Syntheses, structures, and luminescence properties of anion-controlled heterometal modular coordination polymers based on a metalloligand†
Qi-Long Zhang*a,
Guang-Wei Fenga,
Yun-Qian Zhangb and
Bi-Xue Zhu*b
aDepartment of Chemistry, Guiyang Medical College, Guiyang 550004, China. E-mail: gzuqlzhang@126.com
bKey Laboratory of Macrocyclic and Supramolecular Chemistry, Guizhou University, Guiyang 550025, China
First published on 9th January 2014
Abstract
Three new heterometallic coordination polymers (CPs) formulated as [NiAg3(acac)3(NO3)2(H2O)]n (2), [NiAg3(acac)3(ClO4)2(EtOH)2]n (3), and [NiAg2(acac)2(CF3SO3)2(H2O)2]n (4) have been obtained following a synthetic strategy of introducing AgX (X = NO3−, ClO4−, CF3SO3−) to react with a metalloligand [Ni(acac)2(EtOH)2]n (1, Hacac = acetylacetone). Their structures have been determined by single-crystal X-ray diffraction analyses and compounds 2–4 are further characterized by elemental analyses, IR spectra, powder X-ray diffraction (PXRD) and thermogravimetric (TG) analyses. Complex 2 displays an unprecedented three-dimensional (3D) inorganic chiral framework with novel 5-nodal (3,3,4,4,4)-connected (4.82.103)2(42.6)(42.8.103)(43)(44.62)2 topology. Complex 3 possesses 2D inorganic layers holding 7-nodal (3,3,3,3,4,4,4,4)-connected (4.102)2(42.103.12)(42.6)(43)(44.62)2 networks. Complex 4 shows a 2D (4,5)-connected 4,5-L30 layered architecture with (33.4.62.74)(33.42.5) topology. The anions in compounds 2–4 are crucial factors for the formation of the different structures. In addition, the photoluminescent properties of compounds 2–4 are investigated in the solid state at room temperature.
1. Introduction
The assembly of coordination polymers (CPs) with specific network topologies has attracted widespread research interest owing to their tremendous potential applications as functional solid materials.1–3 The diverse structures of such materials are always dependent on many factors, such as metal ions, template, metal–ligand ratio, pH, counteranion, and the number of coordination sites provided by organic ligands.4,5 Anions being a permanent part of the final product can influence the structure either by coordinating directly to the metal centers or by acting as templates that preorganize the organic building blocks used to build up the assembly.6 Therefore, ancillary ligation by different counteranions may cause a significant structural change in the resultant polymers.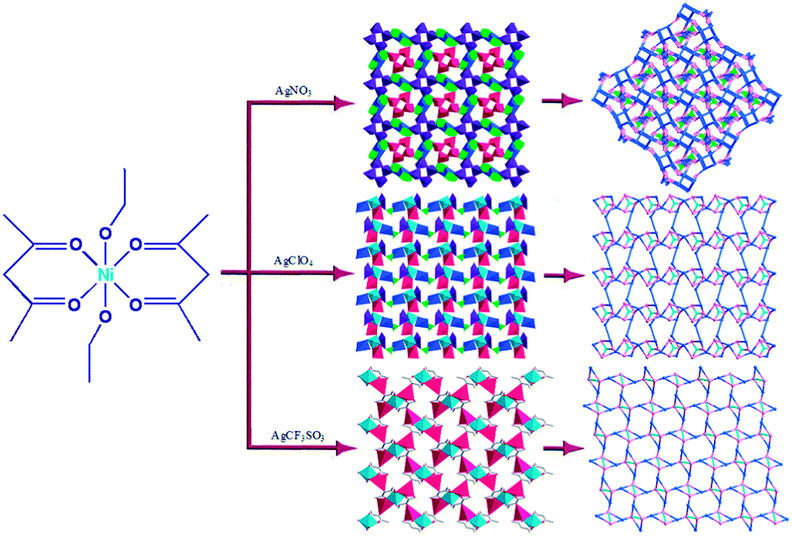 | ||
| Scheme 1 The assembly of three Ag–Ni heterometallic coordination polymers. | ||
There are many ligands which could be rationally designed and reasonably used to compose functional coordination polymers with excellent structures and properties.7 However, the use of metal-containing species, usually called metalloligands, which in place of the organic ligands have suitably oriented peripheral donor groups and are therefore able to direct the formation of the polymeric array, has rarely been reported.8 Heterometallic architectures, often with the ligands linked to different kinds of metal centers, can potentially give secondary building blocks, 1D metal chains, 2D metal networks, or even 3D metal frameworks with additional properties.9 To date, a series of heterometallic architectures, including 2p–3d, 3d–3d, 2p–4f, 3d–4f, 4d–4f, and so on, have been deliberately designed by employing chelating or bridging ligands.10 The introduction of metalloligands has caused the family of heterometal coordination polymers to grow. The metalloligands can serve as excellent candidates for building highly connected, self-penetrating, or helical coordination frameworks due to their bent backbones and versatile bridging characteristics.11 In addition, to the best of our knowledge, Ag–C bonds in acetylacetone (Hacac) based compounds have rarely been documented to date.12
Inspired by the above-mentioned points, we developed a rational synthetic strategy for synthesizing three novel Ni–Ag heterometallic coordination polymers by employing the rigid metalloligand [Ni(acac)2(EtOH)2]n (1) reacted with AgX (X = NO3−, ClO4−, CF3SO3−) (Scheme 1). The results indicated that the counteranions are crucial factors for the formation of the different structures. In this paper, we report the syntheses and characterization of three novel coordination polymers, [NiAg3(acac)3(NO3)2(H2O)]n (2), [NiAg3(acac)3(ClO4)2(EtOH)2]n (3), and [NiAg2(acac)2(CF3SO3)2(H2O)2]n (4), which exhibit a systematic variation of architectures from 2D (4,5)-connected networks to 3D (3,3,4,4,4)-connected inorganic frameworks.
2. Experimental
2.1. Materials and methods
Chemical reagents were purchased commercially and were used as received without further purification. Elemental analyses of C, N, and H were performed on an EA1110 CHNS-0 CE elemental analyzer. IR (KBr pellet) spectra were recorded on a Nicolet Magna 750FT-IR spectrometer. Fluorescent data were collected on an Edinburgh FLS920 TCSPC system. Thermogravimetric measurements were carried out in a nitrogen stream using Netzsch STA449C apparatus with a heating rate of 10 °C min−1. X-ray powder diffraction (XRPD) was carried out on Rigaku DMAX2500 apparatus. Solid-state fluorescence and excitation spectra were recorded at ambient temperature on a Hitachi F4500 fluorescence spectrophotometer.2.2. Synthesis of complexes 1–4
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at room temperature; slow evaporation of this solution yielded green block-shaped single crystals that proved suitable for X-ray analysis. The crystals were filtered off and dried in air. Anal. (%) calcd for C14H26NiO6: C, 48.17; H, 7.51; Ni, 16.82. Found: C, 47.78; H, 7.12; O, 16.28. IR (KBr pellet, cm−1): 3434 (vs), 2926 (m), 1613 (s), 1467 (s), 1390 (s), 1022 (v), 934 (m), 765 (m), 677 (w).
