
An electron-transfer photochromic metal–organic framework (MOF) compound with a long-lived charge-separated state and high-contrast photoswitchable luminescence†
Xiu-Shuang Xingab,
Zi-Wei Chenb,
Li-Zhen Caib,
Cai Sunb,
Lin-Rong Caibc,
Ming-Sheng Wang*b and
Guo-Cong Guo*b
aCollege of Chemistry, Fuzhou University, Fuzhou, Fujian 350108, P. R. China
bState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, P. R. China. E-mail: mswang@fjirsm.ac.cn; gcguo@fjirsm.ac.cn
cCollege of Materials Science and Engineering, Fujian Normal University, Fuzhou, Fujian 350007, P. R. China
First published on 26th February 2016
Abstract
Long-lived charge-separated states and photoswitchable photoluminescence are important for many photochemical and photophysical applications. Recent studies have demonstrated the effectiveness of MOFs to achieve long-lived charge-separated states; however, the related studies are still rather rare owing to the limited numbers of photoresponsive MOFs. In this work, a new electron-transfer photochromic MOF compound, encapsulating photoactive viologen cations, was found to exhibit a charge-separated state with a lifetime value of more than 2 months, exceeding the reported values of the analogues. In addition, the photochromic process can afford a luminescence contrast up to 10 times between two stable states, which is higher than those of most known pyridine derivative-based photochromic compounds.
Introduction
Photochromic compounds have been studied in depth in the past years for their appealing applications on smart windows,1 photomasks,2 data storage,3 photocatalysis,4 solar energy conversion,5 etc.6 Long-lived colored states of these compounds are usually required for these applications. As for electron-transfer (redox) photochromic compounds, their charge-separated states are usually radicals, which are relatively active and unstable in the presence of molecular oxygen and other radical scavengers.5 Therefore, it is of importance to develop effective approaches to stabilize the photogenerated radicals. The key is trying to avoid the contacts of radicals with molecular oxygen and other radical scavengers. One of the reliable methods is to shield the photogenerated radicals by layered matrixes.5,7 For instance, Vermeulen and Thompson reported that viologen-pillared layered zirconium phosphate could form a long-lived charge-separated state in air.5 Another way is to host the radicals in the pores of zeolites. Dutta et al. found that the lifetime of viologen radical cations in zeolite Y could reach the hour time sacle.8 A new method that appeared recently is to include the radicals into MOFs,9 which feature high designability of frameworks and pores. We found that photogenerated radicals in the framework of the electron-transfer (ET) photochromic MOF [Cd2(ic)(mc)(Bpy)3]n·4nH2O (ic = itaconate, mc = mesaconate, Bpy = 4,4′-bipyridine) could be maintained for two weeks in air in the dark.9a Liu et al. also showed some interesting photoresponsive MOFs, where the photogenerated radicals in the cavities could retain for one month in air.9b These results demonstrate the effectiveness of MOFs on the formation of long-lived charge-separated states. To date, the related studies are still rather rare because the known photochromic MOFs were scarcely documented in the literature.9–20In addition, the utilization of photochromism to photoswitch physical properties, such as gas adsorption,13,18,21 conductance,22 magnetism23 and photoluminescence,16,20,24 have aroused intense interests recently. Photoswitching of photoluminescence offers a promising tool for data storage, bioimaging and barcode because of the high sensitivity, selectivity and spatial information of photoluminescence.25 Many literatures have evidenced the successful application of photochromism for this purpose.3,26 Even so, less attention has been paid to the ET photochromic systems comparing with others, such as diarylethene,27 azobenzene28 and spiropyran.29 ET photochromic species are very suitable to solid-state media considering their small structural differences between two switchable states. Moreover, their photogenerated charge-separated states are commonly nonemissive, and thus a high contrast of photoluminescence between two states can be achieved. We and others have found that ET photochromism is relatively effective to photoswitch photoluminescence in the solid state.16,20,24
In this work, we obtained a new ET photochromic MOF compound, {(H2Bpy)[Cd3(BTC)2]·2H2O}n (1, H2Bpy = deprotonated 4,4′-bipyridinium, H4BTC = 1,2,4,5-benzenetetracarboxylic acid), where the photoactive H2Bpy2+ cations were successfully introduced into the pores of a known anionic MOF found in {[K2(H2O)6][Cd3(BTC)2]}n30 and {[Na2(H2O)6][Cd3(BTC)2]}n.31 Its photogenerated charge-separated state can be stable in the dark under ambient environment for more than 2 months. This lifetime is longer than the reported values for known ET photochromic MOF compounds, which are below 1 month.8–11,14,20,32 Moreover, compound 1 also exhibits photoswitchable photoluminescence with a contrast of about 10 times between two states. As for the widely studied pyridine derivative-based photochromic compounds, the recorded values for luminescence contrast are almost below 7 times15,20,24,32,33 and only two examples reach 22.4 and 99 times, respectively.16 Therefore, the luminescence contrast of 1 is higher than those of most known pyridine derivative-based photochromic compounds.
