
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Production of oxymethylene ethers (OME) as sustainable diesel fuel substitutes: continuous synthesis from dimethyl ether and trioxane and evaluation of catalyst stability†
Marius
Drexler
 ,
Victor
Zaghini Francesconi
,
Ulrich
Arnold
*,
Thomas A.
Zevaco
and
Jörg
Sauer
,
Victor
Zaghini Francesconi
,
Ulrich
Arnold
*,
Thomas A.
Zevaco
and
Jörg
Sauer
Karlsruhe Institute of Technology (KIT), Institute of Catalysis Research and Technology (IKFT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: ulrich.arnold@kit.edu
First published on 10th March 2025
Abstract
Oxymethylene ethers (OMEs) are currently being investigated as attractive substitutes for fossil diesel fuel. In particular, the properties of OMEs containing 3–5 formaldehyde units (CH3O(CH2O)nCH3 with n = 3–5) are similar and mostly compliant with current diesel specifications. With their production based on renewable methanol, OMEs can contribute significantly to a future sustainable mobility. This study elaborates an anhydrous, liquid phase OME synthesis based on dimethyl ether (DME). Using a newly designed continuous production plant, the performance of extruded zeolite catalysts based on a commercially available ZSM-5 material is evaluated. The characteristics of the produced catalysts are analyzed extensively and discussed. Comprehensive characterization of the spent catalyst as well as regeneration experiments were performed to investigate catalyst deactivation mechanisms. It was shown that deactivation mechanisms are similar to those observed in methanol-to-hydrocarbon processes. Thus, understanding of these aspects is improved and approaches for further optimization can be identified.
Sustainability spotlightGreenhouse gas emissions from the mobility and transportation sector contribute substantially to the ongoing climate crisis and continue to increase rapidly. As this trend is expected to continue in the coming decades, solutions for a sustainable mobility which can be implemented gradually and immediately are of utmost importance, i.e. renewable fuels compatible to existing infrastructure. Oxymethylene ethers (OMEs) produced from renewable methanol can fulfill this requirement and at the same time reduce emission of pollutants such as particulate matter and NOx. This study elaborates the production of this new fuel, thus contributing to the UN SDG 3 (good health and well-being), SDG 7 (affordable and clean energy), SDG 9 (industry, innovation and infrastructure) and SDG 13 (climate action). |
1. Introduction
While there is a general consensus to reduce humanity's dependency of fossil resources, technical solutions are not readily available for all applications. A major challenge regarding future sustainable mobility is the replacement of liquid fuels. Renewable drop-in solutions such as bio- and e-fuels have the potential to accelerate the shift to sustainability, as they are compatible with the existing infrastructure and reduce overall emissions. This is especially relevant for applications that are hard to electrify, such as aviation, shipping or heavy duty. In this context, oxymethylene ethers (OMEs) of the type CH3O(CH2O)nCH3 (OMEn with n formaldehyde units in the molecule chain) have gained attention as synthetic diesel substitute fuels over the last few years.1 OMEs are oligomeric oxygenates with a varying number of repetition units in their molecular structure and especially the chain lengths n = 3–5 are of interest, as their fuel properties meet the current diesel standard DIN EN 590 to a large extent.2,3 With DIN/TS 51699:2023-11, a pre-standard specifically for OME fuel has been published recently. OMEs are promising synthetic fuels due to two aspects in particular. Due to their molecular structure of alternating carbon and oxygen atoms they burn virtually soot-free like a C1 fuel, although small amounts of particle emissions can be generated due to the influence of engine lube oil, metallic abrasions or radical reactions under certain conditions.4–8 By increasing the exhaust gas recirculation rate, formation of NOx can be suppressed, enabling significant reduction of potentially hazardous emissions without extensive exhaust gas treatment.9,10 Moreover, as their production is based on methanol and its derivatives, they can be produced exclusively from renewable resources, therefore reducing the overall carbon footprint by up to 93% compared to fossil diesel fuel.11,12 In contrast to fossil diesel fuel, OMEs are generally assumed to be non-toxic and not hazardous to the aquatic environment.13 OMEs can be applied as blend with fossil as well as synthetic diesel fuel, allowing for a gradual and therefore easier transformation towards sustainable mobility.12,14,15OME synthesis is acid-catalyzed, e.g. by ion exchange resins, aluminosilicates or zeolites and proceeds even at mild temperatures.16–18 Research regarding OME production has been carried out to improve overall efficiency of the process, including utilizing novel reaction pathways or heat integration.19–21 In general, a source of formaldehyde such as formalin solution, paraformaldehyde or trioxane (TRI) is used for chain elongation. Additionally, a source for the end capping groups, e.g. methanol, dimethoxymethane, which is OME1 according to the OME nomenclature, or dimethyl ether (DME), is needed.22 The production processes can be divided into aqueous and water-free processes depending on the choice of raw materials and the presence of water in the OME synthesis step.23 While aqueous processes in general employ cheaper reactants, their efficiency suffers from the formation of byproducts and water which need to be separated in energy intensive operations.24,25 Water-free processes on the other hand offer a higher selectivity and lower energy demand for product separation due to the absence of water and the reduced amount of byproducts.22,26,27 In an attempt to utilize the advantages of a water-free process while simultaneously cutting down the reactant costs, OME synthesis from DME and a source of dry formaldehyde has gained increasing attention.28,29 Viability of OME synthesis from DME and TRI in a liquid phase reaction has been demonstrated in 2018 and then developed further towards a continuously operating production process.30,31 Recently, a gas phase synthesis process employing DME and gaseous formaldehyde has been demonstrated but selectivity to OME is significantly reduced.32 Regarding OME synthesis from TRI, a stepwise insertion of formaldehyde via TRI decomposition (eqn (1)) is often considered in the literature, resulting in the formation of OME1 (eqn (2)). Subsequent incorporation of formaldehyde leads to chain elongation (eqn (3)).33,34 All of these reactions are considered to be reversible.22,32,35
| (CH2O)3 ⇌ 3CH2O | (1) |
| CH3OCH3 + CH2O ⇌ CH3O(CH2O)1CH3 | (2) |
| CH3O(CH2O)nCH3 + CH2O ⇌ CH3O(CH2O)n+1CH3 | (3) |
In previous studies on OME synthesis in liquid phase, direct incorporation of TRI into DME leading to the initial formation of OME3 (eqn (4)) could be demonstrated, leading to a higher yield of the desired fuel fraction OME3–5 for this process.30,31
| CH3OCH3 + (CH2O)3 ⇌ CH3O(CH2O)3CH3 | (4) |
Transacetalization reactions can also occur (eqn (5)) and after reaching equilibrium, a typical Schulz-Flory distribution can be observed.32,33,36
| CH3O(CH2O)nCH3 + CH3O(CH2O)mCH3 ⇌ CH3O(CH2O)n+1CH3 + CH3O(CH2O)m−1CH3 | (5) |
Additionally, the formation of methyl formate (MeFo) from formaldehyde by a Tishchenko type reaction occurs irreversibly (eqn (6)).29,37
| 2CH2O → HCOOCH3 | (6) |
In the context of green chemistry, OME production starting from DME and TRI aligns well with the principle of avoiding hazardous substances in chemical syntheses, as both DME and TRI are considered safer options compared to e.g. methanol or formaldehyde due to their stability and reduced toxicity.38,39 Zeolites, which are known to be very efficient, regenerable and, moreover, non-hazardous, are employed to catalyze the reaction.31,40,41 OMEs with shorter or longer chain lengths than the desired n = 3–5 fraction could be fed back to the reactor as the transacetalization reactions are reversible, thus avoiding waste streams.28,37,42 Additionally, the only irreversibly formed byproduct MeFo is considered to be a promising substitute for gasoline fuel, making simultaneous production of both types of fuel in an integrated process viable.4,43,44
To progress with this approach of a water-free OME production pathway, evaluation of scalability and reliability, which generally poses a challenge for many new clean energy technologies, is required. Hence, the stability of continuous OME synthesis from DME and TRI in a liquid phase process has been further investigated. Employing a H-ZSM-5 zeolite catalyst powder, which was found to be suitable in previous studies,31,41 the catalyst particles were shaped by extrusion and characterized. Employing a newly designed laboratory scale plant, the performance of shaped ZSM-5 type catalysts was tested over a period of several hours on a kg h−1 scale for the first time. To inquire into the mechanism responsible for catalyst deactivation, used catalysts have been characterized and regeneration procedures evaluated experimentally.
2. Experimental
2.1 Materials
As precursor for the catalysts, commercial zeolite H-ZSM-5 powder with a Si/Al ratio of 80 (CBV8014) was purchased from Zeolyst International. Pural SB-1 was obtained from Sasol and Aerosil 200 was purchased from abcr. Hydroxyethyl cellulose (HEC, Tylose H200 YG4) was obtained from SE Tylose GmbH. Ammonia solution (25%) was purchased from Sigma Aldrich.For continuous OME synthesis experiments, DME (99.9%) was purchased from basi Schöberl GmbH & Co. KG and TRI (>99.9%), as well as n-dodecane (≥99.9%) were purchased from Sigma Aldrich. For the batch experiments, n-dodecane (>99%) was purchased from Alfa Aesar.