:1) at room temperature; slow evaporation of this solution yielded green block-shaped single crystals that proved suitable for X-ray analysis. The crystals were filtered off and dried in air. Anal. (%) calcd for C14H26NiO6: C, 48.17; H, 7.51; Ni, 16.82. Found: C, 47.78; H, 7.12; O, 16.28. IR (KBr pellet, cm−1): 3434 (vs), 2926 (m), 1613 (s), 1467 (s), 1390 (s), 1022 (v), 934 (m), 765 (m), 677 (w).2.3. X-ray crystallography
Single crystals of the complexes 1–4 with appropriate dimensions were chosen under an optical microscope and quickly coated with high vacuum grease (Dow Corning Corporation) before being mounted on a glass fiber for data collection. Intensity data collection was carried out on a Siemens SMART diffractometer equipped with a CCD detector using MoKα monochromatized radiation (λ = 0.71073 Å) at 293(2) K. The absorption correction was based on multiple and symmetry-equivalent reflections in the data set using the SADABS program. The structures were solved by direct methods and refined by full-matrix least-squares using the SHELXTL package.13 The water H atoms were located from difference maps and no constraint was applied. Crystallographic data for complexes 1–4 are given in Table 1. Selected bond lengths and angles for 1–4 are listed in Table 2.| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| a R1 = Σ||Fo| − |Fc||/Σ|Fo|, wR2 = [Σw(Fo2 − Fc2)2]/Σw(Fo2)2]1/2. | ||||
| Formula | C14H26NiO6 | C15H23Ag3N2NiO13 | C19H33Ag3Cl2NiO16 | C12H18Ag2F6NiO12S2 |
| Mr | 349.06 | 821.66 | 970.67 | 808.80 |
| Crystal system | Triclinic | Tetragonal | Monoclinic | Monoclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P41 | P21 | P21/c |
| a (Å) | 5.2750(4) | 15.0637(12) | 10.2550(17) | 10.5956(8) |
| b (Å) | 8.7923(7) | 15.0637(12) | 12.840(2) | 9.9517(7) |
| c (Å) | 9.3895(7) | 10.8379(12) | 12.682(2) | 11.3334(8) |
| α (°) | 74.675(2) | 90.00 | 90.00 | 90.00 |
| β (°) | 81.562(2) | 90.00 | 107.119(5) | 93.328(2) |
| γ (°) | 76.634(2) | 90.00 | 90.00 | 90.00 |
| V (Å3) | 406(9) | 2459(3) | 1595(9) | 1193(0) |
| Z | 4 | 4 | 2 | 2 |
| ρ (g cm−3) | 1.424 | 2.214 | 2.020 | 2.235 |
| μ (mm−1) | 1.215 | 3.171 | 2.627 | 2.682 |
| T (K) | 293(2) | 293(2) | 293(2) | 293(2) |
| Rint | 0.0161 | 0.0703 | 0.0649 | 0.0195 |
| R [I > 2σ(I)] | R1 = 0.0347 | R1 = 0.0381 | R1 = 0.0400 | R1 = 0.0295 |
| wR2 = 0.0984 | wR2 = 0.0755 | wR2 = 0.0859 | wR2 = 0.0751 | |
| R (all data) | R1 = 0.0348 | R1 = 0.0576 | R1 = 0.0642 | R1 = 0.0300 |
| wR2 = 0.0984 | wR2 = 0.0827 | wR2 = 0.0950 | wR2 = 0.0754 | |
| Gof | 1.059 | 0.991 | 0.0961 | 1.051 |
| Complex 1 | |||||||
| O1–Ni1 | 2.0293 (17) | O2–Ni1 | 2.0013 (17) | O3–Ni1 | 2.1313 (17) | Ni1–O2#1 | 2.0013 (17) |
| Ni1–O1#1 | 2.0293 (18) | Ni1–O3#1 | 2.1313 (17) | O2#1–Ni1-O2 | 180.00 | O2#1-Ni1-O1 | 88.28 (7) |
| O2–Ni1–O1 | 91.72 (7) | O2#1–Ni1–O1#1 | 91.72 (7) | O2–Ni1–O1#1 | 88.28 (7) | O1–Ni1–O1#1 | 180.00 |
| O2#1–Ni1–O3 | 92.42 (7) | O2–Ni1–O3 | 87.58 (7) | O1–Ni1–O3 | 90.63 (7) | O1#1–Ni1–O3 | 89.37 (7) |
| O2#1–Ni1–O3#1 | 88.58 (7) | O2–Ni1–O3#1 | 92.42 (7) | O1–Ni1–O3#1 | 90.63 (7) | O1#1–Ni1–O3#1 | 90.63 (7) |
| Symmetry code: #1: −x+2, −y, −z+2 | |||||||
| Complex 2 | |||||||
| C3–Ag3 | 2.372(7) | C8–Ag1 | 2.382(7) | C13–Ag2#1 | 2.306(6) | O1–Ni1 | 2.054(4) |
| O1–Ag2#1 | 2.586(4) | O2–Ni1 | 2.012(5) | O3–Ni1 | 2.061(4) | O3–Ag2 | 2.400(5) |
| O4–Ni1 | 2.056(5) | O4–Ag1 | 2.567(4) | O5–Ni1 | 2.037(5) | O5–Ag3 | 2.592(5) |
| O6–Ni1 | 2.025(4) | O7–Ag1 | 2.407(6) | O7–Ag1#2 | 2.540(6) | O9–Ag1#2 | 2.568(7) |
| O10–Ag3#3 | 2.368(7) | O10–Ag3 | 2.492(7) | O13–Ag2 | 2.275(4) | Ag1–O7#4 | 2.540(6) |
| Ag1–O9#4 | 2.568(7) | Ag2–C13#5 | 2.306(6) | Ag2–O1#5 | 2.586(4) | Ag3–O10#6 | 2.368(7) |
| O2–Ni1–O6 | 88.2(2) | O2–Ni1–O5 | 91.60(19) | O6–Ni1–O5 | 90.72(18) | O2–Ni1–O1 | 89.6(2) |
| O6–Ni1–O1 | 177.68(19) | O5–Ni1–O1 | 90.11(18) | O2–Ni1–O4 | 177.02(19) | O6–Ni1–O4 | 88.80(18) |
| O5–Ni1–O4 | 88.60(19) | O1–Ni1–O4 | 93.39(18) | O2–Ni1–O3 | 91.86(19) | O6–Ni1–O3 | 92.70(17) |