Experimental
Materials and general procedures
All of chemicals were obtained from commercial sources and used without further purification. A PLS-SXE300C 300 W xenon lamp system equipped with an IR filter was used to prepare colored samples for diffuse reflectance (DR) ultraviolet-visible spectra, powder X-ray diffraction (PXRD), photoluminescence and electron spin resonance (ESR) studies, and the distances between these samples and the Xe lamp were around 20 cm. The DR spectra were recorded on a PerkinElmer Lambda 900 UV/vis/near-IR spectrophotometer equipped with an integrating sphere and a BaSO4-coated glass slide as a reference (100% reflection). The PXRD patterns were collected with a Rigaku MiniFlex II diffractometer powered at 30 kV and 15 mA for Cu Kα (λ = 1.54057 Å). The simulated pattern was achieved using Mercury, version 1.4 (http://www.ccdc.cam.ac.uk/products/mercury/). The FT-IR spectrum was recorded using KBr pellets on a VERTEX 70/70v FT-IR-spektrometer in the region of 4000 to 400 cm−1. The ESR spectra were achieved at the X-band frequency (9.854158 GHz) on a Bruker ELEXSYS E500 spectrometer. The in situ photoluminescence determination was conducted on a single-grating Edinburgh EI920 fluorescence spectrometer equipped with a 450 W Xe lamp and a R928P PMT detector. The elemental analyses of C, H, and N were performed on an Elementar Vario EL III microanalyzer. Thermogravimetric analysis (TGA) was done on a NETZSCH STA 449F3 thermal analyzer with Al2O3 crucibles under N2 at a heating rate of 10 °C min−1. All of these measurements except TGA were carried out in air at room temperature.Single-crystal X-ray crystallography
Data collection of a single crystal of 1 was performed on Agilent Technologies SuperNova Dual Wavelength CCD diffractometer with Cu Kα radiation (λ = 1.5418 Å) at 100 K. The structure was solved and refined by full-matrix least squares on F2 using the SHELXL software package,34 with anisotropic thermal parameters for all nonhydrogen atoms. The (H2Bpy)2+ cations are symmetrically disordered. H atoms of lattice water molecules and those riding on the N atoms of Bpy were included from the difference Fourier maps, while others generated geometrically. The structure was finally verified using the Addsym algorithm from the program PLATON.35 Crystallographic data and structural refinements for 1 are summarized in Table S1 (ESI†). More details on the crystallographic studies as well as atomic displacement parameters are given as cif ESI.†The entry of CCDC-1439857 contains the supplementary crystallographic data for 1.
