
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharge-induced isomerization in alkyl imine molecular motors: a reduced energy barrier approach†
Syed Bilal Ahmed a,
Hei Wun Kanb,
King-Cheong Lam*c and
Cho-Tung Yip*a
a,
Hei Wun Kanb,
King-Cheong Lam*c and
Cho-Tung Yip*a
aSchool of Science, Harbin Institute of Technology (Shenzhen), Shenzhen 518055, China. E-mail: h0260416@hit.edu.cn
bKey Laboratory of Quantitative Synthetic Biology, Shenzhen Institute of Synthetic Biology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China
cDivision of Science, Engineering and Health Studies, The School of Professional Education and Executive Development, Hong Kong Polytechnic University, Hung Hom, Hong Kong, China. E-mail: antony.lam@cpce-polyu.edu.hk
First published on 17th March 2025
Abstract
Molecular motors offer promising applications in the fields of nanodevices and biological systems, as the accurate control of directional rotation at the molecular scale holds great potential. In this context, it is highly relevant to study a new class of molecular motors that can undergo isomerization. Since the first report of the chiral N-alkyl imine-based motors, most investigations have focused on the unidirectional rotation process induced by light and heat. However, this work explores an alternative mechanism – the electron-induced stimulating mechanism of the molecular motor. We theoretically investigate how charge injection and extraction can influence molecular rotation. The rotation occurs around the central axle, which is measured as the torsion angle between the rotor and the stator fragments of the molecule against the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond. Our computational study reveals that the introduction of charge reduces the energy barrier, facilitating more favourable molecular rotation than in the neutral singlet state. The charged molecule in a quartet spin state can rotate internally, while that in the doublet state cannot. Our findings provide a molecular scale understanding of the reaction pathways and highlight the significant role of charge in promoting the isomerization and rotational behaviour of the molecular motor.
N double bond. Our computational study reveals that the introduction of charge reduces the energy barrier, facilitating more favourable molecular rotation than in the neutral singlet state. The charged molecule in a quartet spin state can rotate internally, while that in the doublet state cannot. Our findings provide a molecular scale understanding of the reaction pathways and highlight the significant role of charge in promoting the isomerization and rotational behaviour of the molecular motor.
Introduction
The field of molecular machines has experienced significant advancements in recent years, with particular focus on devices capable of performing directional motion at the nanoscale.1–3 These remarkable nanomachines harness energy from their environment to generate mechanical motion, either rotational or translational. Among these, molecular motors have emerged as a subject of intense experimental and theoretical studies,4–8 including biological motors such as myosins and kinesins.9–11 In parallel with the study of biological motors, the development of synthetic molecular motors has gained considerable interest. Pioneering work by Kelly, and their colleagues has spurred a surge of research interest in this area.12–15 These artificial motors utilize a diverse range of external stimulation and rotation mechanisms. Among other external stimuli, these motors are activated by chemical redox processes,16,17 electric fields,18–20 and photoelectron stimulation.21–23 Molecular motors can be engineered to operate on substrates or under solvent environments, and they range from simple organic molecules to complex artificial nanoscale systems.4,24–26The chiral overcrowded alkenes reported by Feringa have paved the way to develop a family of artificial light-driven molecular motors capable of a full 360° unidirectional rotation motion around the CC double bond.27 Following that, there have been different generations of the “Feringa motors” that have been investigated and characterized.18,28–30 Several investigations focusing on enhancing rotation speed and expanding the activation wavelength resulted in the development of new kinds of artificial molecular motors. Among various designs studied, imine-based motors are a distinct family of light-responsive molecules that have attracted significant attention since their introduction by Greb and Lehn in 2014.31 Their work demonstrated that these imine-based structures could operate as either four-step or two-step unidirectional rotors around the CN axle in response to light stimuli, opening new possibilities for the design of efficient, easy-to-synthesize and photosensitive motors.32,33
The activation energy required for rotational movement can be reduced to enhance isomerization reactions. Various strategies, including the manipulation of electronic structures, have been explored to achieve this goal.34–36 Studies have demonstrated that the addition or removal of a single electron can move the reaction to a different potential energy surface.37,38 High-level ab initio calculations have shown that electron attachment or ionization can weaken chemical bonds, such as carbon–carbon double bonds in olefins, leading to barrier-less cis–trans isomerization processes.39 The charge attachment (anion formation) or charge removal (cation formation) to the molecules may significantly reduce the energy barrier for conformational changes compared to traditional light-driven processes. It has been observed in various molecular systems, including modified versions of overcrowded alkenes,4 and in experiments using scanning tunnelling microscopy (STM), where electron transfer between the tip and substrate can induce various chemical reactions, including isomerization and bond-breaking.40–45 The ability to manipulate energy barriers through charge modulation offers a potentially more efficient means of controlling molecular motion, as it does not rely on the absorption of specific wavelengths of light.