| O5–Ni1–O3 | 175.21(19) | O1–Ni1–O3 | 86.60(17) | O4–Ni1–O3 | 88.12(18) | C8-Ag1-O7 | 111.5(2) |
| C8–Ag1–O7#4 | 97.9(2) | O7–Ag1–O7#4 | 111.3(3) | C8–Ag1–O4 | 86.66(19) | O7–Ag1–O4 | 98.54(18) |
| O7#4–Ag1–O4 | 145.29(17) | C8–Ag1–O9#4 | 122.7(2) | O7–Ag1–O9#4 | 123.5(2) | O7#4–Ag1–O9#4 | 49.61(18) |
| O4–Ag1–O9#4 | 99.29(17) | O13–Ag2–C13#5 | 135.7(2) | O13–Ag2-O3 | 91.38(18) | C13#5–Ag2–O3 | 122.9(2) |
| O13–Ag2–O1#5 | 109.02(16) | C13#5–Ag2–O1#5 | 86.27(19) | O3–Ag2–O1#5 | 110.19(15) | O10#6–Ag3–C3 | 118.0(3) |
| O10#6–Ag3–O10 | 116.2(3) | C3–Ag3–O10 | 105.0(2) | O10#6–Ag3–O5 | 105.8(2) | C3–Ag3–O5 | 85.1(2) |
| Symmetry code: #1 −x + 1, −y, z + 1/2; #2 y, −x + 1, z − 1/4; #3 −y + 1, x − 1, z + 1/4; #4 −y + 1, x, z + 1/4; #5 −x + 1, −y, z − 1/2; #6 y + 1, −x + 1, z − 1/4 | |||||||
| Complex 3 | |||||||
| C3–Ag3 | 2.357(8) | C8–Ag2 | 2.282(9) | C13–Ag1 | 2.314(8) | O15–Ag2 | 2.213(7) |
| O1–Ni1 | 2.048(5) | O2–Ni1 | 2.051(5) | O3–Ni1 | 2.048(5) | O3–Ag3#1 | 2.526(5) |
| O4–Ni1 | 2.024(6) | O4–Ag1 | 2.512(5) | O5–Ni1 | 2.056(5) | O5–Ag3 | 2.592(5) |
| O6–Ni1 | 2.042(6) | O6–Ag3#1 | 2.492(5) | O10–Ag1 | 2.455(7) | O16–Ag3 | 2.389(6) |
| O11–Ag1 | 2.356(7) | Ag3–O6#2 | 2.492(5) | Ag3–O3#2 | 2.526(5) | O4–Ni1–O6 | 93.1(2) |
| O4–Ni1–O3 | 89.7(2) | O6–Ni1–O3 | 86.7(2) | O4–Ni1–O1 | 89.5(2) | O6–Ni1–O1 | 177.3(2) |
| O3–Ni1–O1 | 92.5(2) | O4–Ni1–O2 | 179.7(2) | O6–Ni1–O2 | 86.9(2) | O6–Ni1–O2 | 86.9(2) |
| O3–Ni1–O2 | 90.5(2) | O1–Ni1–O2 | 90.5(2) | O4–Ni1–O5 | 87.6(2) | O6–Ni1–O5 | 88.8(2) |
| O3–Ni1–O5 | 174.7(2) | O1–Ni1–O5 | 92.1(2) | O2–Ni1–O5 | 92.1(2) | C13–Ag1–O11 | 147.9(3) |
| C13–Ag1–O10 | 119.5(3) | O11–Ag1–O10 | 88.8(3) | C13–Ag1–O4 | 91.1(2) | O11–Ag1–O4 | 107.5(2) |
| O10–Ag1–O4 | 84.6(2) | O15–Ag2–C8 | 149.0(3) | C3–Ag3–O16 | 135.9(3) | C3–Ag3–O6#2 | 116.2(2) |
| O16–Ag3–O6#2 | 75.3(2) | C3–Ag3–O3#2 | 119.5(2) | O16–Ag3–O3#2 | 104.4(2) | O6#2–Ag3–O3#2 | 68.05(18) |
| C3–Ag3–O5 | 89.9(2) | O16–Ag3–O5 | 96.73(19) | O6#2–Ag3–O5 | 149.95(17) | O3#2–Ag3–O5 | 86.67(16) |
| Symmetry codes: #1 −x, y − 1/2, −z + 1; #2 −x, −y + 1/2, −z + 1 | |||||||
| Complex 4 | |||||||
| C3–Ag1#1 | 2.283(3) | O3–Ag1 | 2.465(3) | O4–Ni1 | 2.016(2) | O4–Ag1 | 2.298(2) |
| O5–Ni1 | 2.001(2) | O6–Ni1 | 2.050(2) | Ni1–O5#2 | 2.001(2) | Ni1–O4#2 | 2.016(2) |
| Ni1–O6#2 | 2.050(2) | Ag1–C3#3 | 2.283(3) | O5#2–Ni1–O5 | 180.00 | O5#2–Ni1–O4 | 86.96(9) |
| O5–Ni1–O4 | 93.04(9) | O5#2–Ni1–O4#2 | 93.04(9) | O5–Ni1–O4#2 | 86.96(9) | O4–Ni1–O4#2 | 180.00 |
| O5#2–Ni1–O6#2 | 91.13(10) | O5–Ni1–O6#2 | 88.87(10) | O4–Ni1–O6#2 | 89.71(9) | O4#2–Ni1–O6#2 | 90.29(9) |
| O5#2–Ni1–O6 | 88.87(10) | O5–Ni1–O6 | 91.13(10) | O4–Ni1–O6 | 90.29(9) | O4#2–Ni1–O6 | 89.71(9) |
| O6#2–Ni1–O6 | 180.00 | C3#3–Ag1–O4 | 167.80(10) | C3#3–Ag1–O3 | 100.71(11) | O4–Ag1–O3 | 89.29(9) |
| Symmetry codes: #1 −x + 2, y − 1/2, −z + 7/2; #2 −x + 2, −y − 1, −z + 3; #3 −x + 2, y + 1/2, −z + 7/2 | |||||||
3. Results and discussion
3.1. Synthesis and characterization
The synthesis of complexes 2–4 was carried out in darkness to avoid photodecomposition. Compounds 2–4 were obtained after compound 1 reacted with AgX (X = NO3−, ClO4−, CF3SO3−) in an organic solution, followed by slow evaporation at room temperature. Their molecular structures were determined by single-crystal X-ray structure analyses and further characterized by elemental analyses, infrared (IR) spectra, and thermogravimetric (TG) analyses. X-ray powder diffraction (XRPD) patterns confirmed the phase purity of the bulk materials (Fig. S1†).The relatively weak absorption bands around ca. 3435 cm−1 are due to the characteristic peaks of water O–H vibrations. The absorption bands with variable intensity in the frequency range 1400–1600 cm−1 correspond to vibrations of the backbones of the ligands. The absorption band of the –CHmethine moieties is observed to be relatively weak at 3240 cm−1 and was significantly shifted toward the lower frequency region, compared to the acac− ligands in compounds 2–4 (ca. 3165 and 3135 cm−1) (Fig. S2†). The relatively low frequency of the band is indicative of Ag–C bonding.
