
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced catalytic activity of MgO-grafted aluminium isopropoxide in heterogeneous H-transfer reduction reactions through surface support modification
Xiao Yua,
Atika Muhammada,
Boya Qiua,
Aristarchos Mavridis a,
Min Hu*a and
Carmine D'Agostino*ab
a,
Min Hu*a and
Carmine D'Agostino*ab
aDepartment of Chemical Engineering, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: min.hu@manchester.ac.uk; carmine.dagostino@manchester.ac.uk
bDipartimento di Ingegneria Civile, Chimica, Ambientale e dei Materiali (DICAM), Alma Mater Studiorum-Università di Bologna, Via Terracini, 28, Bologna, 40131, Italy
First published on 22nd April 2025
Abstract
The heterogenization of aluminium isopropoxide [Al(OiPr)3] on modified magnesium oxide (MgO) supports was investigated to develop efficient catalysts for hydrogen-transfer (H-transfer) reduction reactions. MgO surfaces were functionalized with octadecyltrichlorosilane (OTES) or dibromobutane (DBB) to optimize the surface chemistry of commercial MgO. The OTES-modified MgO exhibited a porous “nest-like” structure with a markedly increased surface area of 84.3 m2 g−1, compared to 2.9 m2 g−1 for the unmodified MgO. In contrast, DBB-modified MgO displayed a “brush-like” morphology attributed to the flexibility of the immobilized carbon chains. The modified heterogenized catalysts demonstrated substantial improvements in activity compared to the unmodified MgO-based systems. Among the heterogenized catalysts, Al–DBB–MgO achieved the highest turnover frequency (TOF), which is attributed to enhanced substrate adsorption and reduced steric hindrance, facilitating efficient reactant access to active sites. The activity of the two modified catalysts after 5 reduction cycles shows no obvious change in terms of the TOF of Al–DBB–MgO, while the TOF of Al–OTES–MgO dropped by around 14%. These findings highlight the critical role of MgO surface modification in enabling effective Al(OiPr)3 immobilization and enhancing catalytic performance for H-transfer reactions, offering a promising strategy for designing advanced heterogenized catalysts for reduction of carbonyl compounds.
1. Introduction
Catalytic transfer hydrogenation has emerged as an attractive alternative for synthesizing alcohols from aldehydes and ketones, offering a safer and more cost-effective approach compared to conventional direct hydrogenation.1,2 This process eliminates the need for pressurized hydrogen, instead utilizing secondary alcohols as hydrogen donors. Furthermore, it exhibits high chemoselectivity, particularly in the reduction of unsaturated aldehydes or ketones to produce allylic alcohols-valuable chiral building blocks in organic synthesis.3,4 Traditionally, the reaction is catalysed by homogeneous metal alkoxide catalysts, such as aluminium (Al), boron (B), or zirconium (Zr) alkoxides, which demonstrate superior catalytic performance due to their Lewis acidic character in combination with ligand exchangeability.5,6 However, these systems often require an excess of alkoxides and neutralization of the residual alkoxide with strong acid, posing practical and environmental challenges.7 To address these limitations, recent efforts have focused on developing heterogeneous catalysts, which can be easily separated from the reaction mixture, offering improved reusability and sustainability.8Initial studies on catalyst design for H-transfer reductions predominantly explored metal oxides and zeolites, capitalizing on their intrinsic Lewis acidic properties as an extension of homogeneous catalytic systems. For example, Lavalley et al.9 systemically investigated heterogeneous acetone reduction over various metal oxide catalysts, demonstrating that the catalytic activity is strongly correlated with surface acid–base properties. Lewis acidic catalysts, such as chloride-modified Al2O3 (Al2O3–Cl), and basic catalysts, including MgO and ZrO2, exhibited comparable activity. Similarly, H van Bekkumn's group10 reported the reduction of substituted cyclohexanones over zeolite BEA, achieving superior seteroselectivity towards cis-4-tert-butylcyclohexanol. This performance was attributed to Lewis acidic aluminium sites located within the zeolite's micropores. Efforts to enhance zeolite catalytic performance have included incorporating elements like Sn and Zr into the framework to increase Lewis acidity.11,12
Heterogenized homogeneous catalysts represent another significant advancement, achieved by covalently grafting metal alkoxide catalysts onto solid supports. Porous silicas are among the most used supports due to their diverse porous structures.13,14 Such heterogenized catalysts can often retain the high activity and selectivity of their homogeneous counterparts, particularly in the synthesis of α,β-unsaturated alcohols. For example, Uysal and Oksal,15 developed a boron-based heterogeneous catalyst by grafting the boron tri-ethoxide onto mesoporous MCM-41 [B(OEt)3-MCM-41], which exhibited comparable catalytic activity to homogeneous B(OiPr)3 and B(OEt)3 catalysts. This system demonstrated excellent stability and recyclability, maintaining catalytic activity over six cycles without significant performance loss. Similarly, in our previous work,16 we immobilised aluminium isopropoxide [Al(OiPr)3] onto various mesoporous supports, including SiO2, TiO2 and γ-Al2O3, and evaluated their performance in H-transfer reductions. The heterogenized catalysts exhibited excellent activity and high selectivity, closely mirroring the performance of homogeneous Al(OiPr)3. Among these, Al(OiPr)3 grafted onto SiO2 outperformed those on γ-Al2O3 and TiO2, owning to its higher surface area and large pore structure, which facilitated improved Al(OiPr)3 dispersion and enhanced accessibility of the carbonyl substrates to active sites. These findings underscore the importance of carefully selecting and engineering support materials to achieve optimal catalytic performance.
