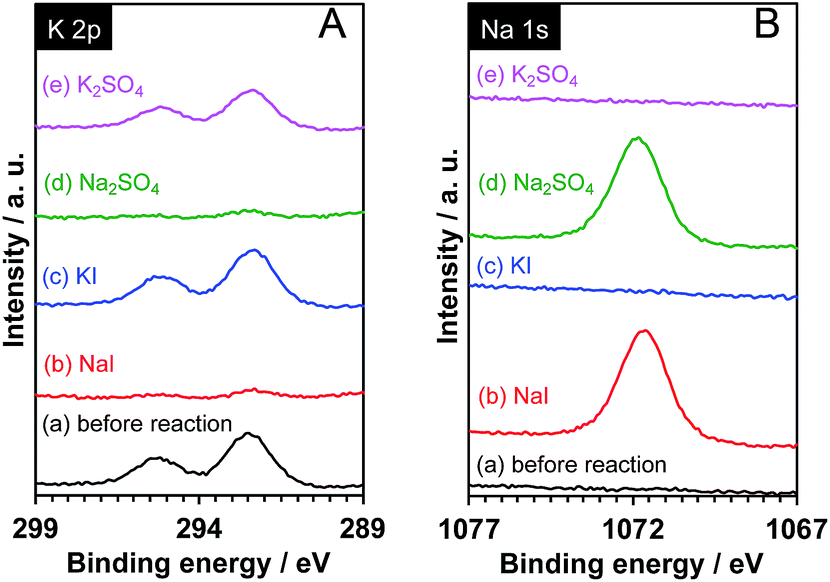 | ||
| Fig. 6 XPS spectra for (A) K 2p and (B) Na 1s of Pt/KCa2Nb3O10 (a) before and after reaction in aqueous (b) NaI (10 mM), (c) KI (10 mM), (d) Na2SO4 (5 mM), and (e) K2SO4 (5 mM) solution. | ||
The ion-exchange, however, does not seem to explain the shift of the diffraction peak position (Fig. 5). The ionic radius of Na+ (1.02 Å) in a 6-coordination environment is smaller than that of K+ (1.38 Å). Thus, one can suppose that the 002 diffraction peak after ion-exchange of K+ for Na+ appears at a higher two-theta position because of the smaller size of Na+ and the resulting shorter (002) plane distance. A plausible explanation for the phenomena opposite to our expectation is more hydration of the interlayer of Pt/NaCa2Nb3O10, concealing the effect of K+/Na+ exchange.
In order to evaluate the correlation between interlayer cations and the affinity for water, we measured H2O and N2 adsorption isotherms of KCa2Nb3O10 and NaCa2Nb3O10 (Fig. S3†). The surface areas estimated from the H2O and N2 isotherms (represented as SH2O and SN2, respectively) are listed in Table 2. In both cases of KCa2Nb3O10 and NaCa2Nb3O10, the specific surface areas estimated from H2O adsorption (SH2O) were larger than those from N2 adsorption (SN2), indicating that H2O molecules could penetrate into the interlayer space of these materials. Although SN2 was not affected by interlayer cations, NaCa2Nb3O10 exhibited obviously larger SH2O than KCa2Nb3O10. It is thus likely that the stronger affinity for H2O promoted the photo-redox reaction in the interlayer space of Na+-exchanged Pt/KCa2Nb3O10, leading to higher photocatalytic performance for overall water splitting.
| Material | S N2/m2 g−1 | S H2O/m2 g−1 | S H2O/SN2 |
|---|---|---|---|
| KCa2Nb3O10 | 10.7 | 30.2 | 2.8 |
| NaCa2Nb3O10 | 10.0 | 48.3 | 4.8 |
Ebina et al. suggested that Na+ ions in the interlayer of restacked nanosheets have stronger affinity toward water than K+.6 Very recently, we also reported that a layered metal oxide photocatalyst, K2CaNaNb3O10, provided better access for reactant(s) in the interlayer space, which led to higher photocatalytic performance.43 Therefore, the enhanced photocatalytic activity in the presence of Na+ is ascribed to better affinity for water in the interlayer of restacked nanosheets, which resulted from the ion-exchange reaction of K+ to Na+. Because interlayer modification is very important for enhancing the activity of nanosheet photocatalysts, we believe that the finding of this work would be an “indirect” means of decorating the interlayer nanospace of ion-exchangeable metal oxide nanosheets that improves photocatalytic activity for overall water splitting.