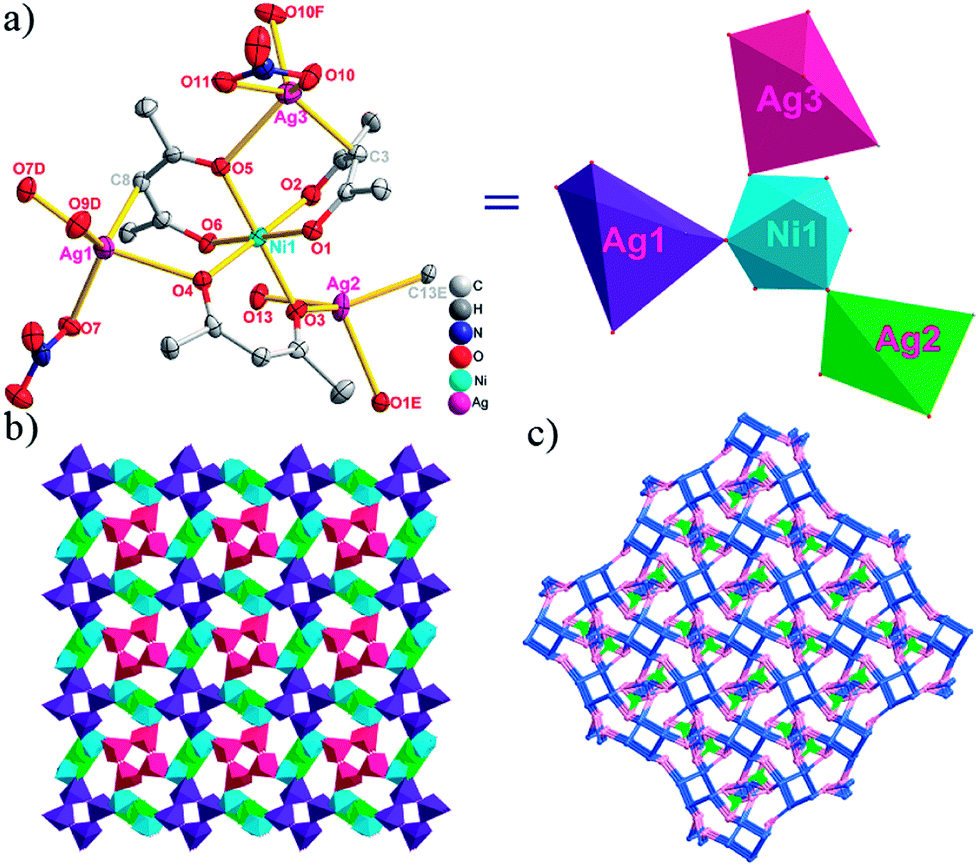 | ||
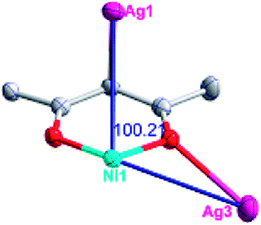
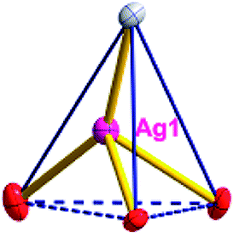
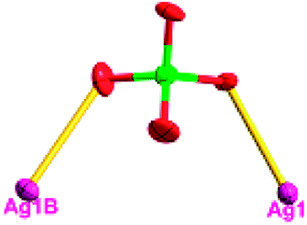
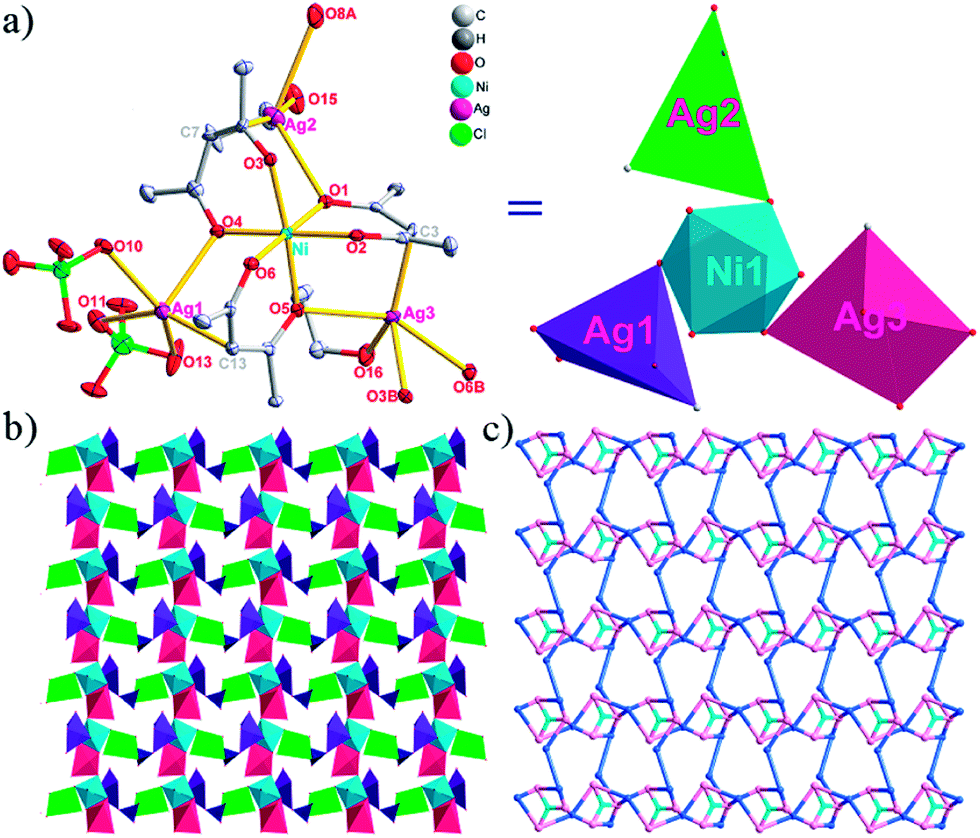
| Fig. 1 (a) A general view of the [NiAg3(acac)3(NO3)2(H2O)]n compound (2) with atoms (30% probability) represented as ellipsoids, and the NiAg3 cluster. The hydrogen atoms were omitted for clarity (symmetry codes: A: 1 − x, −y, 0.5 + z; B: y, 1 − x, −0.25 + z; C: 1 − y, −1 + x, 0.25 + z; D: 1 − y, x, 0.25 + z; E: 1 − x, −y, −0.5 + z; F: 1 + y, 1 − x, −0.25 + z). (b) View of the unprecedented 3D inorganic frameworks. (c) Schematic view of the 5-nodal (3,3,4,4,4)-connected (4.82.103)2(42.6)(42.8.103)(43)(44.62)2 topology of 2 (green spheres: NiII atoms; light blue spheres: AgI atoms; rose spheres: acac− ligands). | ||
| acac− | AgI | X− | |
|---|---|---|---|
a  . . |
|||
| 2 | 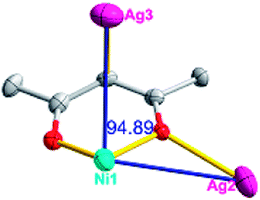 |
 |
 |
 |
 |
||
 |
 |
 |
|
| 3 |  |
 |
 |
 |
 |
||
 |
 |
 |
|
| 4 | 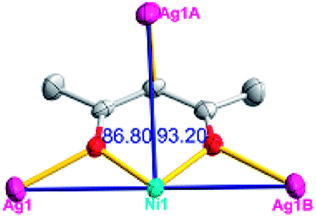 |
 |
 |
There are two coordination modes of acac− ligands: the ∠-shaped ligand and the ⊥-shaped ligand (Table 3). Either the ∠-shaped or the ⊥-shaped acac− ligand is bidentate-chelated with one NiII center and linked to one AgI anion using the methine carbon atom. The difference between the two coordination modes is that there is only one η2 oxygen atom in the ∠-shaped ligand, but two in the ⊥-shaped acac− ligand. The NiAg3 cluster shows a triangle shape and is connected with the acac− ligands, giving a 1D zigzag polymeric [NiAg3(acac)3]n2n+ chain along the crystallographic [001] direction with an adjacent Ni⋯Ni separation of 7.180 Å (Fig. S3†). Of particular interest is that the NO3− ions in compound 2 adopted μ2-η2:η1 coordination modes with linked AgI ions, forming an infinite right-handed 43 helical [Ag(NO3)]n chain along the [100] direction (Fig. S4†). The right-handed 43 helical [Ag(NO3)]n chains linked the 1D polymeric [NiAg3(acac)3]n2n− chains, finally forming a chiral 3D framework (Fig. S5†). With the non-coordinated and mono-coordinated atoms omitted, an unprecedented 3D inorganic [NiAg3O5]n+5n framework appeared (Fig 1b and S6†), which has rarely been reported before as far as we know.15
From a topological view,16 the network of 2 can be rationalized to a novel 5-nodal (3,3,4,4,4)-connected (4.82.103)2(42.6)(42.8.103)(43)(44.62)2 topology by denoting the NiII atoms as three-connected nodes, all of the AgI anions as four-connected nodes, the NO3− as linkers, and the acac− ligands as three- and four-connected nodes (Fig. 1c).