In this study, we explore the impact of MgO surface modification using different functional groups, octyltriethoxysilane, OTES and 1,4-dibromobutane, DBB, to tailor MgO surface chemistry for improved catalytic performance. A comprehensive set of characterization techniques, including FTIR, XRD, SEM, and nitrogen adsorption, were employed to probe the structural and surface chemistry changes induced by the modifications. The catalyst activity of the unmodified and modified supports was evaluated in H-transfer reduction reactions with various substrates including aldehydes and ketones. This work aims to provide insights into the relationship between surface modification and catalytic efficiency, paving the way for the development of advanced heterogenized catalysts with improved performances through relatively simple support modifications.
2. Experimental
2.1 Materials and chemicals
Aluminium isopropoxide (98%), 2-propanol (anhydrous, 99.5%), 1,3,4,5-tertmethylbenze (99%), hexanal (99%), cinnamaldehyde (99%), benzaldehyde (99%), cyclohexanone (99%), MgO catalyst support, 1,4-dibromobutane (DBB) (98%), octyltriethoxysilane (OTES) (97%), chloroform-d (99.8 atom %D) and NaOH were obtained from Sigma Aldrich, UK. All the chemicals were used without further treatment.2.2 MgO modified with OTES (MgO–OTES)
The salinisation of MgO was performed using a chemical grafting method. A 150 mL round-bottom flask was charged with 1.8 mL of octyltriethoxysilane (OTES) and 30 mL of water first, stirring for 3 h. Then 3.0 g of MgO support was added into the mixture and followed by stirring overnight. The modified MgO was then collected by centrifugation and dried at room temperature for 48 h.2.3 MgO modified with DBB (MgO–DBB)
1.38 g of NaOH and 3.0 g of MgO support were added to 30 mL of cyclohexane and stirred for 2 h to get a homogeneous suspension. Then 0.6 mL of 1,4-dibromobutane (DBB) was added and the mixture was refluxed at 90 °C overnight. After that, 5 mL of water was added to quench the reaction and stirred for 1 h. The product was then collected by centrifugation and dried at room temperature for 48 h.2.4 Heterogenized catalysts synthesis with various supports
The immobilization of Al(OiPr)3 onto different supports were achieved by a grafting method as previously reported.16 In a typical synthesis, 3.0 g homogeneous Al(OiPr)3 catalyst was first dissolved in 30 mL anhydrous cyclohexane (theorical Al loading: ∼20 wt%). Then 2.0 g modified or un-modified MgO support was added into the above solution. The suspension was heated to 90 °C and refluxed overnight. Then the product was collected by centrifugation and washed with cyclohexane three times before drying. The resulted catalysts named as Al–MgO, Al–OTES–MgO and Al–DBB–MgO respectively.2.5 Characterization
The X-ray diffraction (XRD) patterns of both supports and catalysts were recorded on a Philips X'Pert X-ray diffractometer using Cu Kα1 radiation (λ = 1.5406 Å) operating at 40 kV, 40 mA with a scanning rate of 2° min−1, a step size of 0.02° s−1, and a 2θ angle from 20° to 80°. The Al amount of the graft catalyst was measured by inductively coupled plasma-optical emission spectroscopy (ICP-OES) on an Analytic Jena Plasma Quant 9000 Elite. The heterogenized catalysts were dissolved in a strong acid solution (VHNO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VHCl = 3:1) in a microwave digest reactor. The resulted solution was diluted and filtered before the measurement. An acid solution containing aluminium was used as a standard reference. The surface area of both supports and catalysts was measured by nitrogen physisorption isotherms at −196 °C on a Micrometrics ASAP 2020 according to the Brunauer–Emmett–Teller (BET) methods. The surface area was calculated using the liner fitting of the Brunauer–Emmett–Teller (BET) equation with the range of P/P0 = 0.05–0.3. Surface morphology and EDX analysis of the samples were obtained using FEI Quanta 250 FEG microscope operating at 15 kV. The FTIR spectra of the samples were collected using a Bruker Vertex 7.0 Fourier transform infrared (FTIR) spectrometer with a scanning wavenumber (ranging from 400 to 4000 cm−1) and a spectral resolution of 2 cm−1. X-ray photoelectron spectrometer (XPS) spectra of fresh and spent catalysts were recorded on a Kratos AXIS Supra spectrometer with a monochromatic Al Kα X-ray (1486.6 eV) source for excitation. The binding energy of the chemical species was calibrated using the C 1s peak at 284.8 eV as an internal standard. The percentages of the individual elements were fitted by analysing the areas of the respective peaks (CasaXPS).