Conclusions
Effects of co-existing ions on the photocatalytic water splitting activity of Pt nanocluster-intercalated KCa2Nb3O10 restacked nanosheets were investigated. The performance was much improved in aqueous NaI solution, compared to the pure water case, in terms of gas evolution rate, stability and H2/O2 stoichiometry. The results strongly suggest that during the water splitting reaction, I− ions suppressed H2/O2 recombination and photo-reduction of O2. Furthermore, K+ ions in the interlayer of Pt/KCa2Nb3O10 underwent ion-exchange for Na+ in the Na+-containing reaction solution, producing Pt/NaCa2Nb3O10. H2O and N2 adsorption measurements demonstrated that NaCa2Nb3O10 had better affinity for H2O in the interlayer space than KCa2Nb3O10. It appears that this character of NaCa2Nb3O10 is the key to improving the accessibility of H2O and the photocatalytic redox reaction in the interlayer space.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Mixed Anion” (Project JP16H06441) from the Japan Society for the Promotion of Science (JSPS). T. O. wishes to acknowledge support by a JSPS Fellowship for Young Scientists (Project JP16J10084). The authors thank Prof. Masashi Hattori and Michikazu Hara (Tokyo Institute of Technology) for assistance in XPS measurements.Notes and references
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12, 237–268 CrossRef CAS.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS PubMed.
- R. E. Schaak and T. E. Mallouk, Chem. Mater., 2000, 12, 2513–2516 CrossRef CAS.
- Y. Ebina, T. Sasaki, M. Haruda and M. Watanabe, Chem. Mater., 2002, 14, 4390–4395 CrossRef CAS.
- Y. Ebina, N. Sakai and T. Sasaki, J. Phys. Chem. B, 2005, 109, 17212–17216 CrossRef CAS PubMed.
- H. Hata, S. Kudo, Y. Kobayashi and T. E. Mallouk, J. Am. Chem. Soc., 2007, 129, 3064–3065 CrossRef CAS PubMed.
- S. Ida, C. Ogata, K. Izawa, T. Inoue, O. Altuntasoglu and Y. Matsumoto, J. Am. Chem. Soc., 2007, 129, 8956–8957 CrossRef CAS PubMed.
- H. Hata, Y. Kobayashi, V. Bojan, W. J. Youngblood and T. E. Mallouk, Nano Lett., 2008, 8, 794–799 CrossRef CAS PubMed.
- S. Ida, C. Ogata, M. Eguchi, W. J. Youngblood, T. E. Mallouk and Y. Matsumoto, J. Am. Chem. Soc., 2008, 130, 7052–7059 CrossRef CAS PubMed.
- R. Ma, Y. Kobayashi, W. J. Youngblood and T. E. Mallouk, J. Mater. Chem., 2008, 18, 5982–5985 RSC.
- K. Maeda, M. Eguchi, W. J. Youngblood and T. E. Mallouk, Chem. Mater., 2008, 20, 6770–6778 CrossRef CAS.
- M. C. Sarahan, E. C. Carroll, M. Allen, D. S. Larsen, N. D. Browning and F. E. Osterloh, J. Solid State Chem., 2008, 181, 1678–1683 CrossRef CAS.
- K. Maeda, M. Eguchi, W. J. Youngblood and T. E. Mallouk, Chem. Mater., 2009, 21, 3611–3617 CrossRef CAS.
- K. Maeda and T. E. Mallouk, J. Mater. Chem., 2009, 19, 4813–4818 RSC.
- M. R. Allen, A. Thibert, E. M. Sabio, N. D. Browning, D. S. Larsen and F. E. Osterloh, Chem. Mater., 2010, 22, 1220–1228 CrossRef CAS.