 | ||
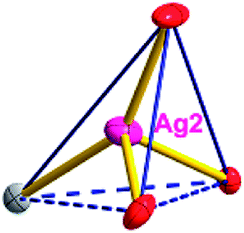
| Fig. 2 (a) A general view of the [NiAg3(acac)3(ClO4)2(EtOH)2]n compound (3) with atoms (30% probability) represented as ellipsoids, and the NiAg3 cluster. The hydrogen atoms were omitted for clarity (symmetry codes: A: −x, y − 1/2, −z + 1; B: −x, −y + 1/2, −z + 1). (b) Polyhedral view of the 2D inorganic networks of 3 along the [101] direction. (c) Schematic view of the 7-nodal (3,3,3,3,4,4,4,4)-connected (4.102)2(42.103.12)(42.6)(43)(44.62)2 topology of 3 (green spheres: NiII atoms; light blue spheres: AgI atoms; rose spheres: acac− ligands). | ||
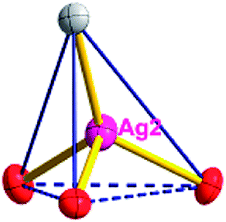
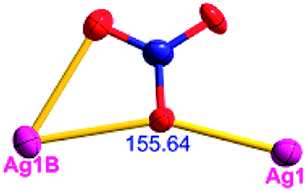
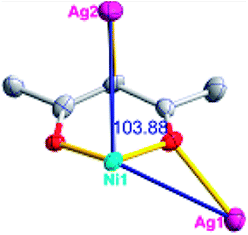
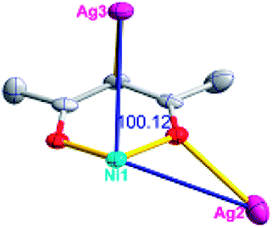
Different from compound 2, all of the acac− ligands in 3 have adopted ∠-shaped coordination modes linked with one NiII center and two AgI anions, and the Ag–Ni–Ag angles are 103.85, 100.12, and 103.87°. There is also a NiAg3 cluster in compound 3, similar but different to that in compound 2. The NiAg3 clusters linked with each other by sharing η2 oxygen atoms, giving a 1D polymeric [NiAg3(acac)3]n2n+ chain along the [010] direction with an adjacent Ni⋯Ni separation of 6.845 Å (Fig. S7†). The bridging ClO4− anions linked the neighboring chains, finally giving a 2D network, with an adjacent Ag⋯Ag separation of 5.072 Å. Omitting the non-coordinated carbon, hydrogen, and oxygen atoms, a 2D inorganic layer was constructed (Fig. 2b and S8†).
From a topological view, the network of 3 can be rationalized to a much more simplified 7-nodal (3,3,3,3,4,4,4,4)-connected (4.102)2(42.103.12)(42.6)(43)(44.62)2 network architecture by denoting the NiII atoms as three-connected nodes, the Ag1 and Ag2 anions as three-connected nodes, the Ag3 anions as four-connected nodes, the ClO4− as linkers, and the acac− ligands as three- and four-connected nodes (Fig. 2c). Furthermore, neighboring layers stack in a parallel fashion along the c crystal direction, giving rise to a 3D supramolecular framework via inter-layer C–H⋯O (C6–H6B⋯O12 (D–A = 3.962(1) Å, ∠D–H⋯A = 123.22(6)°) and C8–H8⋯O14 (D–A = 3.398(5) Å, ∠D–H⋯A = 177.62(8)°)) hydrogen bonds between the methyl of the methanol and methylene of the acac− ligands and the ClO4− anions (Fig. S9†).