:VHCl = 3:1) in a microwave digest reactor. The resulted solution was diluted and filtered before the measurement. An acid solution containing aluminium was used as a standard reference. The surface area of both supports and catalysts was measured by nitrogen physisorption isotherms at −196 °C on a Micrometrics ASAP 2020 according to the Brunauer–Emmett–Teller (BET) methods. The surface area was calculated using the liner fitting of the Brunauer–Emmett–Teller (BET) equation with the range of P/P0 = 0.05–0.3. Surface morphology and EDX analysis of the samples were obtained using FEI Quanta 250 FEG microscope operating at 15 kV. The FTIR spectra of the samples were collected using a Bruker Vertex 7.0 Fourier transform infrared (FTIR) spectrometer with a scanning wavenumber (ranging from 400 to 4000 cm−1) and a spectral resolution of 2 cm−1. X-ray photoelectron spectrometer (XPS) spectra of fresh and spent catalysts were recorded on a Kratos AXIS Supra spectrometer with a monochromatic Al Kα X-ray (1486.6 eV) source for excitation. The binding energy of the chemical species was calibrated using the C 1s peak at 284.8 eV as an internal standard. The percentages of the individual elements were fitted by analysing the areas of the respective peaks (CasaXPS).
2.6 H-transfer reduction reaction
The H-transfer reduction reaction of catalysts with various aldehyde substrates were performed under batch conditions: 1.4 mmol aldehyde was dissolved in 2-propanol (4.6 mL) in a round bottom flask equipped with reflux condenser. Then 0.14 mmol 1,2,4,5-tetramethylbenzene were added as the internal standard. Then the catalyst was then added (∼0.4 mmol Al) and the reaction mixture was stirred at 90 °C. After reaction, 200 μL of the crude solution was extracted when after the reaction mixture cooling down to room temperature and then diluted with chloroform-d (CDCl3) for NMR analysis. The 1H NMR analyses was performed using a Bruker AVIII HD 400 MHZ spectrometer, and the chemical shifts in 1H spectra were referenced to trimethylsilane (TMS). The conversion, selectivity and turnover frequency (TOF) was determined using the following equations:
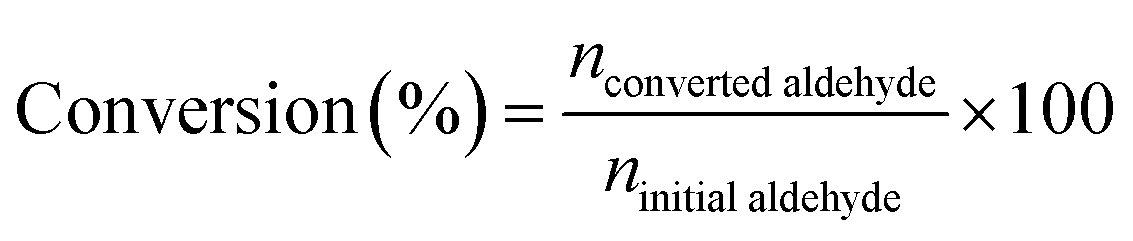 | (1) |
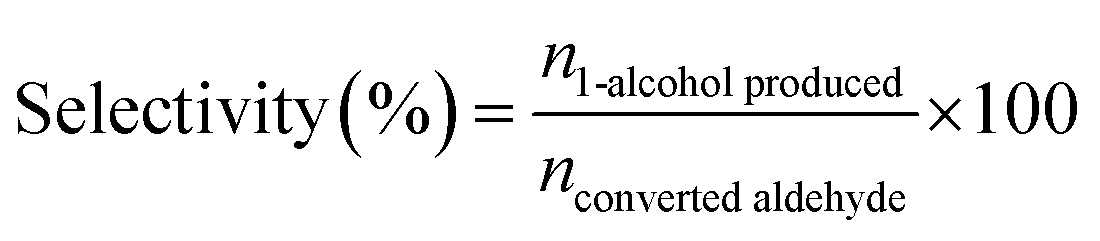 | (2) |
 | (3) |
2.7 Catalysts recyclability tests
The catalyst recyclability tests were conducted with the modified Al–OTES–MgO and Al–DBB–MgO using hexanal reduction as the model reaction. The reaction condition was kept the same and after each reaction cycle, the spent catalysts were washed and dried for the subsequent cycles without any refresh treatment. After 5 reaction cycles, the catalysts were collected and washed for XPS and XRD analysis.3. Results and discussions