- E. M. Sabio, M. Chi, N. D. Browning and F. E. Osterloh, Langmuir, 2010, 26, 7254–7261 CrossRef CAS PubMed.
- S. Ida and T. Ishihara, J. Phys. Chem. Lett., 2014, 5, 2533–2542 CrossRef CAS PubMed.
- S. Ida, A. Takashiba, S. Koga, H. Hagiwara and T. Ishihara, J. Am. Chem. Soc., 2014, 136, 1872–1878 CrossRef CAS PubMed.
- K. Maeda, M. Eguchi and T. Oshima, Angew. Chem., Int. Ed., 2014, 53, 13164–13168 CrossRef CAS PubMed.
- T. Oshima, O. Ishitani and K. Maeda, Adv. Mater. Interfaces, 2014, 1, 1400131 CrossRef.
- K. Maeda, G. Sahara, M. Eguchi and O. Ishitani, ACS Catal., 2015, 5, 1700–1707 CrossRef CAS.
- T. Oshima, D. Lu, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2698–2702 CrossRef CAS PubMed.
- T. Oshima, M. Eguchi and K. Maeda, ChemSusChem, 2016, 9, 396–402 CrossRef CAS PubMed.
- T. Oshima, D. Lu and K. Maeda, ChemNanoMat, 2016, 2, 748–755 CrossRef CAS.
- Y. Xia, W. Chen, S. Liang, J. Bi, L. Wu and X. Wang, Catal. Sci. Technol., 2017, 7, 5662–5669 RSC.
- Y. Xia, S. Liang, L. Wu and X. Wang, Catal. Today, 2018 DOI:10.1016/j.cattod.2018.03.061.
- F. Amano, A. Yamakata, K. Nogami, M. Osawa and B. Ohtani, J. Am. Chem. Soc., 2008, 130, 17650–17651 CrossRef CAS PubMed.
- A. Kudo, A. Tanaka, K. Domen and T. Onishi, J. Catal., 1988, 111, 296–301 CrossRef CAS.
- K. Maeda and T. E. Mallouk, Bull. Chem. Soc. Jpn., 2018 DOI:10.1246/bcsj.20180258.
- K. Sayama and H. Arakawa, J. Chem. Soc., Faraday Trans., 1997, 93, 1647–1654 RSC.
- R. Abe, K. Sayama and H. Arakawa, Chem. Phys. Lett., 2003, 371, 360–364 CrossRef CAS.
- R. Abe, K. Sayama and H. Sugihara, J. Phys. Chem. B, 2005, 109, 16052–16061 CrossRef CAS PubMed.
- H. Kato, Y. Sasaki, A. Iwase and A. Kudo, Bull. Chem. Soc. Jpn., 2007, 80, 2457–2464 CrossRef CAS.
- K. Maeda, H. Masuda and K. Domen, Catal. Today, 2009, 147, 173–178 CrossRef CAS.
- Y. Miseki, H. Kusama, H. Sugihara and K. Sayama, Chem. Lett., 2010, 39, 846–847 CrossRef CAS.
- H. Suzuki, O. Tomita, M. Higashi and R. Abe, J. Mater. Chem. A, 2017, 5, 10280–10288 RSC.
- R. Abe, M. Higashi and K. Domen, ChemSusChem, 2011, 4, 228–237 CrossRef CAS PubMed.
- M. Higashi, R. Abe, K. Teramura, T. Takata, B. Ohtani and K. Domen, Chem. Phys. Lett., 2008, 452, 120–123 CrossRef CAS.
- K. Maeda, M. Higashi, D. Lu, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858–5868 CrossRef CAS PubMed.
- K. Maeda, D. Lu and K. Domen, ACS Catal., 2013, 3, 1026–1033 CrossRef CAS.
- M. Machida, T. Mitsuyama, K. Ikeue, S. Matsushima and M. Arai, J. Phys. Chem. B, 2005, 109, 7801–7806 CrossRef CAS PubMed.
- T. Oshima, T. Yokoi, M. Eguchi and K. Maeda, Dalton Trans., 2017, 46, 10594–10601 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00240a |
| This journal is © The Royal Society of Chemistry 2019 |