In this work, we study the isomerization behaviour of the alkyl-imine-based molecular motor induced by electron attachment and extraction. The unique feature of imine-based motors lies in their two distinct isomerization pathways: photochemical and thermal. The photochemical CN isomerization occurs via out-of-plane rotation,46 while the thermal process involves direct inversion at the nitrogen atom. A study by Martinez and Fang in 2023 demonstrated the mechanism of unidirectional rotation, attributing it to the repulsion between the nitrogen lone pair and the closest hydrogen of the stator.47 After photoisomerization, Wang et al. propose a rotational mechanism consisting of two thermal steps.48 One step involves a slight adjustment in the orientation of the -tBu substituent on the rotor, indicating minimal rotation. The other step features an in-plane nitrogen inversion, allowing the motor to return to its initial cis-isomer configuration, thus completing a rotational cycle. Each of these steps includes a transition state, with the nitrogen inversion step exhibiting a higher energy barrier compared to the adjustment of the -tBu substituent, which involves only a slight twist of CN dihedral angle. Although the imine motor has been studied as an effective light-driven molecular rotary motor,46–48 most investigations focus on the isomerization reaction caused by light and heat. This work aims to explore an alternative mechanism to initiate intramolecular rotation through charge modulation. This approach aligns with the interest in charge-driven isomerization, which has shown promise in reducing energy barriers for chemical reactions.
Computational methodology
Theoretical simulations provide valuable insights into reaction mechanisms that are not easily observable in experiments. While a single-reference method like DFT and time-dependent DFT (TD-DFT) methods are commonly employed for medium to large systems due to their computational efficiency, they can have limitations in accurately describing double excitations and the multiplet character of electronic states. Therefore, multi-reference ab initio approaches such as CASSCF are essential for accurately characterizing the PES, particularly in the vicinity of conical intersections (CI). The choice of basis sets should also have an impact on the overall accuracy of such calculations.49 For that reason, our computation employs complete active space self-consistent field (CASSCF)50,51 calculations using the cc-pVTZ Gaussian basis set of Dunning et al.52 These basis sets are regarded for the accuracy of data, although they come with considerable computational costs,53 as observed in our work. This approach provides a more accurate representation of the electronic structure for the neutral, cationic, and anionic systems.In this study, we have performed a number of single-point energy calculations for the primary analysis of the potential energy surfaces (PES). To benchmark the results obtained from the CASSCF method, we conducted cost-effective DFT calculations with the widely used 6-311+G(d,p) basis set using long-range-corrected hybrid functional ωB97xD,54 as well as the hybrid functional CAM-B3LYP and the variant hybrid functional M06-2X.55–57 These comparisons allow us to assess the reliability and accuracy of the CASSCF results for the imine-based molecular rotor system. All DFT and CASSCF computations were carried out using the Gaussian 16 software package.58 The spin density maps and visualizations of the spatial distributions of HOMO–LUMO were generated using Multiwfn59 and VMD60 software.