3.1 Materials characterization
The modification of MgO support and the immobilization of Al(OiPr)3 are illustrated in Fig. 1. The successful grafting of OTES onto MgO was confirmed by FTIR spectroscopy, as shown in Fig. 2a. For OTES–MgO, distinct peaks were observed at approximately 1080 cm−1, along with a smaller shoulder peak at 1260 cm−1, corresponding to the vibrational modes of Si–O–Si and Si–O–C bond, respectively.17 These peaks were absent in unmodified MgO. Furthermore, the grafted OTES formed densely packed self-assembled layers, which significantly increased the surface area from 2.9 m2 g−1 (for unmodified MgO) to 84.3 m2 g−1 for OTES–MgO (Table 1). This modification altered the surface properties of MgO, creating a “nest-like” structure, showing the characteristics of mesoporous materials. Evidence of this meso-porosity was provided by the N2 adsorption–desorption isotherm of OTES–MgO (Fig. 2c), which transformed from the non-porous nature of unmodified MgO (type II isotherm) to a type IV isotherms with moderate slop at higher P/P0 range (P/P0 > 0.8).18 | ||
| Fig. 1 Processes of (a) MgO support modification followed by Al(OiPr)3 immobilization (a); and (b) Al(OiPr)3 immobilization onto MgO directly (b). | ||
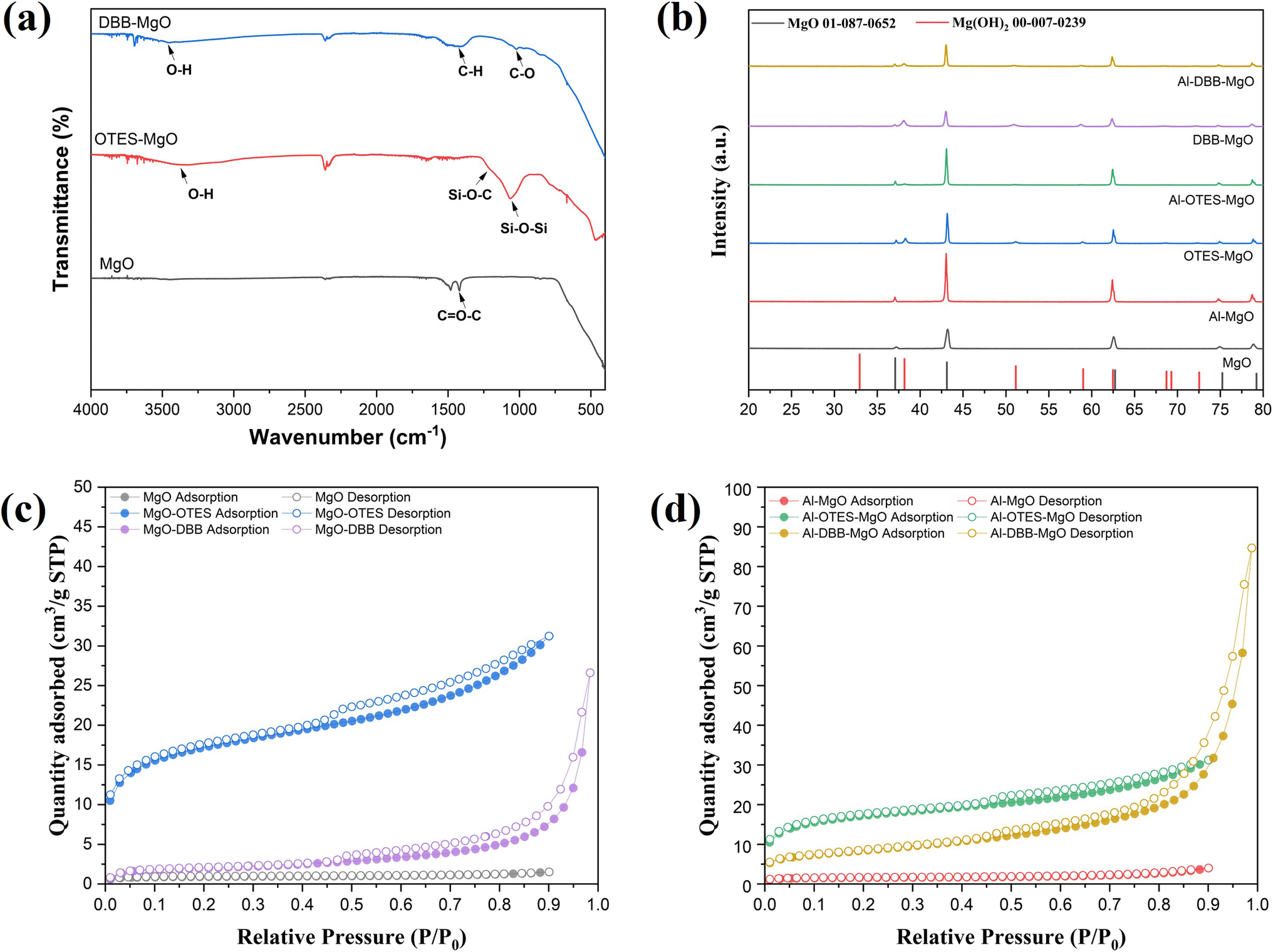 | ||
| Fig. 2 (a) FTIR spectra of MgO and modified MgO supports; (b) XRD patterns of supports and corresponding catalysts with JCPDS standard cards: 01-087-0652 (MgO) and 00-007-0239 [Mg(OH)2]; N2 adsorption–desorption isotherms of supports (c) and catalysts (d). | ||
| Al (wt%) | SBET (m2 g−1) | |
|---|---|---|
| MgO | — | 2.9 |
| OTES–MgO | — | 84.3 |
| DBB–MgO | — | 6.8 |
| Al–MgO | 0.25 | 5.1 |
| Al–MgO–OTES | 3.72 | 57.4 |
| Al–DBB–MgO | 11.42 | 29.5 |
Similarly, the addition of DBB to the MgO surface also increased the surface area, resulting a material with a surface area of 6.8 m2 g−1. The N2 adsorption–desorption isotherm of DBB–MgO (Fig. 2c and Table 1) exhibited a profile similar to that of bare MgO, with the steepest slope at higher relative pressures due to the high difference in volumes of gas adsorbed at the high-pressure points, indicating its microporous nature induced by the presence of the DBB carbon chain layer.19,20 This DBB layer acts as a “brush” on the MgO surface, consisting of extended hydrocarbon chains terminated with hydroxyl groups (–OH). The terminal bromine (Br) of DBB likely undergoes nucleophilic substitution with water (solvent) or surface hydroxyl groups on MgO, forming C–O bond.21 The FTIR spectrum of DBB–MgO (Fig. 2a) showed a characteristic C–O stretching peak at around 1050 cm−1.