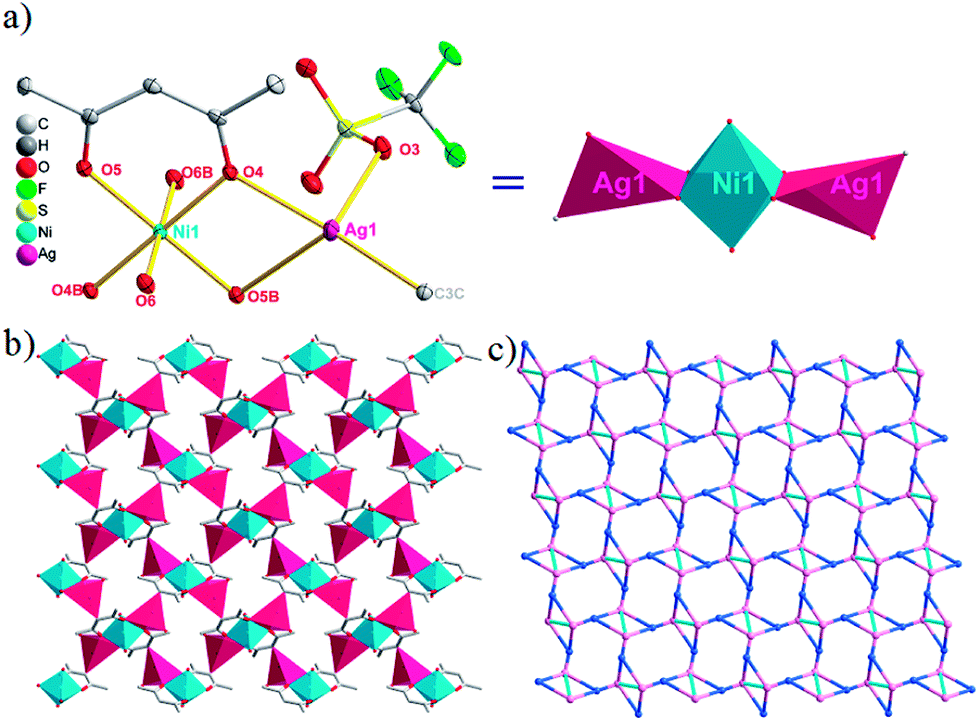 | ||
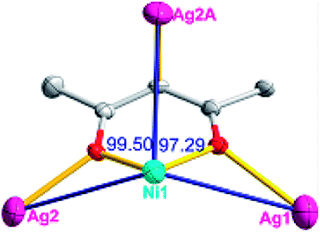
| Fig. 3 (a) A general view of the [NiAg2(acac)2(CF3SO3)2(H2O)2]n compound (4) with atoms (30% probability) represented as ellipsoids, and the NiAg2 cluster. The hydrogen atoms were omitted for clarity (symmetry codes: A: 2 − x, −0.5 + y, 3.5 − z; B: 2 − x, −1 − y, 3 − z; C: 2 − x, 0.5 + y, 3.5 − z). (b) NiAg2 cluster based 2D [NiAg2(acac)2]n2n+ layers. (c) Schematic view of the (4,5)-connected 4,5-L30 topology of 4 with the Schläfli symbol (33.4.62.74)(33.42.5) (green spheres: NiII atoms; light blue spheres: AgI atoms; rose spheres: acac− ligands). | ||
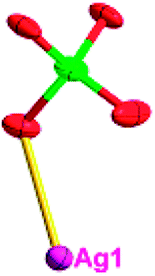
The acac− in compound 4 acts as a ⊥-shaped ligand connecting the NiII centers and AgI anions to generate a 2D network (Fig. S10†), and the Ag–Ni–Ag angles are 93.20 and 86.80°. The NiAg2 clusters in compound 3 are connected with each other through the Ag–C bonds. We found that the neighboring NiAg2 clusters are interlocked, giving 2D polymeric [NiAg2(acac)2]n2n− layers along the [101] direction with an adjacent Ni⋯Ni separation of 7.541 Å (Fig. 3b). The monodentate CF3SO3− anions in compound 4 can't bridge the neighboring layers, only giving a 2D network. From the topological point of view, the overall structure of 4 can be defined as a 2D (4,5)-connected 4,5-L30 layered architecture with the Schläfli symbol (33.4.62.74)(33.42.5) if the NiII atom is considered as a linker, the AgI anions as five-connected nodes, and the acac− ligands as four-connected nodes (Fig. 3c). If we consider the NiAg2 cluster as a node, the whole structure of compound 4 can be regarded as a 4-connected 44-sql net.
3.2. Influence of anions and coordination modes of Hacac ligands on structures
According to the above description of the crystal structures, Hacac exhibits ∠- and ⊥-shaped coordination modes, shown in Table 3. ∠-shaped acac− coordinates to one NiII and two AgI ions through Cmethylene and terminal η1- and η2-O, correspondingly the ⊥-shaped acac− ligand coordinates to one NiII and three AgI ions through Cmethylene and two terminal η2-O atoms. In complex 2, there are two ∠-shaped acac− ligands with Ag–Ni–Ag angles of 94.89 and 100.21° and one ⊥-shaped acac− ligand with Ag–Ni–Ag angles of 99.50 and 97.29°. The acac− ligands shared with the center ions formed a 1D [NiAg3(acac)3]n2n+ chain. For compound 3, all of the acac− ligands adopted ∠-shaped coordination modes, with the angles of Ag–Ni–Ag around 100.12 and 103.85°, here the acac− ligands linked with metal ions, finally generating a 1D zigzag [NiAg3(acac)3]n2n+ polymeric chain. There are only ⊥-shaped acac− ligands in compound 4, and the angles of Ag–Ni–Ag are 86.80 and 93.20°. Bridged by the acac− ligands, a 2D [NiAg2(acac)2]n2n+ polymeric layer was obtained.The NO3− in compound 2 acts as the μ2-η2:η1 linker, connected the neighbouring AgI ions to a right-handed 61 helical [Ag(NO3)]n chain along the c axis, finally giving a 3D chiral framework. One of the ClO4− ions in compound 3 is monodentate with AgI ions, and the other ClO4− ion bridges the 1D zigzag [NiAg3(acac)3]n2n+ polymeric chain, forming a 2D (3,3,3,3,4,4,4,4)-connected network. The CF3SO3− ions in compound 4 occupied one coordination site of the AgI ion, and had no effect on the organisation of the whole structure. Thus, the coordination modes of the ligands and the anions have great influence on the final structure and topology.