Synthesis of {(H2Bpy)[Cd3(BTC)2]·2H2O}n (1)
A mixture of CdCO3 (0.217 g, 0.75 mmol), H4BTC (0.127 g, 0.5 mmol), Bpy (0.078 g, 0.5 mmol), and H2O (10 mL) was added into a 23 mL Teflon-lined autoclave and stirred for 2 h. The vessel was then sealed and heated at 180 °C for 7 d before cooling to room temperature naturally. Pale yellow prismatic crystals of 1 were obtained by filtering the solution, washed by water for several times and dried in air. The calculated content of water per molecule is ∼3.5%, closing the found weight loss of 3.8% from the TGA (Fig. S5, ESI†). Yield: 41% (based on the H4BTC). Anal. calcd (%) for C30H18Cd3N2O18: C, 34.93; H, 1.76; N, 2.72. Found: C, 35.27; H, 1.76; N, 2.71.Results and discussion
The experimental PXRD pattern at room temperature for 1 matches well with the simulated one from single-crystal structure data (Fig. S1, ESI†). This result and the elemental analysis data of C, H, and N demonstrated the phase purity of the obtained crystalline product of 1. The asymmetric and symmetric stretching vibration bands of COO− were observed at 1555 and 1383 cm−1 in the FT-IR spectrum (Fig. S2, ESI†), respectively. These peaks were shifted to lower values compared with the carbonyl frequencies of the free H4BTC ligand. The absence of the characterized absorption band around 1700 cm−1 for a protonated carboxylic group implies that the carboxylate groups in 1 are completely deprotonated.Crystal structure
A single-crystal X-ray crystallographic analysis demonstrated that compound 1 features a 3-D anionic framework [Cd3(BTC)2]n2n− templated by (H2Bpy)2+ cations and lattice water molecules. The framework structure is almost the same to those in {[K2(H2O)6][Cd3(BTC)2]}n30 and {[Na2(H2O)6][Cd3(BTC)2]}n,31 and thus only described briefly here. As illustrated in Fig. 1a, each BTC4− ligand bridges six Cd atoms, and all O atoms join the coordination. The formed 3-D anionic framework contains 1-D rhomboid channels of approximately 11.396 × 7.915 Å along the c direction (Fig. 1b), where each (H2Bpy)2+ cation connects the framework through the Coulomb force, N–H⋯O hydrogen bonds (Table 1), π⋯π stacking interactions with two neighbouring BTC4− ligands (centroid (phenyl)-to-centroid (pyridine), 3.5569(3) Å), and other possible weaker interactions (Fig. 1c). No π⋯π stacking interactions can be observed among the (H2Bpy)2+ cations themselves. | ||
| Fig. 1 (a) Coordination geometry around the BTC4− ligand in 1; (b) view of the 3-D structure of 1 along the c direction; (c) hydrogen bonding and π⋯π stacking interactions around the (H2Bpy)2+ cation. H atoms of C in all diagrams and those of N in (b) are deleted for clarity. Symmetry codes: A, x, y, 1−z; B, 1−x, −y, z; C, 1−x, y, 0.5−z; D, x, −y, 0.5−z; E, 0.5−x, −0.5 + y, z; F, 0.5−x, 0.5−y, 0.5−z; G, 0.5−x, 0.5−y, 0.5 + z; H, 1−x, −y, 1−z; I, 0.5 + x, 0.5−y, 1−z; J, 1−x, 1−y, z; K, x, 1−y, z; L, 0.5−x, 0.5 + y, 1−z. | ||