Notably, the addition of OTES and DBB did not significantly alter the crystal structure of MgO, as indicated by the XRD patterns of both supports and catalysts (Fig. 2b). The diffraction peaks at 2θ values of 37.2°, 43.2°, 62.5°, 74.9° and 78.8° correspond to the (111), (200), (220), (311), and (222) planes of cubic polycrystalline MgO, consistent with the JCPDS No. 01-087-0652 standard.22 However, a minor Mg(OH)2 phase was detected in the modified MgO supports, as evidenced by additional diffraction peaks (marked with black circles) corresponding to the (101), (102), and (110) planes at 2θ = 38.2°, 51.0°, and 58.9° (JCPDS No. 00-007-0239), respectively.23,24 This phase likely resulted from surface hydrolysis induced by water.25 After immobilization, significant differences in crystal structure were not observed between the supports and the catalysts.
The immobilization of Al(OiPr)3 was achieved through exchange reactions between the surface/terminated hydroxyl groups and the isopropyl linkers of Al(OiPr)3.3,26 The amount of immobilized Al(OiPr)3 was determined by measuring the Al loading via ICP-OES, with results summarized in Table 1. Among the samples, Al–MgO exhibited the lowest Al loading, which can be attributed to its minimal surface area. Furthermore, due to the relatively large size of the Al(OiPr)3 molecule, its grafting onto the MgO surface likely hindered access to adjacent hydroxyl groups, thereby limiting further immobilization.
In contrast, the Al loading increased significantly after the MgO surface was modified with OTES or DBB, with Al–DBB–MgO showing the highest Al loading. Both OTES and DBB contain terminal hydroxyl groups that can facilitate the immobilization of Al(OiPr)3. For Al–OTES–MgO, the Al(OiPr)3 was likely grafted primarily onto the outer layer of OTES, as the densely packed OTES near the MgO surface hindered access to inner hydroxyl groups. Additionally, the lower surface area of MgO limited the amount of OTES grafting, reducing the number of hydroxyl groups available for further Al(OiPr)3 immobilization. The grafting of Al(OiPr)3 may have also occupied some of the cavity space within the OTES framework, contributing to a decrease in surface area.
Conversely, the DBB modification significantly increased the number of the terminal hydroxyl groups. The flexibility of carbon chains of DBB also minimized steric hindrance during Al(OiPr)3 grafting, resulting in the highest Al loading among the samples. The densely grafted Al(OiPr)3 molecules formed a onto DBB–MgO might pack a also contributed to the overall surface area, explaining the increased surface area observed for Al–DBB–MgO.
The surface chemical states of the fresh catalysts were further examined using X-ray photoelectron spectroscopy (XPS), with the Al 2p and O 1s spectra presented in Fig. 3. The O 1s peak in all samples appeared as a single peak at approximately 531.7 eV, corresponding to hydroxyl groups on the MgO surface or terminated hydroxyl groups introduced by the OTES and DBB functional groups. Due to the similar binding energies of these two types of hydroxyl groups, distinguishing them from one another is challenging. The Al 2p peak confirmed the successful immobilization of Al(OiPr)3 molecules onto the support surfacers. The Al–MgO exhibited a single Al 2p doublet peak, indicative of Al–O bond formation between Al(OiPr)3 and the surface hydroxyl groups. In contrast, the modified catalyst, Al–OTES–MgO and Al–DBB–MgO, displayed two doublet Al 2p peaks, suggesting that Al(OiPr)3 molecules were anchored both on the MgO surface (at 74.2–74.6 eV) and on the terminated hydroxyls groups from the OTES or DBB modifiers (at 73.7–74.0 eV), with the area ratio of around 0.25 (80% terminated –OH to 20% surface –OH). These findings support our hypothesis that hydroxyl groups introduced by the modification process serve as active sites for catalyst grafting and play a crucial role in facilitating the subsequent hydrogen transfer reactions.27,28
 | ||
| Fig. 3 XPS spectra of O 1s and Al 2p in the fresh catalyst (a) Al–MgO; (b) Al–OTES–MgO and (c) Al–DBB–MgO. | ||
The morphology and distribution of relevant elements were further analysed using SEM and EDX mapping, as shown in Fig. 4. The SEM images revealed an intensive and uniform distribution of carbon on DBB–MgO, whereas only a small amount of silicon was detected on OTES–MgO. This supports the conclusion that limited OTES was grafted onto the MgO surface. As SEM primarily examines surface features, the detected silicon distribution likely originated from the outer layer of the OTES coating. All catalyst samples showed the presence of aluminum, confirming the successful immobilization of Al(OiPr)3.