2.2 Catalyst preparation
Two shaped ZSM-5 type zeolite catalysts have been prepared as described by Wodarz et al.,45 utilizing alumina and silica as a binder, respectively. The compositions of both catalysts are summarized in Table 1. The dry components were mixed thoroughly in a ratio of 80 wt% zeolite and 20 wt% binder (related to the dry mass of the compounds) before adding an aqueous solution of hydroxyethyl cellulose (HEC, 4 wt%). In the case of the catalyst with silica as binder, ammonia solution (25% NH3) has been added as plasticizer. After mixing all components thoroughly, the obtained masses were kneaded in a mixing machine (Mini Mixer, Caleva) to form an extrudable paste. The mixing chamber was tempered to 15 °C using a thermostat (RM6, Lauda) to prevent the paste from drying out. For shaping of the catalysts, an extruder (Mini Screw Extruder, Caleva) equipped with a 4 mm thick nozzle with three openings of 2 mm diameter was used. The extruded catalyst strands have been dried at 60 °C and crushed to particles with a size of 0.8–1.25 mm using analytical sieves (Retsch) to be finally calcined in a furnace (LV 9/11, Nabertherm) at 500 °C in static air. Prior to the experiments, the catalysts were dried at 110 °C and 10 mbar (furnace VO 200, Memmert) to remove any traces of moisture, as the presence of water has been reported to inhibit catalytic activity.34,46,47| Catalyst | m CBV8014 [g] | Binder | m Binder [g] | m HEC, 4% [g] | m NH3, 25% [g] |
|---|---|---|---|---|---|
| KIT-Z80PU | 1611.6 | Pural SB-1 | 500.5 | 1155.0 | 0.0 |
| KIT-Z80AE | 572.0 | Aerosil 200 | 142.0 | 384.3 | 100.0 |
2.3 Experimental procedures
OME synthesis experiments employing the prepared catalysts have been conducted in a continuously operating process module (Fig. 1). The module design is based on previous work carried out using a small-scale setup.31 A fixed bed reactor with heating mantle was used to ensure isothermal conditions in the reaction zone. A thermostat (C35P, Thermo Haake) was used to heat the reactor and a thermocouple (type K) was inserted into the reactor from the top up to the end of the catalyst bed to measure the reactor temperature (values for all experiments are summarized in the ESI Section C†). Electrical heating cables (HS, Horst) controlled by inline installed thermocouples (type K) were used for the peripheral piping. As dilution and as an internal reference, n-dodecane was added to the feed as auxiliary. It remains inert under the chosen reaction conditions and would be either omitted or replaced by a recycle stream in an industrial production process. The feed was prepared in 2 liter stainless steel containers using analytical scales (BJ2200C, Precisa; 1403 MP8, Sartorius) to dose TRI and dodecane and a syringe pump (500D, Teledyne Isco) as well as a high capacity analytical scale (PM34-K, Mettler) to dose DME. After adding all components as well as a magnetic stirring bar, the feed was mixed with a magnetic stirrer (RCT basic, IKA) for several hours before being transferred to the process module. The transferred mass was determined gravimetrically using an industrial bench scale (9530.04.040, Soehnle). During the experiments, a magnetically coupled gear pump (Series 1, Gather) was used to pump the feed through the reactor. Samples could be drawn from the product stream for analysis via a heated 1/16′′ stainless steel capillary tube. Before and after experiments, n-dodecane was used to flush the system due to its inert nature. | ||
| Fig. 1 Simplified flow diagram of the process module for continuous OME synthesis. | ||
Based on previous work,31 the reaction temperature was set to 100 °C and to ensure that all compounds stay in liquid phase, the process module was pressurized to 40 bar using helium as inert gas in all experiments. Pressure transmitters (S-11, Wika) and thermocouples (type K) were used to monitor the operating conditions in the process module. The catalyst bed, positioned in the center of the heating zone, had a length of 500 mm in all experiments and the flow rate of reactants was set to 1 kg h−1. Two feed mixtures, C1 and C2, were applied in the continuous experiments and their compositions can be found in Table 2. In each experiment, the performance of the catalyst was evaluated for several hours by probing the product mixture periodically using online GC analysis at constant reaction conditions. After reaction, samples of the spent catalysts have been collected, ground to a fine powder, washed with OME1 and subsequently dried at ambient air for analysis. Regeneration procedures have been carried out by thermal treatment of the spent catalysts in a tube furnace at various temperatures and a gas flow of 40 ml per min N2 or synthetic air, respectively.
| Feed mixture | Composition [wt%] | n TRI/nDME [mol mol−1] | ||
|---|---|---|---|---|
| DME | TRI | Dodecane | ||
| C1 | 30.0 | 15.0 | 55.0 | 0.256 |
| C2 | 42.0 | 33.0 | 25.0 | 0.400 |
| B1 | 33.0 | 17.0 | 50.0 | 0.256 |
Additional batch experiments have been performed in an autoclave setup to evaluate the activity of catalyst samples. The setup consists of two 75 ml stainless steel autoclaves, both equipped with pressure transmitters (S-11, Wika) and thermocouples (type K). For heating and stirring, two magnetic stirrers (RCT basic, IKA) have been used. The feed for the experiments was prepared using the same equipment as for the continuous experiments. The composition was similar to feed C1 to enable qualitative comparison with the continuous experiments. It can be found in Table 2, designated as feed B1. In each experiment, 40 g of feed was applied and the catalyst loading was chosen to be 2.6 wt%R related to the mass of the reactants. The reaction temperature was set to 100 °C and the reaction time to two hours. After reaction, the autoclaves were quenched in an ice-water bath. The autoclaves were depressurized and a sample was drawn using a syringe filter to remove catalyst particles. The composition of the samples was determined using gas chromatography.
2.4 Analytical methods
The composition of product mixtures has been determined by online analysis with an Agilent 8860 GC system for continuous experiments in the process module. The GC is connected to the process module via a 1/16′′ stainless steel capillary tubing and a liquid sampling valve system (Joint Analytical Systems GmbH), both electrically heated to prevent plugging of the lines with potential long chain, waxy OME species or residual TRI. To prevent particles from entering the valve system and the GC, a 0.5 μm stainless steel sintered metal filter (Swagelok) was installed. For analysis of the mixtures produced in the batch experiments a Hewlett Packard 6890 series GC was used. Samples were prepared by dilution with tetrahydrofuran (≥99.7%, VWR) and adding a known amount of n-octane (≥99%, VWR) as reference for quantification. More detailed information can be found in the ESI (Section A†). As DME evaporates during depressurizing of the autoclaves, the known mass of n-dodecane has been used to calculate the total mass of the liquid fraction (eqn (7)) and the resulting masses of the analytes (eqn (8)). For continuous experiments, the calculations are based on the mass flow of the reactants (eqn (9)). | (7) |
| mi = ωi·mliquid, autoclave | (8) |
| ṁi = ωi·ṁrectants | (9) |
Conversion of TRI and DME has been calculated based on the amount of products (eqn (10)). Selectivity to product species was calculated based on TRI, as it is involved in the formation of both, OMEn and the byproduct MeFo (eqn (11)).
 | (10) |
 | (11) |
As a measure for the chain length distribution within the OMEn fraction the average chain length (ACL) has been calculated (eqn (12)).
 | (12) |
Scanning electron microscopy (SEM) images of the produced catalysts have been taken using a Zeiss Gemini SEM 500 with a Schottky field emission cathode and secondary electron detectors (Everhart-Thornley, Inlens). Energy-dispersive X-ray spectroscopy (EDX) has been performed with an Oxford X-Max System.
Thermogravimetric analysis (TGA) of the catalysts has been performed using a TGA 2 System (Mettler Toledo). The measuring program includes a temperature ramp of 10 °C min−1 from 25 °C to 1000 °C under nitrogen or synthetic air atmosphere.
Nitrogen physisorption experiments were performed using a Quantachrome Nova 2000e analyzer. The samples were degassed for 20 h at 10−2 bar and elevated temperatures. Fresh catalyst samples were treated at 230 °C and samples of the spent catalysts were degassed at 120 °C to avoid removal of carbonaceous species. Adsorption and desorption isotherms were recorded at −196 °C after the pretreatment procedures. The Brunauer–Emmet–Teller (BET) method was used to determine the specific surface area of the samples in a range of p/p0 from 0.004–0.12 (Roquerol plot). Pore area and volume were determined using the t-plot and Barret–Joyner–Halenda (BJH) method.