To investigate the rotational mechanism of the imine motor, we began by fully optimizing the geometry of the (M)-cis and (P)-trans structures (shown in Fig. 1a) in their ground states (S0) using CASSCF/cc-pVTZ, with an active space of 4 electrons in 4 orbitals, hereafter referred to as CASSCF(4,4). Subsequently, we calculated the vertical excitation energies at the ground state (S0) minima of the isomers, representing the energy of the molecule immediately after light absorption. The relative ground state (S0) and the lowest singlet excited state (S1) energies are reported in Table 1. The results from the CASSCF method align well with those obtained from the DFT methods, showing only minor differences. The primary axis of rotation in the imine motor is centered on the CN bond, which effectively functions as the rotational axle, enabling the directional motion of the motor. The geometric structures of the isomers reveal that the key geometric parameter during the isomerization reaction between (M)-cis and (P)-trans is the θCCNC torsion angle. This angle undergoes a substantial change, shifting from 5.10° in the (M)-cis configuration to 178.60° in the (P)-trans configuration at the CASSCF/cc-pVTZ level of theory. Table S1† provides a comparison of key geometric parameters (i.e., θCCNC, θCCCN, CN bond length) for the neutral and charged (M)-cis isomer analyzed using both CASSCF and DFT methods. Notably, the neutral system has a CN bond length of 1.26 Å, while the cationic system has a bond length of 1.24 Å, which is close to that of the neutral system. In contrast, the anionic system exhibits an elongated CN bond length of 1.32 Å, implying lower stability compared to the neutral system. This aspect will be further discussed in subsequent sections, particularly in relation to the HOMO–LUMO energy gaps. To investigate the isomerization reaction, we initiated a systematic approach by performing a relaxed scan of the θCCNC torsion angle. Starting from the constrained geometry optimizations at a fixed θCCNC dihedral, we incrementally increased the dihedral angle, simultaneously calculating the S1 and corresponding S0 energies. Previous works suggest that the imine-based motor has a stronger driving force and better directionality compared to overcrowded-alkene-based motors during the light-driven rotation process.46 Once the direction of rotation is determined, the rotation can be tracked by incrementally turning the rotor a few degrees in the same direction along the central CN axle, followed by decay to the ground state. The PES scan is thus performed at a constrained θCCNC while other parameters are allowed to relax. The resulting geometric structures are then used to obtain the PES of the anionic and cationic charged systems.
 | ||
| Fig. 1 (a) Schematic of the rotary cycle of the N-alkyl imine isomers. (b) The key geometric parameters θCCNC and θCCCN used in this work. | ||
| Methods | State | (M)-cis | (P)-trans |
|---|---|---|---|
| CASSCF(4,4)/cc-pVTZ | S0 | 0 | −0.02 |
| S1 | 5.01 | 5.07 | |
| S1-min | 2.97 | ||
| ωB97xD/6-311+G(d,p) | S0 | 0 | −0.01 |
| S1 | 4.81 | 4.78 | |
| S1-min | 2.84 | ||
| CAM-B3LYP/6-311+G(d,p) | S0 | 0 | 0.01 |
| S1 | 4.73 | 4.71 | |
| S1-min | 2.83 | ||
| M06-2X/6-311+G(d,p) | S0 | 0 | −0.01 |
| S1 | 4.55 | 4.52 | |
| S1-min | 2.67 |
Results and discussions
To elucidate the electronic configurations of the neutral, cationic and anionic systems, the schematic representation of the electron distribution in the frontier molecular orbitals (MO) for each system is presented in Fig. 2. In the case of the neutral molecule in its ground state (S0), spin-up (α) and spin-down (β) electrons are paired in the MO. However, when the molecule is in its first excited state (S1), a single electron is promoted from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). This excitation leads to two unpaired electrons with opposite spins occupying two distinct energy levels within the MOs. The unidirectional motion occurs experimentally in the imine-based motor through the photoisomerization reaction.31,33 This process involves a transition between the S0 and the S1 states, which has an (n,π*) character. In the case of the initial excitation to the higher-energy S2 state (π,π*), it is expected that the molecule will rapidly undergo internal conversion to the lower-energy S1 (n,π*) state.46 Therefore, the subsequent dynamics of the molecule will follow the photoisomerization process from the S1 state back to the ground state S0, regardless of the initial excitation pathway. The anionic and cationic charged systems can be generated by the addition or removal of a single electron from the molecule in its ground state, respectively. As further illustrated in Fig. 2, the two low-energy spin states for both the cationic and anionic systems provide insight into the electronic configurations that arise from these charge alterations. The doublet spin state (D0) features a single unpaired electron, while the quartet spin state (Q0) contains three unpaired electrons. Our objective is to determine whether the charged molecules in their ground states can exhibit rotational behaviour comparable to that of the neutral molecule. | ||
| Fig. 2 Schematic diagram of electron distribution in frontier molecular orbitals (MO) of different electronic states in the anionic, neutral and cationic imine-based molecular motor. | ||
It is important to note that the number of electrons in the anionic and cationic charged molecule is different from that of the neutral system. To facilitate a comparison of the PES across these systems with a varying number of electrons, the energy adjustment is implemented to normalize the energies of the cationic and anionic systems to the neutral system. Following a prior study, the adjustment energy for cationic and anionic systems is described in the following equations.