 | ||
| Fig. 4 SEM images and EDX elemental maps of catalysts: (a) Al–MgO; (b) Al–OTES–MgO; (c) Al–DBB–MgO. Elemental maps of Al (red), Mg (blue), O (green) and Si or C (turquoise blue) are shown. | ||
3.2 H-transfer reaction activity of the heterogenized catalysts
The catalytic activity of the materials for the H-transfer reaction was evaluated using a range of substrates, including saturated and unsaturated aldehydes and ketones, with 2-proponal serving as both the solvent and hydrogen donor. The activity data are summarised in Table 2.| Substrate | Structure | Catalyst | Conversion (%) | Selectivity (%) | TOFa (h−1) |
|---|---|---|---|---|---|
| a TOF was calculated as the average value (over 3 h). | |||||
| Hexanal |  |
Al–DBB–MgO | 46.6 | 92.7 | 0.21 |
| Al–OTES–MgO | 22.9 | 87.2 | 0.14 | ||
| Al–MgO | 12.8 | >99 | 0.06 | ||
| Cyclohexanone |  |
Al–DBB–MgO | 28.1 | >99 | 0.13 |
| Al–OTES–MgO | 22.3 | 92.1 | 0.10 | ||
| Al–MgO | 5.6 | >99 | 0.03 | ||
| Benzaldehyde |  |
Al–DBB–MgO | 23.9 | 84.4 | 0.23 |
| Al–OTES–MgO | 13.0 | 87.5 | 0.12 | ||
| Al–MgO | 7.8 | 94.6 | 0.07 | ||
| Cinnamaldehyde |  |
Al–DBB–MgO | 12.6 | 96.5 | 0.12 |
| Al–OTES–MgO | 6.7 | >99 | 0.05 | ||
| Al–MgO | 4.6 | >99 | 0.02 | ||
As previously reported,29 the reaction proceeds via a cyclic six-membered transition state involving direct hydrogen transfer from the H-donor (alcohol) to the H-acceptor (aldehyde or ketone). Among the substrates tested, saturated aldehydes were generally easier to reduce than ketones, particularly liner chain aldehydes. For example, all heterogenized catalysts demonstrated the highest activity in reducing hexanal compared to cyclohexanone. The reduced activity observed for cyclohexanone is likely due to the steric hindrance, which restricts its coordination of the ketones to the Al centre.30 In the case of unsaturated aldehydes, such as cinnamaldehyde, a typical α,β-unsaturated aldehyde with a conjugated aromatic ring and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, exhibited the poorest conversion. This reduced activity can be attributed to the combined influence of steric and electronic effects, resulting in the lowest observed activity.
C bond, exhibited the poorest conversion. This reduced activity can be attributed to the combined influence of steric and electronic effects, resulting in the lowest observed activity.
Catalyst based on the unmodified MgO demonstrated the lowest activity among various substrates, owning to the lowest Al(OiPr)3 grafting. In contrast, the modified catalysts demonstrated significantly improved activity across all the substrates, achieving approximately 2- to 4-fold increase compared to unmodified catalyst. Moreover, the heterogeneous catalysis followed the same substrate-specific trends as the homogeneous process, suggesting a similar reaction mechanism for the classical H-transfer reduction.
When normalised by turnover frequency (TOF, last entry in Table 2), Al–DBB–MgO outperformed the other solid catalysts. This superior performance can be attributed to the flexibility of the DBB layer grafted onto MgO, where rotation of the carbon chains reduces steric limitations, facilitating coordination between the Al canter and the substrates. Additionally, the structural similarity between DBB and certain substrates, such as hexanal, enhances reactant adsorption through stronger interaction.
For Al–OTES–MgO, an activity increase was also observed compared to the unmodified catalyst. The enhanced surface area of Al–OTES–MgO is thought to improve contact between the reactants and active sites. However, the densely packed OTES layer induces significant steric hindrance, which was particularly detrimental for substrates with ring structures. This may explain the comparatively lower activity of catalyst Al–OTES–MgO for such substrates.
To further evaluate the impact of the support modification on catalytic performance, we conducted recyclability tests on the modified catalysts Al–DBB–MgO and Al–OTES–MgO using hexanal reduction as a model reaction. As shown in Fig. 5, the turnover frequency (TOF) of Al–DBB–MgO remained stable after 5 reaction cycles, indicating excellent catalytic stability. While the TOF of Al–OTES–MgO dropped by around 14%, likely due to the leaching of Al(OiPr)3.
 | ||
| Fig. 5 Recycling performance of modified Al–MgO catalysts (Al–DBB–MgO and Al–OTES–MgO) for the H-transfer reduction of hexanal. | ||
XPS and XRD analyse were performed on the spent catalysts after the recyclability tests investigate potential structural and surface property changes during the reaction. As depicted in Fig. 6a and b, an additional peak emerged at approximately 530.5 eV with an area of ∼20%, suggesting the formation of the more reduced oxygen species, which can be attributed to Mg(OH)2. The XRD patterns of the spent catalysts, as shown in Fig. 6c, further supported these fundings, showing an increased intensity of the diffraction peaks at 2θ = 38.2°, 51.0°, and 58.9°, corresponding to the (101), (102), and (110) planes of Mg(OH)2. This increment is likely due to accumulated surface hydrolysis accelerated by water during the catalyst recovery and washing. Additionally, oxidation state of Al remained largely unchanged. These observations confirm that the modified catalysts retain their structural integrity upon recycling, with only limited surface changes.