Temperature-programmed desorption of NH3 (NH3-TPD) was performed on an Altamira AMI-300 using the standard ground configuration with 3 MFCs (carrier, treatment & blend) and the internal TCD to quantify the amount of NH3 liberated during the TPD procedure. A 5% NH3 in helium gas mixture (Air Liquide, CRYSTAL mixture) was used for the experiments. The samples were preliminarily dried in situ, in the dedicated quartz U-tube, under pure helium flow (30 ml min−1) at 450 °C for 120 min (from RT to 450 °C with a 10 K min−1 ramp). Afterwards, the samples were cooled to 100 °C (450 °C to 100 °C with a 10 K min−1 ramp; holding 15 min) and loaded with NH3 for 60 min. Excess NH3 was removed by heating to 120 °C and flushing with pure helium for 100 min. NH3-TPD was conducted at ambient pressure (3 K min−1 heating rate up to 750 °C; holding 60 min). The measurement program always includes a calibration step in order to correctly determine the amount of desorbed NH3. Five discrete pulses of the 5% NH3 in helium mixture (30 ml min−1) are sent through the TCD onboard with the average area of all five pulses being used for calibration.
To inquire into the nature of acid sites pyridine-FTIR experiments were conducted at the Leibniz Institute for Catalysis (LIKAT Rostock). 50 mg of catalyst were pressed into a self-supporting wafer with a diameter of 20 mm and initial pretreatment was performed at 400 °C for 1 h under vacuum atmosphere. The pyridine desorption spectra were collected in steps of 50 K starting at 400 °C using a Tensor 27 spectrometer (Bruker) and for background correction spectra recorded at 200 °C were used. For quantification, adsorption bands at 1545 cm−1 (PyH+, Brønsted acid) and 1455 cm−1 (PyL, Lewis acid) at 250 °C were considered and the integrated molar extinction coefficients of Emeis were used for calculations.48
Extraction of soluble carbonaceous species was performed to evaluate the type of deposits formed during the experiment. The method used is based on the standard procedure described by Guisnet et al.49 1 g of sample was dissolved in 5 ml of hydrofluoric acid (40%, Merck) for 1 h in a PTFE container. Next, 3 ml of CH2Cl2 (≥99.8%, Merck) were added and the solution was mixed for 1 min. After a waiting period of 2 h, the separated organic phase was recovered and subsequently washed with an aqueous solution of NaHCO3 (≥99%, Merck). After a neutral pH value in the aqueous phase was reached, the organic phase was separated and subjected to further analysis.
Analysis of the recovered species was performed with an Agilent 6890 gas chromatograph equipped with an Agilent 5973 mass selective detector. The system uses a Restek RTX-5MS column and helium as carrier gas. Mass spectra of the compounds were compared to NIST libraries for identification.
NMR spectra were acquired on a JNM-ECZ400R series spectrometer (JEOL), equipped with a 9.4 T cryomagnet (Oxford Instruments; resonance: 1H @ 399.905 MHz and 13C @ 100.556 MHz), using a 3.2 mm AutoMAS Solid State probe head for the measurement of the solid samples and a “royal” 5 mm probe head for the measurement in the liquid phase (combination of broadband and inverse detection probe head). Measurements were carried out using the Jeol software Delta 5.3.3. with the standard Jeol pulse sequences and optional optimization (see ESI Section A for more precise information†). The NMR spectra are evaluated using MestReNova 15 (vers. 15.0.0-34764 © 2023, Mestrelab Research S.L).
3. Results and discussion
3.1 Catalyst characterization
After calcination, samples of the two catalysts have been subjected to various characterization methods. SEM imaging was used to evaluate the physical appearance of the materials as well as the distribution of zeolite and binder in the shaped catalysts (Fig. 2). For the KIT-Z80PU catalyst with alumina as binder a good intermixing of zeolite and binder particles can be seen at high magnification. This might be due to a phase transition of the original AlOOH binder to γ-Al2O3 during calcination. At lower magnification however, larger particles can be found, indicating zones of de-mixing or agglomeration. The KIT-Z80AE catalyst on the other hand seems to be very uniform at low magnification, but at high magnification, a clear distinction between zeolite and binder particles is possible and the material appears like a physical mix of both components. This can be attributed to a weaker interaction between the zeolite and binder as the SiO2 binder does not undergo a phase transition during calcination. | ||
| Fig. 2 SEM images of the calcined KIT-Z80PU (left) and KIT-Z80AE (right) catalyst. | ||
EDX analysis further confirms these findings, showing clearly distinguishable regions of higher aluminum concentration for the KIT-Z80PU catalyst (Fig. 3). For the KIT-Z80AE catalyst on the other hand, a uniform elemental distribution can be observed, which is dominated by silica. Overall, the physical appearance of the catalysts agrees very well with the results reported by Wodarz et al.50
 | ||
| Fig. 3 EDX mapping of the elemental composition of KIT-Z80PU (left) and KIT-Z80AE (right) with aluminum in orange and silica in green. | ||
TGA of the catalysts shows some weight loss at temperatures below 200 °C, which can be attributed to water from ambient air adsorbed during the preparation of the samples. No further weight loss was detected for both catalysts until the final measuring temperature of 1000 °C, regardless of the gas type used during the measurements. This indicates that the hydroxyethyl cellulose used during production of the extrudates decomposed completely and no carbon containing residues remained in the shaped catalysts (see ESI Fig. B1†).
The results of nitrogen physisorption analyses (Table 3) indicate similar values for specific surface area and pore volume compared to commercial products used in earlier studies.31 Compared to the zeolite powders, the specific surface area and especially the micropore surface area is reduced considerably, which is due to the binder in the extrudates.41 Comparing both catalysts, KIT-Z80AE exhibits a larger value for the micropore surface area and a lower share of meso-/macropore and external surface, likely because of the smaller particle size of the areosil binder compared to the alumina binder.
| Catalyst | Surface areaa [m2 g−1] | Pore volume [cm3 g−1] | Pore diameterc [nm] | |||
|---|---|---|---|---|---|---|
| BET | Micropore | Meso/externalb | Microa | Mesoc | ||
| a Based on the t-plot method. b Difference of total BET surface area and micropore area. c Based on the Barret–Joyner–Halenda (BJH) method using the desorption branch. | ||||||
| KIT-Z80PU | 394 | 275 | 119 | 0.115 | 0.166 | 3.81 |
| KIT-Z80AE | 386 | 298 | 88 | 0.128 | 0.260 | 3.82 |
Regarding the acidic properties of the produced catalysts, NH3 and pyridine desorption experiments have been performed. The determination of the nature of the acidic sites and their quantification (weak vs. strong acidic sites) have been carried out by NH3-TPD analysis and the results indicate the presence of predominantly strong acidic sites for both catalysts (Fig. 4). For the alumina containing KIT-Z80PU catalyst, a larger amount of weakly acidic sites can be found, resulting in a larger total number of acidic sites. These additional acidic sites most likely stem from the aluminum binder, which was reported to form weakly Lewis acidic sites during calcination.45,50
 | ||
| Fig. 4 NH3-TPD analysis for KIT-Z80PU (measurement: blue, fit: cyan) and KIT-Z80AE (measurement: olive, fit: green), catalyst bed temperature in red. Deconvolution in low (∼200 °C) and high (∼385 °C) temperature peak performed with OriginPro peak analysis. | ||
The results of pyridine desorption experiments corroborate these findings, as an increased amount of Lewis acidic sites can be found for KIT-Z80PU compared to KIT-Z80AE, which confirms that the alumina binder forms weakly Lewis acidic sites during calcination (Table 4). Consequently, the ratio of Brønsted to Lewis acidic sites is reduced for this catalyst compared to previously used native ZSM-5 powder catalysts. However, KIT-Z80AE displays a high B/L ratio, even higher compared to the pure zeolite powder.41 In conclusion, both catalysts display predominantly strong Brønsted acidic sites with additional weak Lewis acidic sites for the catalyst with the alumina binder.