| Anion system: adjustment energy = total energy (ET) − electron energy in HOMO (EHOMO) |
| Cation system: adjustment energy = total energy (ET) + electron energy in LUMO (ELUMO) |
Before analyzing the PES of the charged molecules, it is essential to first consider the key characteristics of the neutral system. The neutral imine motor system contains 190 electrons and exists in a singlet state. The PES of the photoisomerization step (M → P) of the neutral molecule is depicted in Fig. 3a, providing a baseline for comparison with charged states. The relative energies corresponding to the θCCNC angles of the fully relaxed ground state isomers are highlighted in the coloured squares (M)-cis (green) and (P)-trans (blue), respectively. The S1 state energy profile (black) obtained by constrained geometry optimization, exhibits a downward slope, indicative of a barrierless and directional rotary process for the photoisomerization. The energy difference between the highest (θCCNC = 5.10°) and lowest (θCCNC = 105°) points on the S1 state energy profile is found to be 0.92 eV, in line with the study by Liu et al.46 This energy difference reflects the magnitude of the driving force experienced by the motor as it relaxes along the energy profile. Furthermore, the energy barrier of the S0 state is determined to be 2.33 eV, which is lower than the lowest point on the S1 state energy profile. This observation suggests that the directionality and driving force of the photoisomerization process are sufficiently robust to facilitate the desired rotational behaviour of the molecular motor. It can be further deduced that there exists an energy gap of approximately 0.54 eV between the lowest point of the S1 state and the highest point of the S0 state. This suggests the presence of a conical intersection (CI) in the region around θCCNC = 95–105°. Previous computational studies on the photoisomerization of light-driven motors have established that the decay processes from the excited state to the ground state undergoes a conical intersections at highly twisted geometries, where the two states become degenerate.22,61 Consequently, in the following calculations, we will determine the CI-optimized structure using CASSCF; however we first analyze the PES of the charged systems.
 | ||
| Fig. 3 Relative energy profiles of neutral and charged molecules. (a) Neutral molecule in the singlet excited state (S1) and ground state (S0) for the photoisomerization process. The optimized molecular structures are displayed in the inset without hydrogens for clarity. (b) Charged molecule in doublet spin state (D0) and quartet spin state (Q0). | ||
The PES of the charged anionic and cationic systems are presented in Fig. 3b, where relative adjustment energies are used. The CASSCF method was utilized with the active space of (5,4) and (3,4) for anionic and cationic molecules, respectively. The energy profiles of both the cationic and anionic systems with quartet (Q0) spin multiplicity demonstrate a consistent decrease in energy as the torsion angle increases from 5° to 105°. This trend is analogous to the energy curve of the neutral molecule in the S1 state, as illustrated in Fig. 3a. Therefore, it suggests that the high spin state of charged molecules can induce a rotational behaviour along the cation Q0 and anion Q0 potential energy surfaces from θCCNC = 5°–105°. On the other hand, the energy of charged systems with low spin states rises steadily with the dihedral angle in the range of 30° to 105°. This energy variation trend is contrary to that of the S1 state curve of the neutral molecule. Hence, the positively and negatively charged systems that are in a low spin state cannot generate the required torque to rotate the imine motor molecule.