| D–H⋯A | d(D⋯A) | d(H⋯A) | d(D–H) | ∠(D–H⋯A) |
|---|---|---|---|---|
| N21–H21⋯O11 | 3.114(2) | 2.295(5) | 0.859(1) | 169.4(7) |
| N21–H21⋯O13 | 2.817(1) | 2.595(3) | 0.859(1) | 95.9(7) |
Photochromism
Upon continuous irradiation with a 300 W xenon lamp at room temperature in air, the as-synthesized crystalline sample of 1 (denoted 1A) underwent a clear color change from pale yellow to blue (Fig. 2 top). The coloration could be observed within 1 min. An ESR study showed that 1A had no signals but the blue sample (denoted as 1B) emerged a strong single resonance at g = 2.0035 (Fig. 2 bottom), which is close to that of free electrons at 2.0023. After coloration, two new absorption bands around 393 and 602 nm appeared in the DR spectrum (Fig. 3). Such new bands resemble those of N,N′-disubstituted-4,4′-bipyridinium monoradicals.36 Therefore, the illumination resulted in the reduction of the (H2Bpy)2+ cations and the formation of (H2Bpy)+˙ radicals. The O atoms in the carboxylate groups should be electron donors according to the literature.32c,37 | ||
| Fig. 2 Photochromic phenomena (top) and ESR data (bottom) of 1. | ||
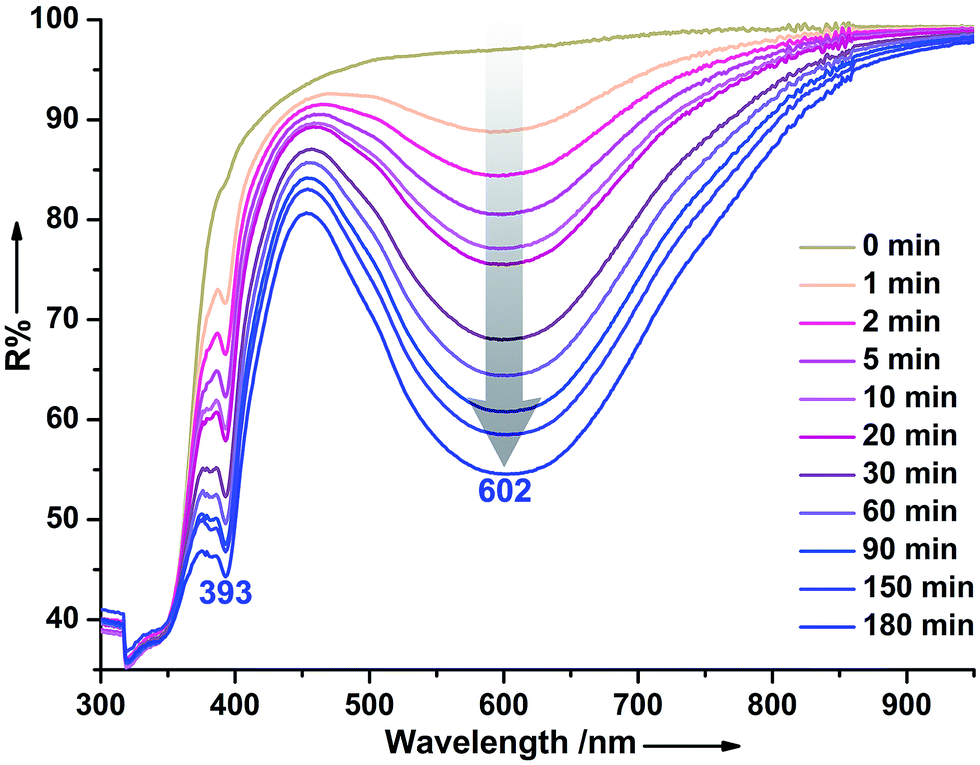 | ||
| Fig. 3 Time-dependent DR spectra of 1 upon irradiation at room temperature in air. | ||
The blue color of 1B can be kept in the dark under ambient environment for more than two months (Fig. S3, ESI†), indicating the formation of a long-lived charge-separated state. This lifetime value is larger than the reported values of ET photochromic MOF compounds, creating a new record.8–11,14,20,32 The effective shielding of radicals by the host–guest structure and the presence of π⋯π stacking interactions around the (H2Bpy)+˙ radicals38 account for the long life of the charge-separated state in the air. The blue color could be bleached by annealing 1B at 150 °C for 4 h in air. The ESR data confirmed that the radical signal almost disappeared after decoloration (Fig. 2 bottom). The coloration–decoloration process can be repeated at least three times. PXRD data revealed that the crystal structure of 1 showed no clear variation during the coloration–decoloration process (Fig. S1, ESI†), and thus the possibility of a photoinduced dissociation or rearrangement reaction can be excluded. All these experimental results demonstrate the ET photochromic characteristic of 1.6
Photoswitching of photoluminescence