| Catalyst | Acid site density by strengtha [μmolNH3 gCatalyst−1] | Acid site distribution by typed [%] | ||||
|---|---|---|---|---|---|---|
| Weakb | Strongc | Total | Brønstede | Lewisf | Ratio B/L | |
| a Based on NH3-TPD experiments. b Low temperature peak at ∼200 °C. c High temperature peak at ∼385 °C. d Based on pyridine desorption experiments monitored by FTIR. e Peak at 1545 cm−1. f Peak at 1455 cm−1. | ||||||
| KIT-Z80PU | 99 | 139 | 238 | 76 | 24 | 3.1 |
| KIT-Z80AE | 54 | 135 | 189 | 90 | 10 | 8.9 |
3.2 OME synthesis
To evaluate the catalysts performance, continuous OME synthesis experiments have been conducted. The reactor temperature was set at 100 °C and the mass flow of the reactants at 1 kg h−1. This corresponds to a weight hourly space velocity (WHSV) of 37.7 h−1 for the catalyst KIT-Z80PU and 39.5 h−1 for KIT-Z80AE. The small difference is due to small deviations in the catalyst densities. Results for the experiments with feed C1 can be found in Fig. 5 for both catalysts. Numerical values for all experiments can be found in ESI Section C.† | ||
| Fig. 5 Experimental results from continuous OME synthesis with KIT-Z80PU (left) and KIT-Z80AE (right) employing feed C1. Reaction conditions: T = 100 °C, p = 40 bar. | ||
Initially, the catalysts exhibit a similar performance with high conversion and selectivity to OME. With almost complete TRI conversion, initial combined reactant conversions of about 46.5% and 44.0% can be reached. The high initial conversion of both catalysts is in line with previous studies, as the presence of strong Brønsted acid sites was found to be decisive, especially for the initial steps of TRI decomposition (eqn (1)) and DME activation (eqn (4))41,51,52 and both catalysts display a similar amount of acid sites of this type. The presence of additional weak Lewis acid sites for the KIT-Z80PU catalyst seems to be of minor influence on OME formation, leading to a slightly increased formation of the byproduct MeFo and consequently increased conversion as well as slightly lower ACL values. These observations can be explained by Lewis acid sites being active for MeFo formation (eqn (6)) but neither TRI decomposition (eqn (1)) nor DME activation (eqn (4)), therefore leading to a shift away from OME chain elongation (eqn. (2)) due to competing consumption of free formaldehyde and consequently decreasing ACL values.41,51 However, for both catalysts a steady decline in activity can be observed during the experiment. The experiment was terminated after about 5.5 h time on stream, with final reactant conversions of about 23.5% and 23.0%. Regarding the OME distribution, a broad spectrum with a large share of long-chain OME oligomers was found. These oligomers form waxy solids at room temperature and could not be detected in previous studies as they precipitate from the product mixture during the cooling down phase. With increasing time on stream, a shift in the product spectrum can be observed. The overall selectivity to OME and the share of long-chain oligomers increase, resulting in a higher ACL value. The OME3–5 share slightly increases and reaches a maximum at about 40%. On the other hand, the selectivity for MeFo decreases. This indicates a faster deactivation of Lewis acid sites, which are active for MeFo formation, in comparison to Brønsted acid sites, which are active for OME formation. While the product spectrum seems to stabilize in the course of the experiments, no steady level of conversion could be reached indicating an ongoing deactivation of the catalysts.
So far, no detailed description for the deactivation mechanism of zeolite catalysts in liquid phase OME synthesis has been given and H-ZSM-5 has been reported to be stable in OME synthesis employing OME1 and TRI at similar temperatures.53–55 However, in previous work we observed a darkening of the catalysts, which was attributed to coke formation.41,56 Furthermore, in a study on TRI synthesis with H-ZSM-5 at 100 °C, Ye et al. reported a strong deactivation by coking after ca. 150 h of time on stream.57 These observations indicate a deactivation of zeolites by formation of carbonaceous species in the presence of monomeric formaldehyde, despite the low reaction temperatures.
To evaluate if the observed deactivation is related to the composition of the feed, e.g. the share of TRI, a second set of experiments has been performed using feed C2 (Fig. 6). The share of TRI has been increased in this specific feed, resulting in a molar ratio of TRI to DME of 0.4. Simultaneously, the dilution with dodecane has been reduced to avoid miscibility issues when preparing the feed.
 | ||
| Fig. 6 Experimental results from continuous OME synthesis with KIT-Z80PU (left) and KIT-Z80AE (right) employing feed C2. Reaction conditions: T = 100 °C, p = 40 bar. | ||
The results of this set of experiments qualitatively match those of the first set, indicating high initial reactant conversions of about 43.5% and 47.5% with a steady decline in the course of the experiments. At the end of the experiments, the conversion was 26–27% for the combined reactants. This indicates that the mechanism for deactivation is not directly influenced by the ratio of the reactants. The product distribution, however, deviates significantly due to the larger amount of TRI in the feed, with higher ACL values indicating a shift in the OME oligomer distribution to longer chain lengths. The selectivity to the target fraction of OME3–5 increases only slightly with maximum values of about 41.5% and 43%. Overall, the level of the reactant conversion could be slightly improved by increasing the share of TRI in the feed, especially during the later stages of the experiments, leading to higher OME productivity. This is in line with previous findings regarding this reaction system.41 Counterintuitively, a slight decrease in selectivity to the byproduct MeFo could be observed for feed C2 (from 18.9% to 15%) compared to feed C1 (from 30% to 18.7%) despite the increased share of TRI in the feed. Moreover, due to the high concentration and incomplete conversion of formaldehyde, 1,3,5,7-tetroxane can be found as an additional byproduct, a compound also described in literature in regard to OME synthesis.58,59 In addition, the formation of larger shares of long-chain OME oligomers requires caution, due to the possible formation of material clogs in insufficiently heated parts of the process module.
Although the conversion of the reactants seems to stabilize towards the end of the experiments, no steady level could be reached again. Therefore, a second run using the same catalyst bed was performed to increase the time on stream of the catalysts (Fig. 7). After the first run, the process module was flushed with dodecane and cooled down while the feed was replenished. The catalysts were stored in the reactor under dodecane to avoid exposition to the atmosphere and especially moisture.
 | ||
| Fig. 7 Experimental results of continuous OME synthesis with KIT-Z80PU (left) and KIT-Z80AE (right) employing feed C2. Reaction conditions: p = 40 bar. Catalysts have not been changed after prior reactions with feed C2 and temperature has been set to 120 °C for a period of 90 min. | ||
In the second run, the activity of the catalysts was significantly reduced in relation to the end of the first run, indicating further deactivation of the catalysts during cooling down, storage or heat up of the system. The reactant conversions dropped down to 14% and 9.5%, respectively. Investigating the product spectrum, a rise in MeFo selectivity can be detected. On the other hand, the ACL remains on a similar level as at the end of the first run. MeFo formation occurs on the external surface area while OME formation is thought to take place within the micropores.41 Hence, the aforementioned findings indicate pore blocking and therefore a reduced amount of accessible active sites of the catalysts as the reason for the reduced activity. A possible reason for blocking of the pores might be unreacted formaldehyde present within the catalysts forming paraformaldehyde during the cooling down or storage phase. Comparing both catalysts, KIT-Z80PU exhibits higher levels of TRI conversion by forming significantly more MeFo, which is probably due to the alumina binder offering additional weak Lewis acid sites outside of the micropores.
In an attempt to reactivate the catalysts, the reactor temperature was set to 120 °C for 90 minutes. With rising temperature, the conversion for TRI and DME increases. The amount of the byproduct MeFo formed increases as well, especially for KIT-Z80PU. The OME distribution shifts to lower chain lengths, indicated by a drop in the ACL. This behavior matches the results of previous studies.41 However, after reducing the temperature back to 100 °C the conversion drops again to the level before the temperature increase, indicating neither reactivation nor further deactivation of the catalysts. At the time of termination of the experiment, the reactant conversions were determined to be 11% and 9.5%, respectively.
In summary, OME synthesis was successfully demonstrated in a continuously operating plant on kg h−1 scale. At a reaction temperature of 100 °C and pressure of 40 bar a comparison of two H-ZSM-5-based catalysts with alumina or silica as binders was performed. Both catalysts are highly active in the initial phase of the experiment followed by severe deactivation, while OME selectivity remains relatively high. The catalyst containing alumina tends to form slightly more of the byproduct MeFo and OME oligomers with lower chain length due to additional Lewis acid sites in the binder phase. The findings indicate pore blocking, reducing the accessible active sites for OME synthesis. This deactivation is possibly induced by formaldehyde-containing compounds, such as longer-chain OME or paraformaldehyde, which could produce a waxy coating on the catalyst, thus reducing the accessibility of active sites. In an attempt to remove such materials from the catalyst, the reaction temperature was elevated during operation. However, since no effective increase in conversion levels was observed, the formation of carbonaceous compounds on the catalyst surface is assumed to contribute to catalyst deactivation. Hence, further analysis of the spent catalysts was performed in order to elucidate the deactivation mechanism.