Further analysis of the PES for the S0 of the neutral molecule revealed that an energy barrier of 2.33 eV must be overcome when the torsion angle increases from M to P isomer. In contrast, the energy barriers associated with twisting the dihedral angle of CN double bond in the anionic (D0) and cationic (D0) systems were significantly lower, measured at 1.94 eV and 2.13 eV, respectively. In particular, the introduction of the charge to the neutral system reduces the energy barrier near the dihedral angle of 105° by 0.4 eV for the anionic system (D0) and 0.2 eV for the cationic system (D0), compared to the neutral case. These results are consistent with previous reports, highlighting the significant influence of charge on the energy barrier of the fluorene-based motor.4
In addition to the PES analysis, we examined the spin density maps and the energy gap between the HOMO and LUMO for the imine-based molecular rotor. The spin density maps provide insights into the distribution of unpaired electrons within the charged species. For both the anionic and cationic systems, the distributions of α- and β-electron densities exhibit distinct characteristics (Fig. S2†). In the anionic system, unpaired electrons are significantly concentrated around the central CN bond and on the carbon atoms within the stator region of the molecular motor. In contrast, the cationic system shows a localization of unpaired electrons primarily around the central CN atoms and the adjacent carbon atoms in the rotator region.
Furthermore, we calculated the HOMO–LUMO energy gap to assess the electronic transitions associated with the molecular rotor (see Fig. S3†). A smaller energy gap typically indicates less stability and lower energy barriers.62 The singly occupied molecular orbital (SOMO) is a half-filled HOMO of radicals and is associated with open-shell molecules. The gaps between the alpha-HOMO (or SOMO) and alpha-LUMO of the neutral, anionic, and cationic systems are 8.96, 4.87, and 7.63 eV, respectively, suggesting that the neutral motor is more stable than its charged counterparts. This observation aligns with the findings from the PES energy barriers. Additionally, the spin density maps indicate a relatively enhanced reactivity of the anionic system compared to the cationic molecule, attributed to the increased electron density around the central CN bond and the localization of unpaired electrons. This characteristic may facilitate bond breaking and forming during isomerization, correlating with lower energy barriers for these reactions.
As mentioned above, earlier studies have proposed that nitrogen out-of-plane movement occurs during the photoisomerization process. This implies that another key dihedral angle, i.e., θCCCN, should be twisted “up” or “down” while scanning the θCCNC dihedral. The nitrogen out-of-plane motion in the “up” direction is away from the stator, while the motion in the “down” direction is toward the stator. Thus, the excited state nature of the imine-based motor encompasses two conical intersections in the up or down direction of nitrogen motion. To further investigate this aspect for the charged systems, we performed an additional scan of the PES in the direction of the degeneracy of the S0 and S1 states. For this scan, we obtained the CASSCF-optimized CI structure, with θCCCN and CN bond length values are measured at 35.81° and 1.40 Å, respectively. The nitrogen was twisted out-of-plane in the direction towards the stator. While keeping fixed values for θCCCN and CN bond length, we scanned θCCNC along the reaction pathway from the M to P isomerization in the near the relevant conical intersection, and the resulting PES is presented in Fig. 4. The S1 relative energy profile, as depicted in Fig. 4a, displays a downward trend, with a point of degeneracy around 105°. This is consistent with the PES observed in Fig. 3a. The calculated ground state S0 energy barrier is 3.92 eV, which can be easily overcome by the S1 excited state. Moreover, the corresponding PES of the charged systems of cation and anion are presented in Fig. 4b. The reduction of the energy barrier near the dihedral angle of 105° is observed by 0.54 eV for the anionic system (D0) and 0.87 eV for the cationic system (D0). To further validate the accuracy of the results obtained using the CASSCF/cc-pVTZ method, we performed additional calculations of the potential energy surfaces (PES) using three representative and well-established DFT functionals, ωB97xD, M06-2X and CAM-B3LYP, with the 6-311+G(d,p) basis set. For the DFT calculation, we determined the values of θCCCN and CN bond obtained from the minimization of S1-state. While keeping θCCCN and CN fixed, we scanned the PES in the region of θCCNC = 50–140°. The PES profiles in the region close to the conical intersection are generated, by three DFT functionals and are in good agreement with CASSCF/cc-pVTZ, as shown in Fig. 4b and S1.† Table 2 presents a summary of the energy barriers observed in the ground state neutral system (S0) and the low spin charged (Anion_D0 and Cation_D0) systems. Correspondingly, the lowest point on the energy profile of the high spin states (Anion_Q0 and Cation_Q0) of the charged systems calculated with various DFT functionals are consistent with acceptable variations in the data. This consistency across different computational methods validates the approaches employed in this study and indicates that these DFT methods can effectively generate results comparable to those obtained from the higher-level CASSCF with cc-pVTZ basis sets, thus saving computational time. The PES scans for the charged systems further suggest the tendency of the motor to induce rotation in the high spin state, similar to the S1 state of the neutral molecule. These findings, combined with the reduced energy barriers for the charged systems compared to the neutral case, highlight the significant role of charge in promoting the isomerization and rotational behaviour of the molecular motor.