 | ||
| Fig. 6 XPS analysis of spent Al–OTES–MgO (a) and Al–DBB–MgO (b); XRD patterns of fresh and spent catalyst for comparison (c). | ||
4. Conclusion
This study investigated the effect of surface modification of MgO supports and subsequent immobilization of Al(OiPr)3 to produce catalytic materials H-transfer reductions. The surface modifications with OTES and DBB significantly altered the surface properties of MgO, as evidenced by the increased surface areas, engineered porosity, and surface functionalization with terminal hydroxyl groups, providing active sites for efficient Al(OiPr)3 grafting.Among all the solid catalysts tested, Al–DBB–MgO demonstrated the highest catalytic activity, which can be attributed to the flexibility of the DBB carbon chains, facilitating enhanced reactant coordination. In comparison, Al–OTES–MgO showed moderate improvements in activity; however, the steric hindrance from densely packed OTES layers limited its performance, particularly with bulky substrates. Additionally, both Al–DBB–MgO and Al–OTES–MgO demonstrated remarkable stability during the cycling tests with minor structure change. These findings highlight the critical role of optimizing the surface chemistry of support materials to enhance catalytic performance.
Data availability
The data that support the findings of this study are available within the article.Author contributions
Xiao Yu: conceptualisation, investigation, formal analysis, visualization, writing – original draft; Atika Muhammad: investigation, visualization, writing – original draft; Boya Qiu: methodology, validation, writing; Aristarchos Mavridis: methodology, investigation; Min Hu: supervision, writing – review & editing; Carmine D'Agostino: conceptualization, supervision, resources, funding acquisition, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors sincerely acknowledge the valuable contributions of Jamie Tibble-Howlings, Shahla Khan, and Otis Leahair from the Analytical Laboratory in the Department of Chemical Engineering and Ben Spencer in the Faculty of Science and Engineering at the University of Manchester. Their expertise and support in FTIR, ICP-OES XPS measurements, as well as their insightful discussions on data analysis, were instrumental to the success of this work. We would also like to acknowledge the EPSRC, grant no. EP/S019138/1, grant no. EP/V026089/1 and grant no EP/V519613/1.References
- B. Taleb, R. Jahjah, D. Cornu, M. Bechelany, M. Al Ajami, G. Kataya, A. Hijazi and M. H. El-Dakdouki, Exploring hydrogen sources in catalytic transfer hydrogenation: a review of unsaturated compound reduction, Molecules, 2023, 28, 7541 CrossRef CAS.
- J. F. Miñambres and J. Čejka, Meerwein-ponndorf-verley reduction in current heterogeneous catalysis research: a review, Catal. Rev., 2024, 66(5), 2111–2152 CrossRef.
- M. De bruyn, D. E. De Vos and P. A. Jacobs, Chemoselective hydrogen transfer reduction of unsaturated ketones to allylic alcohols with solid Zr and Hf catalysts, Adv. Synth. Catal., 2002, 344(10), 1120–1125 CrossRef CAS.
- A. Lumbroso, M. L. Cooke and B. Breit, Catalytic asymmetric synthesis of allylic alcohols and derivatives and their applications in organic synthesis, Angew. Chem., Int. Ed., 2013, 52(7), 1890–1932 CrossRef CAS PubMed.
- T. Ooi, Y. Itagaki, T. Miura and K. Maruoka, Simultaneous functional group manipulation in the Meerwein–Ponndorf–Verley reduction process catalyzed by bidentate aluminum reagent, Tetrahedron Lett., 1999, 40(11), 2137–2138 CrossRef CAS.
- Y. Ishii, T. Nakano, A. Inada, Y. Kishigami, K. Sakurai and M. Ogawa, Meerwein–Ponndorf–Verley type reduction of ketones and oppenauer type oxidation of alcohols under the influence of bis(cyclopentadienyl)zirconium dihydride, J. Org. Chem., 1986, 51(2), 240–242 CrossRef CAS.
- C. F. de Graauw, J. A. Peters, H. van Bekkum and J. Huskens, Meerwein–Ponndorf–Verley reductions and oppenauer oxidations: an integrated approach, Synthesis, 1994, 1994(10), 1007–1017 CrossRef.
- E. J. Creyghton; J. Huskens; J. C. van der Waal and H. van Bekkum, Meerwein–Ponndorf–Verley and oppenauer reactions catalysed by heterogeneous catalysts, in Studies in Surface Science and Catalysis, ed. H. U. Blaser, A. Baiker and R. Prins, Elsevier, 1997, vol. 108, pp. 531–537 Search PubMed.
- V. A. Ivanov, J. Bachelier, F. Audry and J. C. Lavalley, Study of the Meerwein—Pondorff—Verley reaction between ethanol and acetone on various metal oxides, J. Mol. Catal., 1994, 91(1), 45–59 CrossRef CAS.
- E. J. Creyghton, S. D. Ganeshie, R. S. Downing and H. van Bekkum, Stereoselective Meerwein–Ponndorf–Verley and oppenauer reactions catalysed by zeolite BEA1Communication presented at the first francqui colloquium, brussels, 19–20 February 1996.1, J. Mol. Catal. A: Chem., 1997, 115(3), 457–472 CrossRef CAS.
- N. O. Popovych, P. I. Kyriienko, Y. Millot, L. Valentin, J. Gurgul, R. P. Socha, J. Żukrowski, S. O. Soloviev and S. Dzwigaj, Sn-BEA zeolites prepared by two-step postsynthesis method: physicochemical properties and catalytic activity in processes based on MPV reduction, Microporous Mesoporous Mater., 2018, 268, 178–188 CrossRef CAS.