As shown in Fig. 4, the maximum emission of 1 is at 458 nm when excited by 384 nm light. The maximum emission bands of (H2Bpy)(NO3)2 39 (Fig. S4, ESI†) and H4BTC peak at 466 (λex = 306 nm) and 340 nm (λex = 310 nm),40 respectively. The emission peak of 1 is very close to that of (H2Bpy)(NO3)2, implying that the (H2Bpy)2+ cation contributes mainly to the photoluminescence of 1. It has been reported that viologen cations are typical luminescence quencher.41 An in situ time-dependent photoluminescence study revealed that the luminescent intensity did decrease gradually with the increase of irradiation time, and reached 10% of the original intensity when the irradiation time is 180 min (Fig. 4). This luminescent contrast before and after coloration is larger than those of most known pyridine derivative-based photochromic compounds15,16,20,24,32,33 and only smaller than those of two examples.16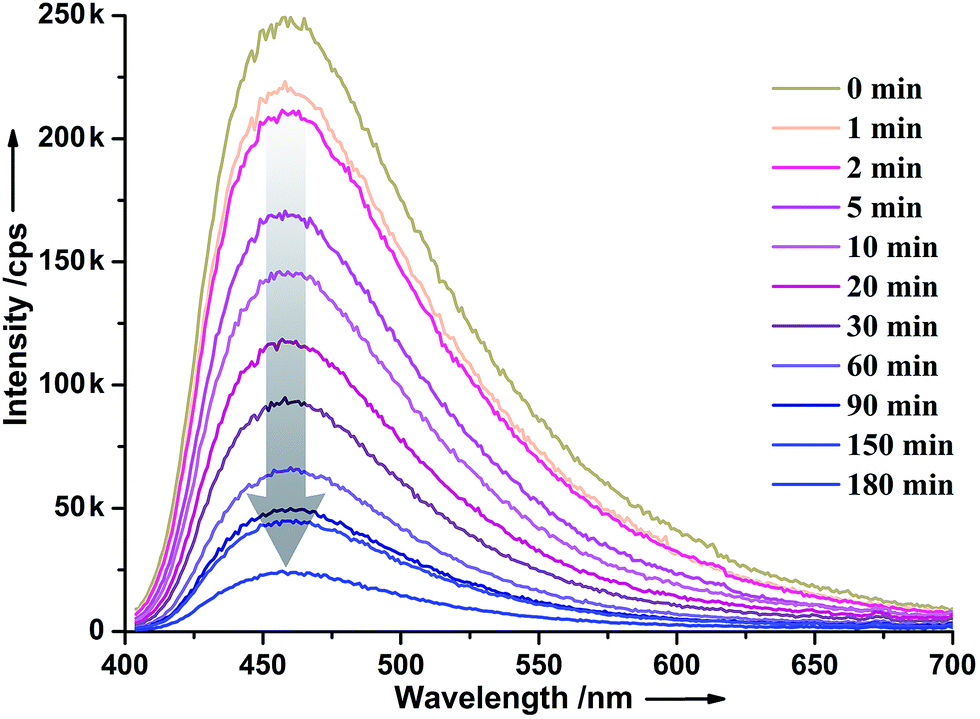 | ||
| Fig. 4 Time-dependent photoluminescence spectra of 1 upon irradiation (λex = 384 nm) at room temperature in air. | ||
Conclusions
We have prepared a new electron-transfer photochromic MOF compound with the photoactive viologen cations as guests. The charge-separated state is relatively stable under ambient conditions. Its lifetime value represents a new record for electron-transfer photochromic MOF compounds. Moreover, the luminescence contrast of about 10 times before and after coloration is also higher than those of most known pyridine derivative-based photochromic compounds. Our results demonstrate again the effectiveness of MOFs on the stability of charge-separated states and the potential of MOFs on the development of new materials with photoswitchable luminescence for data storage, bioimaging, barcode and other possible applications.Acknowledgements
We gratefully acknowledge the financial support by the 973 program of China (2013CB933200), the NSF of China (21373225, 21221001, 21471149), the NSF of Fujian Province (2014J07003, 2014J01065), and Youth Innovation Promotion Association, CAS.Notes and references
- C. Bechinger, S. Ferrere, A. Zaban, J. Spragure and B. A. Gregg, Nature, 1996, 383, 608–610 CrossRef CAS.
- T. L. Andrew, H.-Y. Tsai and R. Menon, Science, 2009, 324, 917–921 CrossRef CAS PubMed.
- S. Kawata and Y. Kawata, Chem. Rev., 2000, 100, 1777–1788 CrossRef CAS PubMed.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS PubMed.
- L. A. Vermeulen and M. E. Thompson, Nature, 1992, 358, 656–658 CrossRef CAS.
- H. Bouas-Laurent and H. DÜrr, Pure Appl. Chem., 2001, 73, 639–665 CrossRef CAS.
- (a) G. Villemure, C. Detellier and A. G. Szabo, J. Am. Chem. Soc., 1986, 108, 4658–4659 CrossRef CAS; (b) T. Nakato, K. Kuroda and C. Kato, Chem. Mater., 1992, 4, 128–132 CrossRef CAS.
- P. K. Dutta and J. A. Incavo, J. Phys. Chem., 1987, 91, 4443–4446 CrossRef CAS.