3.3 Analysis and regeneration of the spent catalysts
After the experiments, samples of the catalysts were recovered. The initially white catalyst particles appear black in colour, indicating the formation of carbonaceous species. Prior to TGA analysis the samples were ground in a mortar to a fine powder and washed with OME1 to remove dodecane and remaining reactants followed by drying at ambient conditions. Results for the catalyst samples of all four runs can be found in the ESI (Fig. B2†). Detailed analysis and regeneration procedures have been performed for the KIT-Z80PU catalyst used in the two consecutive experiments with feed C2 and will be discussed in the following section. The TGA weight profile of the used catalyst (Fig. 8) clearly shows four peaks with different intensities.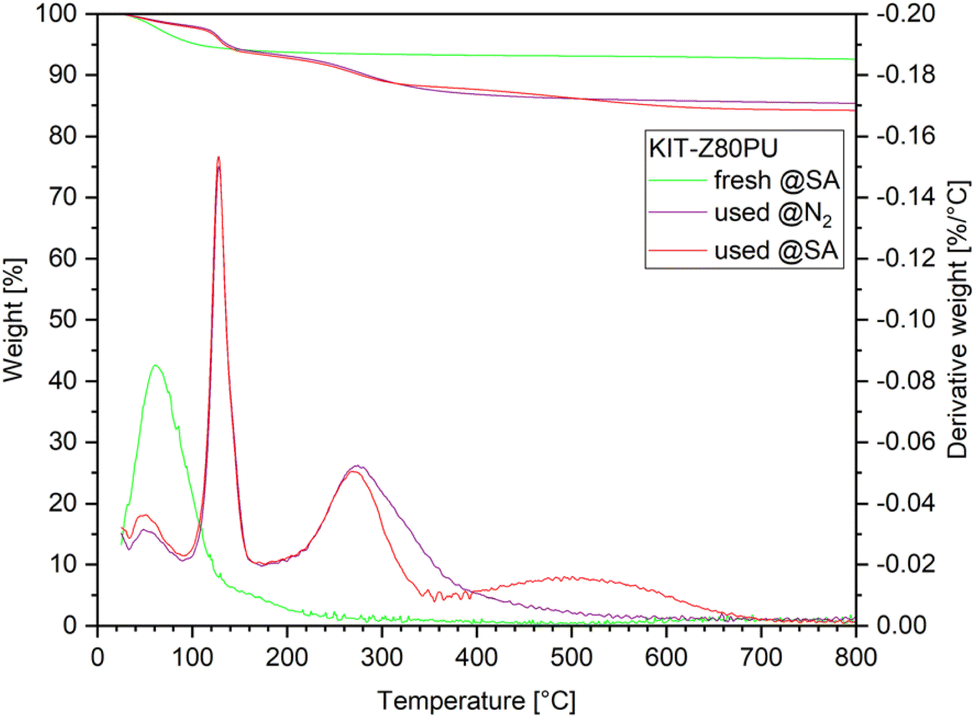 | ||
| Fig. 8 TGA results for the fresh catalyst KIT-Z80PU and used in two consecutive runs with feed C2, measured under synthetic air (@SA) and nitrogen (@N2) atmosphere, respectively. | ||
The lowest temperature peak below 100 °C can be attributed to the loss of water, although the intensity is reduced compared to the fresh catalysts as the active sites are partially blocked. Two additional peaks at 130 °C and 250 °C were identified as paraformaldehyde depolymerizing to formaldehyde gas and n-dodecane respectively. Peak assignment has been confirmed by comparison with samples of the fresh catalyst spiked with the suspected compounds. Mass loss occurring in the range of 300–650 °C can be attributed to carbonaceous species, with a higher loss for the measurement with synthetic air indicating the presence of graphitic species.
Based on the TGA results, three regeneration procedures by thermal treatment in a tube furnace under gas flow have been evaluated. Details on regeneration are as follows: first, a low temperature regeneration at 120 °C under inert atmosphere, which corresponds to the maximum temperature of the process module and should be sufficient to remove organic residues including paraformaldehyde from the catalyst (R1). Second, a high temperature regeneration at 500 °C under inert atmosphere to remove carbonaceous compounds except of the graphitic species as indicated by the high temperature peak at 580 °C (R2). Third, the regeneration at 500 °C under synthetic air for complete removal of organic species (R3). TGA of the regenerated catalysts has been performed in order to verify that the desired effect was achieved (Fig. 9).
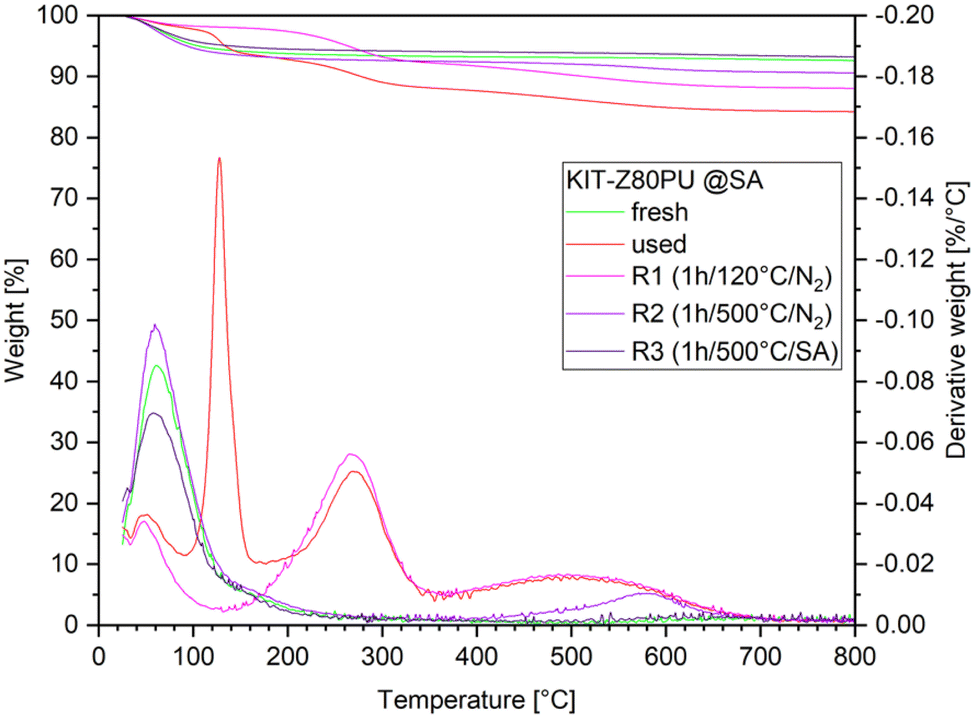 | ||
| Fig. 9 TGA results for the fresh catalyst KIT-Z80PU, used in two consecutive runs with feed C2 and regenerated with three different procedures (SA = synthetic air). All measurements have been performed under synthetic air atmosphere. | ||
N2 physisorption experiments have been performed with the regenerated catalyst (Table 5). The used catalyst without treatment could not be analyzed, as the large quantity of organics would interfere with the degassing and thermal treatment procedures. The results show a greatly reduced surface area and pore volume for the micropores for R1 compared to the fresh material. By treating the catalyst at higher temperatures initial values could be restored, especially for the sample treated under synthetic air atmosphere (R3). The results indicate that a blocking of the micropores is the cause for the observed deactivation.
| Catalyst | Regeneration treatment | Surface area [m2 g−1] | Pore volume [cm3 g−1] | Pore diameterc [nm] | |||
|---|---|---|---|---|---|---|---|
| BET | Microa | Meso/externalb | Microa | Mesoc | |||
| a Based on the t-plot method. b Difference of total BET surface area and micropore area. c Based on the Barret–Joyner–Halenda (BJH) method using the desorption branch. | |||||||
| KIT-Z80PU | — | 394 | 275 | 119 | 0.115 | 0.166 | 3.81 |
| R1 | 1 h @ 120 °C N2 | 103 | 23 | 80 | 0.010 | 0.138 | 3.90 |
| R2 | 1 h @ 500 °C N2 | 370 | 255 | 115 | 0.107 | 0.170 | 3.86 |
| R3 | 1 h @500 °C SA | 393 | 274 | 119 | 0.115 | 0.173 | 3.90 |
To evaluate the effect of the regeneration procedures, a series of batch experiments have been performed using feed B1 (Fig. 10). Double determination was performed to ensure consistency of the observations. For the fresh KIT-Z80PU catalyst, 45% of TRI conversion and 90% selectivity to OMEn could be observed. The used catalyst performed poorly as it is severely deactivated, with only 1.5% TRI conversion and 80% selectivity to OMEn. This loss of selectivity can be attributed to the blocking of micropores, as previous findings indicated that OME formation takes place predominantly within the micropores.41
 | ||
| Fig. 10 Results of batch experiments employing fresh, used and regenerated KIT-Z80PU. Experimental conditions: T = 100 °C, t = 2 h, 2.6 wt%R catalyst, feed B1 (50 wt% n-dodecane). | ||
Evaluation of the regenerated catalysts reveals that the low temperature thermal treatment at 120 °C is not sufficient for reactivation, as R1 performs very similar to the used catalyst sample. Evidently, the presence of paraformaldehyde is not the cause of the deactivation of the catalyst. High temperature thermal treatment at 500 °C however proves to be efficient to reactivate the catalysts R2 and R3. Overall, regarding the regenerated samples, R3 performed best with about 35% TRI conversion and 90% selectivity to OMEn. However, while no decline in selectivity was observed for the reactivated catalysts, initial levels of conversion could not be restored.