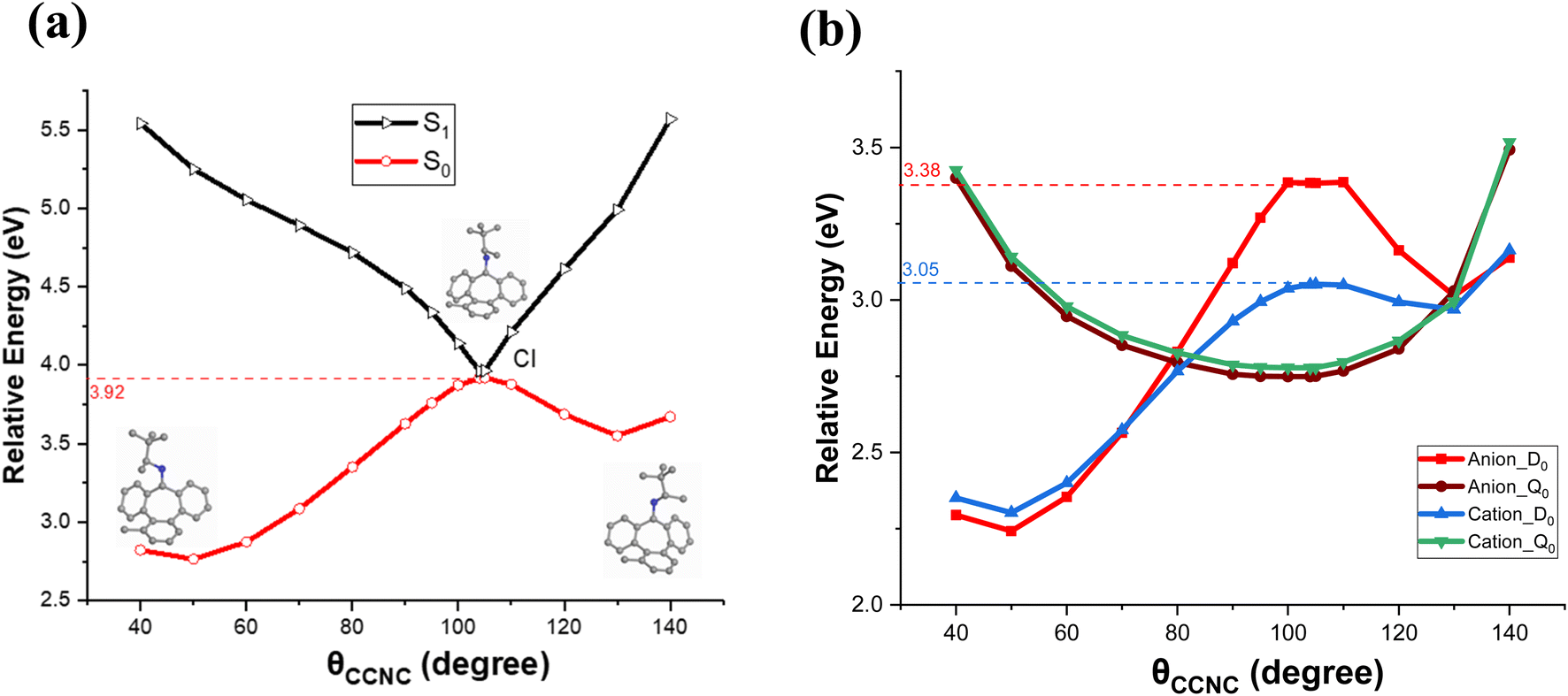 | ||
| Fig. 4 Relative potential energy profiles of (a) the neutral molecule and (b) the charged molecule in the region near the conical intersection. The neutral molecule features three insets showing the molecular structures corresponding to the dihedral angles used in the scan. The geometries are CI-optimized with constrains of θCCCN = 35.81° and C–N = 1.40 Å. | ||
| Method | S0 | Anion_D0 | Cation_D0 | Anion_Q0 | Cation_Q0 |
|---|---|---|---|---|---|
| CASSCF/cc-pVTZ | 3.92 | 3.38 | 3.05 | 2.74 | 2.77 |
| ωB97xD/6-311+G(d,p) | 3.05 | 2.61 | 2.23 | 1.93 | 1.96 |
| CAM-B3LYP/6-311+G(d,p) | 2.96 | 2.95 | 2.57 | 2.28 | 2.29 |
| M06-2X/6-311+G(d,p) | 3.03 | 2.84 | 2.51 | 2.26 | 2.29 |
Conclusion
In this work, we investigate the electron-induced cis–trans isomerization reaction of the light-driven imine-based molecular motor. The results propose that the quartet spin multiplicity can effectively induce the internal rotation of the molecular motor. However, changes in the spin multiplicity of charged molecules do not produce a similar effect. Analysis of the energy profile reveals that the higher spin state of both anionic and cationic molecules can drive the motor molecules to rotate, enabling molecular isomerization. Interestingly, when the dihedral angle approaches 105°, the energy surface reaches a minimum value in the high spin state of the charged molecules, which is analogous to the character of the S1 state in the neutral molecule. In contrast, the low spin i.e., doublet state is unable to rotate the motor due to the increasing energy as the dihedral angle changes, with the maximum of the energy barrier reaching around 105°, which is analogous to the S0 state of the neutral molecule. Energy analysis further reveals that the introduction of charge to the neutral molecule substantially lowers the energy barrier by about 0.5 eV for the negatively charged system and around 0.9 eV for the positively charged system at CASSCF/cc-pVTZ level of theory. Additionally, HOMO–LUMO