- (a) M.-S. Wang, G.-C. Guo, W.-Q. Zou, W.-W. Zhou, Z.-J. Zhang, G. Xu and J.-S. Huang, Angew. Chem., Int. Ed., 2008, 47, 3565–3567 CrossRef CAS PubMed; (b) Q.-K. Liu, J.-P. Ma and Y.-B. Dong, J. Am. Chem. Soc., 2010, 132, 7005–7017 CrossRef CAS PubMed; (c) Y. Zeng, Z. Fu, H. Chen, C. Liu, S. Liao and J. Dai, Chem. Commun., 2012, 48, 8114–8116 RSC.
- Y.-Y. Jiang, S.-K. Ren, J.-P. Ma, Q.-K. Liu and Y.-B. Dong, Chem.–Eur. J., 2009, 15, 10742–10746 CrossRef CAS PubMed.
- F. Zhang, X. Zou, W. Feng, X. Zhao, X. Jing, F. Sun, H. Ren and G. Zhu, J. Mater. Chem., 2012, 22, 25019–25026 RSC.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 124, 3420–3423 CrossRef.
- R. Lyndon, K. Konstas, B. P. Ladewig, P. D. Southon, P. C. J. Kepert and M. R. Hill, Angew. Chem., Int. Ed., 2013, 52, 3695–3698 CrossRef CAS PubMed.
- L. Han, L. Qin, L. Xu, Y. Zhou, J. Sun and X. Zou, Chem. Commun., 2013, 49, 406–408 RSC.
- Y.-N. Gong and T.-B. Lu, Chem. Commun., 2013, 49, 7711–7713 RSC.
- (a) J.-K. Sun, L.-X. Cai, Y.-J. Chen, Z.-H. Li and J. Zhang, Chem. Commun., 2011, 47, 6870–6872 RSC; (b) H. Chen, G. Zheng, M. Li, Y. Wang, Y. Song, C. Han, J. Dai and Z. Fu, Chem. Commun., 2014, 50, 13544–13546 RSC.
- J. Park, D. Feng, S. Yuan and H.-C. Zhou, Angew. Chem., Int. Ed., 2014, 53, 1–6 CrossRef.
- F. Luo, C. B. Fan, M. B. Luo, X. L. Wu, Y. Zhu, S. Z. Pu, W.-Y. Xu and G.-C. Guo, Angew. Chem., Int. Ed., 2014, 53, 9298–9301 CrossRef CAS PubMed.
- A. Mallick, B. Garai, M. A. Addicoat, P. S. Petkov, T. Heine and R. Banerjee, Chem. Sci., 2015, 6, 1420–1425 RSC.
- H.-Y. Li, Y.-L. Wei, X.-Y. Dong, S.-Q. Zang and T. C. W. Mak, Chem. Mater., 2015, 27, 1327–1331 CrossRef CAS.
- M. Baroncini, S. d'Agostino, G. Bergamini, P. Ceroni, A. Comotti, P. Sozzani, I. Bassanetti, F. Grepioni, T. M. Hernandez, S. Silvi, M. Venturi and A. Credi, Nat. Chem., 2015, 7, 634–640 CrossRef CAS PubMed.
- (a) K. Uchida, Y. Yamanoi, T. Yonezawa and H. Nishihara, J. Am. Chem. Soc., 2011, 133, 9239–9241 CrossRef CAS PubMed; (b) T. Naito, T. Karasudani, S. Mori, K. Ohara, K. Konishi, T. Takano, Y. Takahashi, T. Inabe, S. Nishihara and K. Inoue, J. Am. Chem. Soc., 2012, 134, 18656–18666 CrossRef CAS PubMed.
- (a) S. Venkataramani, U. Jana, M. Dommaschk, F. D. Sönnichsen, F. Tuczek and R. Herges, Science, 2011, 331, 445–448 CrossRef CAS PubMed; (b) L.-Z. Cai, Q.-S. Chen, C.-J. Zhang, P.-X. Li, M.-S. Wang and G.-C. Guo, J. Am. Chem. Soc., 2015, 137, 10882–10885 CrossRef CAS PubMed.