3.3. Thermal analysis
The thermal behaviours of 2–4 were studied by Thermogravimetric Analysis (TGA). The experiments were performed on samples consisting of numerous single crystals under a N2 atmosphere with a heating rate of 10 °C min−1, as shown in Fig. 4. For complex 2, the first weight loss of 3.07% (calc. 2.20%) occurred below 110 °C and can be attributed to the removal of water molecules. Next, the framework of compound 2 begins to collapse with the residue of about 50.13% (calc. 51.52%). For complex 3, with ethanol loss the whole structure started to collapse, finally giving a residue of about 48.87% (calc. 49.16%). For complex 4, the first weight loss of 4.71% (calc. 4.48%) occurred at around 120 °C and can be attributed to the removal of coordinated water molecules. The second weight loss at around 240–270 °C and the third loss at around 400 °C finally broke the framework of compound 4 with the residue of about 40.06% (calc. 39.17%). | ||
| Fig. 4 TGA curves for compounds 2–4. | ||
3.4. Photoluminescent properties of 1–4
The photoluminescence spectra of the metalloligand (1) and complexes 2–4 are shown in Fig. 5. Compound 1 displayed photoluminescence with an emission maximum at 512 nm (λex = 380 nm). It can be presumed that the peak originates from the π* → n or π* → π transitions.17 It can be observed that intense emissions occur at 532 nm (λex = 380 nm) for 2, 402 nm (λex = 320 nm) for 3, and 489 nm (λex = 380 nm) for 4. The resemblance between the emissions of 2 and 4 and that of the metalloligand indicates that the emissions of 2 and 4 are probably attributable to the intraligand (IL) π → π* transitions modified by metal coordination.18 For compound 3, there is also a peak at 488 nm, corresponding to the intraligand (IL) π → π* transitions, at the same time, the peak at 402 nm has a blue shift of 110 nm, which can mainly be attributed to the MLCT (metal-to-ligand charge transfer) characteristics.19 The enhancement of luminescence of all complexes was attributed to ligand coordination to the metal center, which effectively increases the rigidity of the ligand and reduces the loss of energy by radiationless decay.20 | ||
| Fig. 5 Photoluminescence spectra of complexes 1–4. | ||
4. Conclusions
In summary, we have developed a rational synthetic strategy for synthesizing three new heterometallic coordination polymers ([NiAg3(acac)3(NO3)2(H2O)]n (2), [NiAg3(acac)3(ClO4)2(EtOH)2]n (3), and [NiAg2(acac)2(CF3SO3)2(H2O)2]n (4)) based on the metalloligand [Ni(acac)2(EtOH)2]n (1). The ions in compounds 2–4 are crucial factors for the formation of the final structures. Complex 2 displays an unprecedented 3D chiral inorganic framework with novel 5-nodal (3,3,4,4,4)-connected architecture. Complex 3 possesses 2D inorganic 7-nodal layers. Complex 4 shows 2D (4,5)-connected 4,5-L30 networks. This metalloligand-based synthetic strategy significantly expands the pool of inorganic building blocks and is clearly useful for the construction of porous MOFs with high gas uptake capacity.Acknowledgements
The support of the Natural Science Foundation of China (Grant no. 21061003) and the Joint fund of Guizhou Province of PR China (no. [2012]016), the Australian Research Council and the Australian partnership are gratefully acknowledged.References
- (a) J. P. Zhang, Y. B. Zhang, J. B. Lin and X. M. Chen, Chem. Rev., 2012, 112, 1001 CrossRef CAS PubMed; (b) B. S. Zheng, J. F. Bai, J. G. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2011, 133, 748 CrossRef CAS PubMed; (c) H. Wang, Z. Chang, Y. Li, R. M. Wen and X. H. Bu, Chem. Commun., 2013, 49, 6659 RSC; (d) P. Q. Liao, D. D. Zhou, A. X. Zhu, L. Jiang, R. B. Lin, J. P. Zhang and X. M. Chen, J. Am. Chem. Soc., 2012, 134, 17380 CrossRef CAS PubMed; (e) Y. Q. Chen, S. J. Liu, Y. W. Li, G. R. Li, K. H. He, Z. Chang and X. H. Bu, CrystEngComm, 2013, 15, 1613 RSC; (f) B. Li, R. J. Wei, J. Tao, R. B. Huang, L. S. Zheng and Z. Zheng, J. Am. Chem. Soc., 2010, 132, 1558 CrossRef CAS PubMed; (g) Y. S. Wei, K. J. Chen, P. Q. Liao, B. Y. Zhu, R. B. Lin, H. L. Zhou, B. Y. Wang, W. Xue, J. P. Zhang and X. M. Chen, Chem. Sci., 2013, 4, 1539 RSC; (h) X. Zhang, L. Fan, W. Zhang, Y. Ding, W. Fan and X. Zhao, Dalton Trans., 2013, 42, 16562 RSC.
- (a) Y. W. Li, J. R. Li, L. F. Wang, B. Y. Zhou, Q. Yang, S. J. Liu, Y. Li and X. H. Bu, Chem. Commun., 2013, 49, 871 RSC; (b) J. B. Lin, W. Xue, B. Y. Wang, J. Tao, W. X. Zhang, J. P. Zhang and X. M. Chen, Inorg. Chem., 2012, 51, 9423 CrossRef CAS PubMed; (c) L. Fan, X. Zhang, D. Li, D. Sun, W. Zhang and J. Dou, CrystEngComm, 2013, 15, 349 RSC; (d) R. G. Xiong, X. Xue, H. Zhao, B. F. Abrahams, X. Z. You and Z. Xue, Angew. Chem., Int. Ed., 2002, 41, 3800 CrossRef CAS; (e) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed; (f) D. Sun, G. G. Luo, N. Zhang, R. B. Huang and L. S. Zheng, Chem. Commun., 2011, 47, 1461 RSC; (g) X. T. Zhang, D. Sun, B. Li, L. M. Fan, B. Li and P. H. Wei, Cryst. Growth Des., 2012, 12, 3845 CrossRef CAS; (h) D. Sun, C. F. Yang, H. R. Xu, H. X. Zhao, Z. H. Wei, N. Zhang, L. J. Yu, R. B. Huang and L. S. Zheng, Chem. Commun., 2010, 46, 8168 RSC.
- (a) J. B. Lin, J. P. Zhang and X. M. Chen, J. Am. Chem. Soc., 2010, 132, 6654 CrossRef CAS PubMed; (b) Y. W. Li, K. H. He and X. H. Bu, J. Mater. Chem. A, 2013, 1, 4186 RSC; (c) Y. W. Li, J. R. Li, L. F. Wang, B. Y. Zhou, Q. Cheng and X. H. Bu, J. Mater. Chem. A, 2013, 1, 495 RSC; (d) D. Sun, N. Zhang, R. B. Huang and L. S. Zheng, Cryst. Growth Des., 2010, 10, 3699 CrossRef CAS; (e) D. Sun, H. Wang, H. F. Lu, S. Y. Feng, Z. W. Zhang, G. X. Sun and D. F. Sun, Dalton Trans., 2013, 42, 6281 RSC; (f) X. T. Zhang, L. M. Fan, X. Zhao, D. Sun, D. C. Li and J. M. Dou, CrystEngComm, 2012, 14, 2053 RSC; (g) L. Fan, X. Zhang, W. Zhang, Y. Ding, L. Sun, W. Fan and X. Zhao, CrystEngComm, 2014 10.1039/C3CE42203H.
- (a) Z. X. Chen, S. X. Xiang, T. B. Liao, Y. T. Yang, Y. S. Chen, Y. M. Zhou, D. Y. Zhao and B. L. Chen, Cryst. Growth Des., 2010, 10, 2775 CrossRef CAS; (b) L. M. Fan, X. T. Zhang, Z. Sun, W. Zhang, Y. S. Ding, W. L.Fan, L. M. Sun, X. Zhao and H. Lei, Cryst. Growth Des., 2013, 13, 2462 CrossRef CAS; (c) X. T. Zhang, L. M. Fan, Z. Sun, W. Zhang, D. C. Li, J. M. Dou and L. Han, Cryst. Growth Des., 2013, 13, 792 CrossRef CAS; (d) B. X. Zhu, Q. L. Zhang, Y. Q. Zhang, T. Zhu, J. K. Clegg, L. F. Lindoy and G. Wei, Inorg. Chem., 2008, 47, 10053 CrossRef CAS PubMed; (e) Q. L. Zhang, B. X. Zhu, Y. Q. Zhang, Z. Tao, J. K. Clegg, L. F. Lindoy and G. Wei, Inorg. Chem. Commun., 2010, 13, 992 CrossRef CAS PubMed.