To identify the compounds responsible for catalyst deactivation samples of the catalysts, the used one as well as R1, were dried and packed in zirconia rotors for MAS 13C NMR spectroscopy. The 13C MAS spectra (Fig. 11) revealed patterns with the used catalyst clearly displaying the presence of remaining dodecane and, more interestingly, of –CH2–O– moieties present in paraformaldehyde species and in short-chain OME. In addition, the presence of methyl formate with two clear signals around 170 ppm (carboxylate signal) and 55 ppm (O–CH3 signal) could also be ascertained. The attribution of all the NMR signals was confirmed via spiking the zeolite with the related pure compounds and recording comparative spectra. This observation indicates entrapment of the reactant phase within the pore system due to pore blocking, as the samples have been thoroughly washed with OME1 and dried prior to analysis. In comparison, the regenerated catalyst R1 only displays with certainty the presence of dodecane. The thermal treatment at 120 °C apparently allows for the removal of paraformaldehyde partially unblocking some of the pore network, hence allowing the volatile OME as well as a large share of MeFo to evaporate. Interestingly, the recorded NMR spectra did not show specific signals that might be attributed to the deposition of aromatic carbonaceous species as one might expect considering the grayish color of the spent catalysts (corresponding signals would be expected around 120 ppm according to literature60), suggesting that the amount of aromatic species present in the solid remains low.
 | ||
| Fig. 11 13C MAS NMR spectra of (a) spent catalyst and (b) R1 (regenerated at 120 °C under N2). | ||
To better understand these results, the extraction of organic soluble species was performed after dissolving the inorganic zeolitic material using a method based on the standard procedure described in literature.49,61 A sample of R1 was used, as the paraformaldehyde already had been ruled out to be the cause of the deactivation of the catalyst. The extracted phase was subjected to GC-MS (Fig. 12) and (standard) liquid NMR spectroscopy (Fig. 13). During extraction, the presence of insoluble coke species in the form of fine black particles was observed after dissolving the catalyst. However, due to the small amount no analysis could be performed.
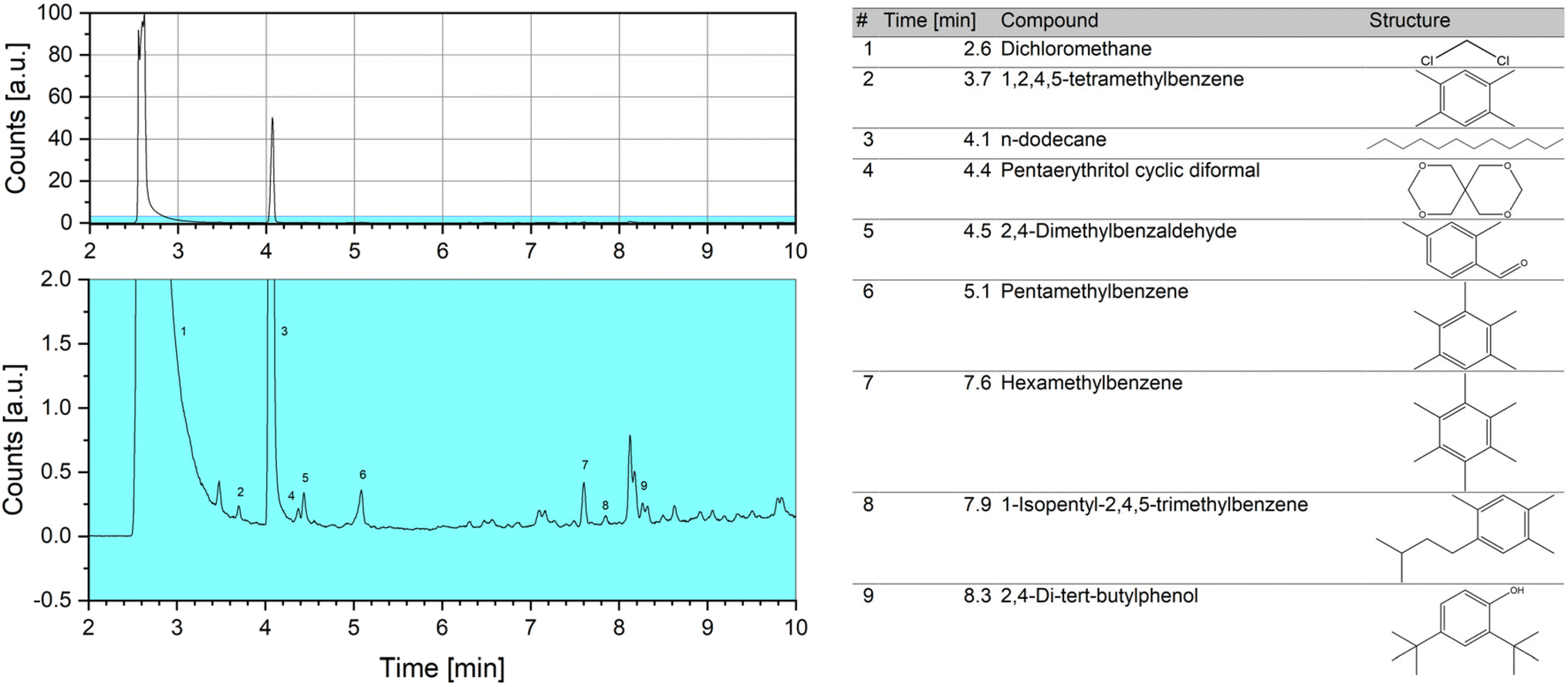 | ||
| Fig. 12 Gas chromatogram of the organic extract of the soluble carbonaceous species (full spectrum on top, enlarged section as indicated below). Identification of the compounds has been performed by mass spectroscopy using NIST libraries. | ||
 | ||
Fig. 13 2D 13C, 1H-correlated NMR spectrum (HSQC) of the organic extract of the soluble carbonaceous species, recorded with CD2Cl2 (ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture). :1 mixture). | ||
In the GC-MS chromatogram, the peak with the highest abundancy besides the solvent dichloromethane could be attributed to dodecane, matching the results of the NMR analysis. Additionally, several peaks for alkylbenzene species, in particular methylated benzenes as well as 2,4-dimethylbenzaldehyde and 2,4-di-tert-butylphenol, could be identified. These compounds are considered as coke precursors in processes following the hydrocarbon-pool mechanism and employing H-ZSM-5 catalysts, like methanol-to-hydrocarbon (MTH) processes.62–64 Conversely, for such processes no formation of hydrocarbons or aromatics is expected to occur at the applied reaction temperature of 100 °C.65,66 It has been reported, however, that formaldehyde plays a key role for the formation of coke in MTH processes and adding it to the feed drastically reduces the catalyst lifetime.66,67 This might be related to a more pronounced formation of aromatics in the presence of formaldehyde.68 The high concentration of formaldehyde in the OME synthesis at hand might therefore contribute to the formation of carbonaceous species. Additionally, it has been shown for methanol conversion processes, that low temperatures of 250–300 °C can lead to increased deactivation rates due to low mobility of the formed methylbenzene compounds and subsequently, to an overloading of the catalyst with those species.69,70 This effect might be especially pronounced, as the applied reaction temperature of 100 °C is even lower.
The presence of alkylated aromatics in the extracted organic phase was confirmed using two-dimensional 13C, 1H NMR spectroscopy, more precisely a HSQC pulse sequence (Heteronuclear Single Quantum Correlation experiment). This allows to associate directly 1H to the bearing 13C and to differentiate at first glance methyl, methylene, methine and quaternary carbon atoms via the phasing of the cross peak (CH2: negative, CH + CH3: positive, quaternary: no cross peak). The 2D spectrum of this phase was recorded after adding some CD2Cl2 to have a stable lock signal and to get spectra that are well resolved. Despite the sample being highly diluted and the strong signals of dodecane and CH2Cl2, the 2D spectrum displayed in Fig. 13 reveals some remarkable information supporting the data gathered from MAS solid state spectra and GC-MS analytics. However, the high dilution leads to a strong f2 respective f1 noise. Dodecane and CH2Cl2 (extraction solvent and deuterated) are clearly the main molecules present (in 1H dimension: 0.8–1.5 ppm for dodecane, 5.3 ppm for CH2Cl2 and in 13C dimension: 15–30 ppm for dodecane, 50 ppm for CH2Cl2/CD2Cl2). The presence of numerous alkylated aromatics can be clearly seen in the 1H region at 2.0–2.5 ppm (alkyl groups) and 6.6–7.5 ppm (aromatics) whereas the connected 13C signals are found at 10–40 ppm and 120–130 ppm, respectively. Methyl groups can be found around 2.2 (1H) and 15 ppm (13C) whereas methylenes are located between 1 and 2.5 ppm for the alkylated aromatics and between 3.0 and 4.0 ppm for the OME derivatives (–O–CH2– fragments). The related 13C signals can be found between 25 and 35 ppm for the aromatic-bound substituents and 60–80 ppm for the OME-related compounds. As often found in the spectra of aromatics, the NMR signals generated by CH present in the aromatic core are noticeably less intensive than the signals of the aromatic-bound alkyl groups, owing to different relaxation times and contributions of the nuclear Overhauser effect (NOE) during 1H-decoupling.