- (a) A. W.-H. Mau, J. M. Overbeek, J. W. Loder and W. H. F. Sasse, J. Chem. Soc., Faraday Trans. 2, 1986, 82, 869–876 RSC; (b) G. Xu, G.-C. Guo, J.-S. Guo, S.-P. Guo, X.-M. Jiang, C. Yang, M.-S. Wang and Z.-J. Zhang, Dalton Trans., 2010, 39, 8688–8692 RSC.
- J. Zhang, W. Tan, X. Meng and H. Tian, J. Mater. Chem., 2009, 19, 5726–5729 RSC.
- (a) T. J. Wiggleworth and N. R. Branda, Chem. Mater., 2005, 17, 5473–5480 CrossRef; (b) J. Lai, Y. Zhang, N. Pasquale and K.-B. Lee, Angew. Chem., Int. Ed., 2014, 53, 14419–14423 CrossRef CAS PubMed.
- (a) K. Matsuda and M. Irie, J. Am. Chem. Soc., 2000, 122, 8309–8310 CrossRef CAS; (b) H. Tian, B. Chen, H. Tu and K. Müllen, Adv. Mater., 2002, 12, 918–923 CrossRef; (c) P. H.-M. Lee, C.-C. Ko, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2007, 129, 6058–6059 CrossRef CAS PubMed.
- (a) M. Han, S. J. Cho, Y. Norikane, M. Shimizu, A. Kimura, T. Tamagawa and T. Seki, Chem. Commun., 2014, 50, 15815–15818 RSC; (b) S. P. Parua, P. Mondal, P. Pattanayak and S. Chattopadhyay, Polyhedron, 2015, 89, 142–148 CrossRef CAS.
- (a) R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC; (b) S. Goswami, A. K. Das, A. K. Maity, A. Manna, K. Aich, S. Maity, P. Saha and T. K. Mandal, Dalton Trans., 2014, 43, 231–239 RSC.
- W.-J. Ji, Q.-G. Zhai, M.-C. Hu, S.-N. Li, Y.-C. Jiang and Y. Wang, Inorg. Chem. Commun., 2008, 11, 1455–1458 CrossRef CAS.
- J. Wu, F.-M. Wang, J.-Q. Liu, K.-B. Li and D.-H. Xu, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 1231–1235 CrossRef CAS.
- (a) J. Wu, Y. Yan, B. Liu, X. Wang, J. Li and J. Yu, Chem. Commun., 2013, 49, 4995–4997 RSC; (b) O. Toma, N. Mercier and C. Botta, Eur. J. Inorg. Chem., 2013, 2013, 1113–1117 CrossRef CAS; (c) Z.-W. Chen, G. Lu, P.-X. Li, R.-G. Lin, L.-Z. Cai, M.-S. Wang and G.-C. Guo, Cryst. Growth Des., 2014, 14, 2527–2531 CrossRef CAS.
- Z. Fu, Y. Chen, J. Zhang and S. Liao, J. Mater. Chem., 2011, 21, 7895–7897 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- T. M. Bockman and J. K. Kochi, J. Org. Chem., 1990, 55, 4127–4135 CrossRef CAS.
- X.-H. Jin, C. Chen, C.-X. Ren, L.-X. Cai and J. Zhang, Chem. Commun., 2014, 50, 15878–15881 RSC.
- L. Sanhueza, J. Castro, E. Urzúa, L. Barrientow, F. Oyarzun-Ampuero, H. Pesenti, Y. Shibue, N. Sugimura, W. Tomita, H. Nishide and I. Moreno-Villoslada, J. Phys. Chem. B, 2015, 119, 13208–13217 CrossRef CAS PubMed.
- D. J. Barker, J. S. Buckleton, G. R. Clark, R. P. Cooney and C. E. F. Rickard, J. Mol. Struct., 1990, 239, 249–255 CrossRef CAS.
- Q. Hua, Y. Zhao, G.-C. Xu, M.-S. Chen, Z. Su, K. Cai and W.-Y. Sun, Cryst. Growth Des., 2010, 10, 2553–2562 CAS.
- A. Beneduci, S. Cospito, M. L. Deda and G. Chidichimo, Adv. Funct. Mater., 2015, 25, 1240–1247 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD, FT-IR spectrum, crystal data and structure refinements. CCDC 1439857. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra25707g |
| This journal is © The Royal Society of Chemistry 2016 |