gap analysis and spin density maps indicate that charged systems exhibit reduced energy barriers for isomerization. It is further supported by the potential energy surface scans in the region near the conical intersection, which reveal a tendency for the motor to rotate in the high spin state. Hence, these findings on charge-induced imine-based molecular motors offer valuable insight by providing a theoretical framework for understanding how charge injection and extraction can influence molecular rotation. The reduced energy barriers observed in the charged systems, along with the tendency for rotation in the high spin state, offer potential advantages in terms of control and efficiency compared to light-driven systems as well as open up new possibilities for the design of multi-responsive molecular machines.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and ESI.† Raw data that support the findings are available from the corresponding author, upon reasonable request.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 17 March 2025, which included an incorrect Fig. 1.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 2174079), Guangdong Basic and Applied Basic Research Foundation (Grant No. 2214050004792), Shenzhen Municipal Science and Technology projects (Grant No. JCYJ20200109113003946), Guangdong Provincial Key Laboratory of Semiconductor Optoelectronic Materials and Intelligent Photonic Systems, Harbin Institute of Technology, Shenzhen, People's Republic of China and the College of Professional and Continuing Education, The Hong Kong Polytechnic University, Hong Kong SAR, China (Grant No. SEHS-2024-331(J)).References
- B. L. Feringa, J. Org. Chem., 2007, 72, 6635–6652 CrossRef CAS PubMed.
- M. Schliwa and G. Woehlke, Nature, 2003, 422, 759–765 CrossRef CAS PubMed.
- K. Kinbara and T. Aida, Chem. Rev., 2005, 105, 1377–1400 CrossRef CAS PubMed.
- L. Chen, F. Qi, K. Jitapunkul, Y. Zhao, R. Zhang and M. A. Van Hove, J. Phys. Chem. A, 2018, 122, 7614–7619 CrossRef CAS PubMed.