In summary, the formation of carbonaceous species seems to be the reason for the observed strong deactivation. This might be caused by the high concentration of formaldehyde and the reaction temperature being too low to remove the formed coke precursors from the catalyst during the reaction. Catalyst lifetime might be improved by modifying the reaction temperature to avoid the formation of methylbenzene species or increase their mobility to remove them from the catalyst while simultaneously adjusting the residence time to maintain a high selectivity to OME and high reactant conversion. However, due to the fast deactivation it seems more promising to modify or change the catalyst system. Previous catalyst screening studies already demonstrated general suitability of zeolite catalysts with various Si/Al ratios and framework types, including zeolite Y and β.31,41 For the DME-to-hydrocarbons (DTH) process, a recent study reported successful suppression of the formation of aromatics by utilizing a *MRE type zeolite, which could be an interesting candidate for OME synthesis as well.71 Literature also suggests modification of the zeolite catalyst such as the introduction of meso- or macroporosity to enhance catalyst lifetime.45,62,72–74 Besides zeolites, a montmorillonite clay material, K10, has been identified to catalyze OME synthesis efficiently.31,75 Due to its layered structure, it should be more resistant to deactivation by coking as there is no extended pore network which could be blocked. In addition, its structure exhibits an increased amount of mesoporosity compared to zeolite materials, which has been reported to enhance lifetimes of zeolite catalysts.45,73,74
4. Conclusions
To evaluate feasibility of the production of OMEs as a renewable synthetic fuel for a future sustainable mobility, scalability and reliability of the DME and TRI based synthesis process have been evaluated. Two shaped H-ZSM-5 catalysts have been prepared and characterized extensively to study OME synthesis at 100 °C in a continuously operating process module on a kg h−1 scale. While the upscaling was successful and general feasibility of the synthesis pathway could be proven with high selectivity to OME3–5 of about 40%, fast deactivation of the catalysts has been observed. Initially, the KIT-Z80PU catalyst demonstrated almost full TRI conversion and after a few hours of operation showcased a decline in conversion to a value of about 55%. Increased shares of TRI in the feed lead to similar results with a slightly increased OME3–5 selectivity of 41.5% and a drop in TRI conversion from 69% to 46%, indicating no direct influence of feed composition in regard to catalyst deactivation. Similar results were obtained for the KIT-Z80AE catalyst, hence no strong influence of the binder is suspected either. Thermal regeneration procedures have been employed, proving the presence of carbonaceous species to be the reason for the observed deactivation and reactivation of the catalyst has been successfully demonstrated at high temperatures. While full regeneration could not be achieved, the sample regenerated in synthetic air at 500 °C performed well with about 35% TRI conversion and 90% selectivity to OMEs compared to 45% TRI conversion and 90% selectivity to OMEs for the fresh catalyst in contrast to 1.5% TRI conversion and 80% selectivity to OME for the deactivated sample. Extensive analysis of the deactivated catalyst revealed the presence of methylbenzene species, suggesting a deactivation mechanism similar to mechanisms for methanol conversion processes known from literature.Evaluation of long-term catalyst stability as well as outlining the mechanism responsible for catalyst deactivation represent important steps in the development of this novel clean fuel production process. Future studies on this OME synthesis pathway should focus on the improvement of catalyst lifetime to increase overall stability of the process. This could be achieved, either by modification of known catalysts or by employing alternative systems. A promising candidate for upcoming catalyst stability studies is K10 due to its layered, mesoporous structure and already demonstrated suitability for the reaction.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Marius Drexler – conceptualization, methodology, investigation, data curation, formal analysis, visualization, writing – original draft. Victor Zaghini Francesconi – investigation, writing – review & editing. Ulrich Arnold – funding acquisition, supervision, writing – review & editing. Thomas A. Zevaco – methodology, investigation, visualization, writing – review & editing. Jörg Sauer – funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the German Federal Ministry of Education and Research (BMBF) within the NAMOSYN Project (FKZ03SF0566K0). The authors would like to thank Dr M. C. Zimmermann (SEM/EDX imaging), Dr T. N. Otto (N2-physisorption), N. A. Slaby (N2-physisorption), D. Neumann-Walter (N2-physisorption) and A. Lautenbach (HF-extraction). The authors also acknowledge Zeolyst International for supplying the base zeolite catalyst powder.References
- K. Hackbarth, P. Haltenort, U. Arnold and J. Sauer, Chem. Ing. Tech., 2018, 90, 1520–1528 CrossRef CAS.
- D. Deutsch, D. Oestreich, L. Lautenschütz, P. Haltenort, U. Arnold and J. Sauer, Chem. Ing. Tech., 2017, 89, 486–489 CrossRef CAS.
- L. Lautenschütz, D. Oestreich, P. Seidenspinner, U. Arnold, E. Dinjus and J. Sauer, Fuel, 2016, 173, 129–137 CrossRef.
- E. Jacob, M. Stark, M. Härtl and G. Wachtmeister, in 11. Tagung Einspritzung Und Kraftstoffe 2018: Diesel Benzin Gas Alternative Kraftstoffe Medien Für SCR Wasser, ed. H. Tschöke and R. Marohn, Springer Fachmedien Wiesbaden, Wiesbaden, 2019, pp. 17–55 Search PubMed.
- A. D. Gelner, D. Rothe, C. Kykal, M. Irwin, A. Sommer, C. Pastoetter, M. Härtl, M. Jaensch and G. Wachtmeister, Environ. Sci.: Atmos., 2022, 2, 291–304 CAS.
- Y. R. Tan, M. Salamanca, L. Pascazio, J. Akroyd and M. Kraft, Fuel, 2021, 283, 118769 CrossRef CAS.
- K. De Ras, T. Panaget, Y. Fenard, J. Aerssens, L. Pillier, J. W. Thybaut, G. Vanhove and K. M. Van Geem, Combust. Flame, 2023, 253, 112792 CrossRef CAS.
- K. De Ras, M. Kusenberg, G. Vanhove, Y. Fenard, A. Eschenbacher, R. J. Varghese, J. Aerssens, R. Van De Vijver, L.-S. Tran, J. W. Thybaut and K. M. Van Geem, Combust. Flame, 2022, 238, 111914 CrossRef CAS.
- D. Pélerin, K. Gaukel, M. Härtl, E. Jacob and G. Wachtmeister, Fuel, 2020, 259, 116231 CrossRef.
- M. Virt and U. Arnold, CogSust, 2022, 1(3) DOI:10.55343/cogsust.20.
- C. Hank, L. Lazar, F. K. Mantei, O. M. Salem, R. J. White, T. Smolinka, A. Schaadt, C. Hebling and H.-M. Henning, Sustainable Energy Fuels, 2019, 3, 3219–3233 RSC.
- S. Voelker, S. Deutz, J. Burre, D. Bongartz, A. Omari, B. Lehrheuer, A. Mitsos, S. Pischinger, A. Bardow and N. Von Der Assen, Sustainable Energy Fuels, 2022, 6, 1959–1973 RSC.
- H. Pitsch, D. Goeb, L. Cai and W. Willems, Prog. Energy Combust. Sci., 2024, 104, 101173 CrossRef.
- A. Holzer, M. Günthner and P. Jung, Automot. Engine Technol., 2022, 7, 369–383 CrossRef.
- A. Omari, B. Heuser, S. Pischinger and C. Rüdinger, Appl. Energy, 2019, 239, 1242–1249 CrossRef CAS.
- F. K. Mantei, S. Kopp, A. Holfelder, E. Flad, D. Kloeters, M. Kraume and O. M. Salem, React. Chem. Eng., 2023, 8, 917–932 RSC.
- A. Fink, C. H. Gierlich, I. Delidovich and R. Palkovits, ChemCatChem, 2020, 12, 5710–5719 CrossRef CAS.
- Z. Xue, C. Lu, H. Shang, G. An, J. Zhang, S. Zhao and Y. Liu, New J. Chem., 2020, 44, 2788–2796 RSC.
- F. Mantei, M. Kraume and O. Salem, Chem. Ing. Tech., 2023, 95, 912–924 CrossRef CAS.
- F. K. Mantei, R. E. Ali, C. Baensch, S. Voelker, P. Haltenort, J. Burger, R.-U. Dietrich, N. Assen, A. Schaadt, J. Sauer and O. M. Salem, Sustainable Energy Fuels, 2022, 6, 528–549 RSC.
- M. Held, Y. Tönges, D. Pélerin, M. Härtl, G. Wachtmeister and J. Burger, Energy Environ. Sci., 2019, 12, 1019–1034 RSC.
- C. J. Baranowski, A. M. Bahmanpour and O. Kröcher, Appl. Catal., B, 2017, 217, 407–420 CrossRef CAS.
- A. Peter, H. Scherer, E. Jacob and I. Krossing, in Internationaler Motorenkongress 2020, ed. J. Liebl, C. Beidl and W. Maus, Springer Vieweg, Wiesbaden, 2020, pp. 405–414 Search PubMed.