- M. Boronat, A. Corma and M. Renz, Mechanism of the Meerwein–Ponndorf–Verley–Oppenauer (MPVO) redox equilibrium on Sn– and Zr–beta zeolite catalysts, J. Phys. Chem. B, 2006, 110(42), 21168–21174 CrossRef CAS PubMed.
- M. De bruyn, M. Limbourg, J. Denayer, G. V. Baron, V. Parvulescu, P. J. Grobet, D. E. De Vos and P. A. Jacobs, Mesoporous Zr and Hf catalysts for chemoselective MPV reductions of unsaturated ketones, Appl. Catal., A, 2003, 254(2), 189–201 CrossRef CAS.
- Y. Zhu, S. Jaenicke and G. K. Chuah, Supported zirconium propoxide—a versatile heterogeneous catalyst for the Meerwein–Ponndorf–Verley reduction, J. Catal., 2003, 218(2), 396–404 CrossRef CAS.
- B. Uysal and B. S. Oksal, New heterogeneous B(OEt)3-MCM-41 catalyst for preparation of α,β-unsaturated alcohols, Res. Chem. Intermed., 2015, 41(6), 3893–3911 CrossRef CAS.
- A. Muhammad, A. Ismaila, B. J. Usman, G. Di Carmine and C. D'Agostino, Activity and stability studies of H-transfer reduction reactions of aldehydes and ketones over aluminium isopropoxide heterogenised catalysts, RSC Adv., 2022, 12(52), 33970–33980 RSC.
- V. G. Parale, D. B. Mahadik, S. A. Mahadik, M. S. Kavale, P. B. Wagh, S. C. Gupta and A. V. Rao, OTES modified transparent dip coated silica coatings, Ceram. Int., 2013, 39(1), 835–840 CrossRef CAS.
- B. Hazra, D. A. Wood, V. Vishal, A. K. Varma, D. Sakha and A. K. Singh, Porosity controls and fractal disposition of organic-rich Permian shales using low-pressure adsorption techniques, Fuel, 2018, 220, 837–848 CrossRef CAS.
- K. S. W. Sing, Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984), Pure Appl. Chem., 1985, 57(4), 603–619 CrossRef CAS.
- J. Hu, S. Tang and S. Zhang, Investigation of pore structure and fractal characteristics of the Lower Silurian Longmaxi shales in western Hunan and Hubei Provinces in China, J. Nat. Gas Sci. Eng., 2016, 28, 522–535 CrossRef CAS.
- K. Ha, Y. J. Lee, H. J. Lee and K. B. Yoon, Facile assembly of zeolite monolayers on glass, silica, alumina, and other zeolites using 3-halopropylsilyl reagents as covalent linkers, Adv. Mater., 2000, 12(15), 1114–1117 CrossRef CAS.
- J. Musafir, R. Kumar and R. Kumar, Temperature-dependent conduction and transport mechanisms in pure and Zn-doped MgO nanomaterials, J. Mater. Sci.: Mater. Electron., 2024, 35, 681 CrossRef.
- Q. Zhang, L. He, Z. Zhao, W. Han, G. Tan, L. Guo and F. Wang, Synthesis of MgO nanomaterials using ultrasonic electrodeposition and their adsorption performance for Pb(II), J. Indian Chem. Soc., 2024, 101(4), 101146 CrossRef CAS.
- I. D. E. Ramos, E. Ling Sham, O. C. Barrios, L. I. Barbosa and P. R. Orosco, Synthesis of magnesium aluminate spinel by calcination of aluminum recycled from discarded PV solar panels with bischofite, J. Therm. Anal. Calorim., 2025, 25, 14100 Search PubMed.
- M. Chen and D. A. Dixon, Structure and stability of hydrolysis reaction products of MgO nanoparticles leading to the formation of brucite, J. Phys. Chem. C, 2017, 121(39), 21750–21762 CrossRef CAS.
- P. Dubois, M. Krishnan and R. Narayan, Aliphatic polyester-grafted starch-like polysaccharides by ring-opening polymerization, Polymer, 1999, 40(11), 3091–3100 CrossRef CAS.
- K. Sun, P. Wang, Z. Bian and W. Huang, An investigation into the effects of different existing states of aluminum isopropoxide on copper-based catalysts for direct synthesis of dimethyl ether from syngas, Appl. Surf. Sci., 2018, 428, 534–540 CrossRef CAS.
- J. van den Brand, W. G. Sloof, H. Terryn and J. H. W. de Wit, Correlation between hydroxyl fraction and O/Al atomic ratio as determined from XPS spectra of aluminium oxide layers, Surf. Interface Anal., 2004, 36, 81–88 CrossRef CAS.
- R. Cohen, C. R. Graves, S. T. Nguyen, J. M. L. Martin and M. A. Ratner, The mechanism of aluminum-catalyzed Meerwein–Schmidt–Ponndorf–Verley reduction of carbonyls to alcohols, J. Am. Chem. Soc., 2004, 126(45), 14796–14803 CrossRef CAS.
- R. Anwander, G. Gerstberger, C. Palm, O. Groeger and G. Engelhardt, Enhanced catalytic activity of MCM-41-grafted aluminium isopropoxide in MPV reductions, Chem. Commun., 1998,(17), 1811–1812 RSC.
| This journal is © The Royal Society of Chemistry 2025 |