- (a) Q. Chen, J. B. Lin, W. Xue, M. H. Zeng and X. M. Chen, Inorg. Chem., 2011, 50, 2321 CrossRef CAS PubMed; (b) Y. H. Wei, D. Sun, D. Q. Yuan, Y. J. Liu, Y. Zhao, X. Y. Li, S. N. Wang, J. M. Dou, X. P. Wang, A. Y. Hao and D. F. Sun, Chem. Sci., 2012, 3, 2282 RSC; (c) Y. W. Li, H. Ma, Y. Q. Chen, K. H. He, Z. X. Li and X. H. Bu, Cryst. Growth Des., 2012, 12, 189 CrossRef CAS; (d) D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033 CrossRef CAS PubMed; (e) X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2009, 131, 2159 CrossRef CAS PubMed; (f) S. Q. Ma, J. P. Simmons, D. F. Sun, D. Q. Yuan and H. C. Zhou, Inorg. Chem., 2009, 48, 5263 CrossRef CAS PubMed.
- (a) A. Mitra, C. T. Hubley, D. K. Panda, R. J. Clark and S. Saha, Chem. Commun., 2013, 49, 6629 RSC; (b) C. Latouche, S. Kahlal, E. Furet, P. K. Liao, Y. R. Lin, C. S. Fang, J. Cuny, C. W. Liu and J. Y. Saillard, Inorg. Chem., 2013, 52, 7752 CrossRef CAS PubMed; (c) S. I. Noro, K. Fukuhara, K. Sugimoto, Y. Hijikata, K. Kubo and T. Nakamura, Dalton Trans., 2013, 42, 11100 RSC; (d) J. M. Wang, S. G. Li, P. J. Yang, X. J. Huang, X. J. Yang and B. Wu, CrystEngComm, 2013, 15, 4540 RSC; (e) W. Q. Kan, J. Yang, Y. Y. Liu and J. F. Ma, CrystEngComm, 2012, 14, 6271 RSC.
- (a) Q. L. Zhang, B. X. Zhu, L. F. Lindoy and G. Wei, Inorg. Chem. Commun., 2008, 11, 678 CrossRef CAS PubMed; (b) L. F. Ma, L. Y. Wang, M. Du and S. R. Batten, Inorg. Chem., 2010, 49, 365 CrossRef CAS PubMed; (c) D. S. Li, Y. P. Wu, P. Zhang, M. Du, J. Zhao, C. P. Li and Y. U. Wang, Cryst. Growth Des., 2010, 10, 2037 CrossRef CAS.
- (a) S. M. Zhang, R. Pattacini and P. Braunstein, Dalton Trans., 2011, 40, 5711 RSC; (b) S. Ghosh, S. Biswas, A. Bauza, M. Barcelo-Oliver, A. Frontera and A. Ghosh, Inorg. Chem., 2013, 52, 7508 CrossRef CAS PubMed; (c) C. Archambault, R. Bender, P. Braunstein, S. Bouaoud, D. Rouag, S. Golhen and L. Ouahab, Chem. Commun., 2001, 849 RSC.
- (a) J. H. Zhang, S. M. Cheng, X. F. Wang, L. M. Yuan, M. Q. Xue, Y. Wang and W. L. Liu, CrystEngComm, 2013, 15, 6074 RSC; (b) Y. Y. Xu, Y. Q. Sun, S. Yang, G. Y. Zhang and D. Z. Gao, RSC Adv., 2013, 3, 11640 RSC; (c) S. T. Meally, S. M. Taylor, E. K. Brechin, S. Piligkos and L. F. Jones, Dalton Trans., 2013, 42, 10315 RSC; (d) C. J. Chen, N. Wang, Y. Long, J. Y. Gao, W. P. Xie, X. R. Ran and S. T. Yue, CrystEngComm, 2013, 15, 4611 RSC.
- (a) X. T. Zhang, L. M. Fan, Z. Sun, W. Zhang, W. L. Fan, L. M. Sun and X. Zhao, CrystEngComm, 2013, 15, 4910 RSC; (b) X. J. Gu and D. F. Xue, Cryst. Growth Des., 2006, 6, 2551 CrossRef CAS; (c) Y. Y. Liu, C. B. Ma, H. Chen, M. Q. Hu, H. M. Wen, H. H. Cui and C. N. Chen, Dalton Trans., 2013, 42, 3787 RSC.
- (a) D. Y. Du, J. S. Qin, C. X. Sun, X. L. Wang, S. R. Zhang, P. Shen, S. L. Li, Z. M. Su and Y. Q. Lan, J. Mater. Chem., 2012, 22, 19673 RSC; (b) A. D. Burrows, M. F. Mahon, C. L. Renouf, C. Richardson, A. J. Warren and J. E. Warren, Dalton Trans., 2012, 41, 4153 RSC; (c) A. D. Burrows, C. G. Frost, M. F. Mahon, P. R. Raithby, C. L. Renouf, C. Richardson and A. J. Stevenson, Chem. Commun., 2010, 46, 5067 CAS.
- (a) E. Alonso, J. Fornies, C. Fortuno, A. Mattin and A. G. Orpen, Organometallics, 2003, 22, 5011 CrossRef CAS; (b) J. Fronies, R. Navarro, M. Tomas and E. P. Urriolabeitia, Organometallics, 1993, 12, 940 CrossRef.
- (a) G. M. Sheldrick, SHELXTL, version 5.1, Bruker Analytical X-ray Instruments Inc., Madison, WI, 1998 Search PubMed; (b) G. M. Sheldrick, SHELX-97, PC Version, University of Gottingen, Gottingen, Germany, 1997 Search PubMed.
- (a) L. Carlucci, G. Ciani, S. Maggini, D. M. Proserpio and M. Visconti, Chem. – Eur. J., 2010, 16, 12328 CrossRef CAS PubMed; (b) K. Akhbari and A. Morsali, Cryst. Growth Des., 2007, 7, 2024 CrossRef CAS.
- X. J. Gu and D. F. Xue, Inorg. Chem., 2006, 45, 9257 CrossRef CAS PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2002 Search PubMed.
- W. J. Chen, Y. Wang, C. Chen, Q. Yue, H. M. Yuan, J. S. Chen and S. N. Wang, Inorg. Chem., 2003, 42, 944 CrossRef CAS PubMed.
- (a) L. Yi, L. N. Zhu, B. Ding, P. Cheng, D. Z. Liao, S. P. Yan and Z. H. Jiang, Inorg. Chem. Commun., 2003, 6, 1209 CrossRef CAS; (b) F. J. Liu, D. Sun, H. J. Hao, R. B. Huang and L. S. Zheng, Cryst. Growth Des., 2012, 12, 354 CrossRef CAS.
- (a) V. W. W. Yam, Cryst. Growth Des., 2010, 10, 3709 Search PubMed; (b) D. Sun, N. Zhang, R. B. Huang and L. S. Zheng, Cryst. Growth Des., 2010, 10, 3699 CrossRef CAS.
- (a) J. Y. Wu, T. C. Chao and M. S. Zhong, Cryst. Growth Des., 2013, 13, 2953 CrossRef CAS; (b) M. L. Tong, J. X. Shi and X. M. Chen, New J. Chem., 2002, 26, 814 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Powder XRD patterns, IR spectra, and X-ray crystallographic data for 1–4. CCDC 949651–949654. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra47303a |
| This journal is © The Royal Society of Chemistry 2014 |