- Q. Wu, Y. Guo, J. Shang, D. Shi, Y. Zhang, K. Chen, H. Li, Y. Zhao and Q. Jiao, Chem. Eng. Sci., 2022, 253, 117521 CrossRef CAS.
- O. M. Salem, F. K. Mantei, M. Elmehlawy, R. J. White, H. Klein and S.-E. K. Fateen, React. Chem. Eng., 2018, 3, 277–292 RSC.
- L. Lautenschütz, D. Oestreich, P. Haltenort, U. Arnold, E. Dinjus and J. Sauer, Fuel Process. Technol., 2017, 165, 27–33 CrossRef.
- A. Peter, G. Stebens, J. F. Baumgärtner, E. Jacob, F. K. Mantei, O. M. Salem and I. Krossing, ChemCatChem, 2020, 12, 2416–2420 CrossRef CAS.
- C. F. Breitkreuz, N. Schmitz, E. Ströfer, J. Burger and H. Hasse, Chem. Ing. Tech., 2018, 90, 1489–1496 CrossRef CAS.
- C. F. Breitkreuz, N. Hevert, N. Schmitz, J. Burger and H. Hasse, Ind. Eng. Chem. Res., 2022, 61, 7810–7822 CrossRef CAS.
- P. Haltenort, K. Hackbarth, D. Oestreich, L. Lautenschütz, U. Arnold and J. Sauer, Catal. Commun., 2018, 109, 80–84 CrossRef CAS.
- M. Drexler, P. Haltenort, U. Arnold and J. Sauer, Chem. Ing. Tech., 2022, 94, 256–266 CrossRef CAS.
- A. Billion, J. Schulte, A. Vogel, F. Hilfinger and I. Krossing, Small, 2023, 2306862 Search PubMed.
- Y. Zhao, Z. Xu, H. Chen, Y. Fu and J. Shen, J. Energy Chem., 2013, 22, 833–836 CrossRef CAS.
- R. Peláez, P. Marín and S. Ordóñez, Chem. Eng. J., 2020, 396, 125305 CrossRef.
- Y. Zheng, Q. Tang, T. Wang and J. Wang, Chem. Eng. J., 2015, 278, 183–189 CrossRef CAS.
- P. Haltenort, L. Lautenschütz, U. Arnold and J. Sauer, Top. Catal., 2019, 62, 551–559 CrossRef CAS.
- J. Voggenreiter and J. Burger, Ind. Eng. Chem. Res., 2021, 60, 2418–2429 CrossRef CAS.
- A. W. Franz, H. Kronemayer, D. Pfeiffer, R. D. Pilz, G. Reuss, W. Disteldorf, A. O. Gamer and A. Hilt, in Ullmann's Encyclopedia Of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2016, pp. 1–34 Search PubMed.
- M. Müller and U. Hübsch, in Ullmann's Encyclopedia Of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- P. T. Anastas and R. H. Crabtree, Handbook of Green Chemistry, Wiley-VCH, Weinheim, 2009 Search PubMed.
- M. Drexler, P. Haltenort, U. Arnold, J. Sauer, S. A. Karakoulia and K. S. Triantafyllidis, Catal. Today, 2023, 424, 113847 CrossRef CAS.
- N. Schmitz, E. Ströfer, J. Burger and H. Hasse, Ind. Eng. Chem. Res., 2017, 56, 11519–11530 CrossRef CAS.
- S. Blochum, B. Gadomski, M. Retzlaff, F. Thamm, C. Kraus, M. Härtl, R. Gelhausen, S. Hoppe and G. Wachtmeister, SAE Int. J. Adv. Curr. Pract. Mobil., 2022, 4(1), 15–31 CrossRef.
- T. Maier, M. Härtl, E. Jacob and G. Wachtmeister, Fuel, 2019, 256, 115925 CrossRef CAS.
- S. Wodarz, N. A. Slaby, M. C. Zimmermann, T. N. Otto, J. Holzinger, J. Skibsted, T. A. Zevaco, S. Pitter and J. Sauer, Ind. Eng. Chem. Res., 2020, 59, 17689–17707 CrossRef CAS.
- C. J. Baranowski, T. Fovanna, M. Roger, M. Signorile, J. McCaig, A. M. Bahmanpour, D. Ferri and O. Kröcher, ACS Catal., 2020, 10, 8106–8119 CrossRef CAS.
- F. Liu, R. Wei and T. Wang, Chem. Eng. Sci., 2022, 248, 117057 CrossRef CAS.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- P. Magnoux, P. Roger, C. Canaff, V. Fouche, N. S. Gnep and M. Guisnet, Catalyst Deactivation 1987, in Proceedings Of The 4th International Symposium, Elsevier, 1987, pp. 317–330 Search PubMed.
- S. Wodarz, PhD thesis, Karlsruher Institut für Technologie (KIT), 2021.
- C. J. Baranowski, M. Roger, A. M. Bahmanpour and O. Kröcher, ChemSusChem, 2019, 12, 4421–4431 CrossRef CAS PubMed.
- F. Liu, T. Wang, Y. Zheng and J. Wang, J. Catal., 2017, 355, 17–25 CrossRef CAS.
- J. Wu, S. Wang, H. Li, Y. Zhang, R. Shi and Y. Zhao, Nanomaterials, 2019, 9, 1192 CrossRef CAS PubMed.
- J. Wu, H. Zhu, Z. Wu, Z. Qin, L. Yan, B. Du, W. Fan and J. Wang, Green Chem., 2015, 17, 23532357 Search PubMed.
- C. J. Baranowski, A. M. Bahmanpour, F. Héroguel, J. S. Luterbacher and O. Kröcher, Catal. Sci. Technol., 2019, 9, 366–376 RSC.
- P. Haltenort, PhD thesis, Karlsruher Institut für Technologie (KIT), 2022.
- Y. Ye, M. Yao, H. Chen and X. Zhang, Catal. Lett., 2020, 150, 1445–1453 CrossRef CAS.
- F. Mantei, C. Schwarz, A. Elwalily, F. Fuchs, A. Pounder, H. Stein, M. Kraume and O. Salem, React. Chem. Eng., 2023, 8, 2876–2893 RSC.
- I. Bogatykh, T. Osterland, H. Stein and T. Wilharm, Energy Fuels, 2020, 34, 3357–3366 CrossRef CAS.
- G. D. Pirngruber, K. Seshan and J. A. Lercher, Microporous Mesoporous Mater., 2000, 38, 221–237 CrossRef CAS.
- A. P. Antunes, M. F. Ribeiro, J. M. Silva, F. R. Ribeiro, P. Magnoux and M. Guisnet, Appl. Catal., B, 2001, 33, 149–164 CrossRef CAS.
- Y. Zhang, M. Li, E. Xing, Y. Luo and X. Shu, Catal. Commun., 2019, 119, 67–70 CrossRef CAS.
- S. Lee and M. Choi, J. Catal., 2019, 375, 183–192 CrossRef CAS.
- M. T. Arslan, B. Ali, S. Z. A. Gilani, Y. Hou, Q. Wang, D. Cai, Y. Wang and F. Wei, ACS Catal., 2020, 10, 2477–2488 CrossRef CAS.
- C. D. Chang and A. J. Silvestri, J. Catal., 1977, 47, 249–259 Search PubMed.
- Y. Liu, F. M. Kirchberger, S. Müller, M. Eder, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2019, 10, 1462 CrossRef PubMed.
- N. Zhang, R. Zhu, S.-L. Chen, N. Chen, Y. Cao, L. Ma, T. Wu and W. Sun, Energy Fuels, 2020, 34, 742–748 CrossRef CAS.
- S. Müller, Y. Liu, F. M. Kirchberger, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, J. Am. Chem. Soc., 2016, 138, 15994–16003 CrossRef PubMed.
- H. Schulz, Catal. Today, 2010, 154, 183–194 CrossRef CAS.
- H. Schulz and M. Wei, Top. Catal., 2014, 57, 683–692 CrossRef CAS.
- B. Niethammer, U. Arnold and J. Sauer, Appl. Catal., A, 2023, 651, 119021 CrossRef CAS.
- T. Weissenberger, B. Reiprich, A. G. F. Machoke, K. Klühspies, J. Bauer, R. Dotzel, J. L. Casci and W. Schwieger, Catal. Sci. Technol., 2019, 9, 3259–3269 RSC.
- M. Bjørgen, F. Joensen, M. Spangsberg Holm, U. Olsbye, K.-P. Lillerud and S. Svelle, Appl. Catal., A, 2008, 345, 43–50 CrossRef.
- P. N. R. Vennestrøm, M. Grill, M. Kustova, K. Egeblad, L. F. Lundegaard, F. Joensen, C. H. Christensen and P. Beato, Catal. Today, 2011, 168, 71–79 CrossRef.
- C. J. Baranowski, A. M. Bahmanpour, F. Héroguel, J. S. Luterbacher and O. Kröcher, ChemCatChem, 2019, 11, 3010–3021 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00818a |
| This journal is © The Royal Society of Chemistry 2025 |