- S. B. Ahmed, N. Ullah, Y. Zhao, R. Zhang and M. A. Van Hove, J. Phys. Chem. C, 2021, 125, 17612–17621 CrossRef CAS.
- R. Q. Zhang, Y. L. Zhao, F. Qi, K. Hermann and M. A. Van Hove, Phys. Chem. Chem. Phys., 2016, 18, 29665–29672 RSC.
- H. L. Tierney, C. J. Murphy, A. D. Jewell, A. E. Baber, E. V Iski, H. Y. Khodaverdian, A. F. McGuire, N. Klebanov and E. C. H. Sykes, Nat. Nanotechnol., 2011, 6, 625 CrossRef CAS PubMed.
- X. Li, F. Qi, R. Zhao, Z. Qiu, Y. Li, M. Long and G. Zhou, J. Mater. Chem. C, 2022, 10, 5292–5302 RSC.
- T. R. Kelly, Angew. Chem., Int. Ed., 2005, 44, 4124–4127 CrossRef CAS PubMed.
- B. B. McIntosh and E. M. Ostap, J. Cell Sci., 2016, 129, 2689–2695 CrossRef CAS PubMed.
- K. Jitapunkul, Y. Zhao, L. Lawtrakul, M. A. Van Hove and R. Zhang, J. Biomol. Struct. Dyn., 2023, 41, 10368–10376 CrossRef CAS PubMed.
- T. R. Kelly, I. Tellitu and J. P. Sestelo, Angew. Chem., Int. Ed. Engl., 1997, 36, 1866–1868 CrossRef CAS.
- T. R. Kelly, Acc. Chem. Res., 2001, 34, 514–522 CrossRef CAS PubMed.
- J. P. Sestelo and T. R. Kelly, Appl. Phys. A, 2002, 75, 337–343 CrossRef CAS.
- T. R. Kelly, M. C. Bowyer, K. V. Bhaskar, D. Bebbington, A. Garcia, F. Lang, M. H. Kim and M. P. Jette, J. Am. Chem. Soc., 1994, 116, 3657–3658 CrossRef CAS.
- S. P. Fletcher, F. Dumur, M. M. Pollard and B. L. Feringa, Science, 2005, 310, 80–82 CrossRef CAS PubMed.
- T. Ye, A. S. Kumar, S. Saha, T. Takami, T. J. Huang, J. F. Stoddart and P. S. Weiss, ACS Nano, 2010, 4, 3697–3701 CrossRef CAS PubMed.
- T. Kudernac, N. Ruangsupapichat, M. Parschau, B. MacIá, N. Katsonis, S. R. Harutyunyan, K. H. Ernst and B. L. Feringa, Nature, 2011, 479, 208–211 CrossRef CAS PubMed.
- H. Jian and J. M. Tour, J. Org. Chem., 2003, 68, 5091–5103 CrossRef CAS PubMed.
- Y.-L. Zhao, W. Lin, K. Jitapunkul, R. Zhao, R.-Q. Zhang and M. A. Van Hove, ACS Omega, 2022, 7, 35159–35169 CrossRef CAS PubMed.
- J. Conyard, K. Addison, I. A. Heisler, A. Cnossen, W. R. Browne, B. L. Feringa and S. R. Meech, Nat. Chem., 2012, 4, 547–551 CrossRef CAS PubMed.
- K. Kuntze, D. R. S. Pooler, M. Di Donato, M. F. Hilbers, P. van der Meulen, W. J. Buma, A. Priimagi, B. L. Feringa and S. Crespi, Chem. Sci., 2023, 14, 8458–8465 RSC.
- E. M. Arpa, S. Stafström and B. Durbeej, Chem.–Eur. J., 2024, 30, e202303191 CrossRef CAS PubMed.
- M. Quick, F. Berndt, A. L. Dobryakov, I. N. Ioffe, A. A. Granovsky, C. Knie, R. Mahrwald, D. Lenoir, N. P. Ernsting and S. A. Kovalenko, J. Phys. Chem. B, 2014, 118, 1389–1402 CrossRef CAS PubMed.
- R. Zhao, Y. L. Zhao, F. Qi, K. E. Hermann, R. Q. Zhang and M. A. Van Hove, ACS Nano, 2018, 12, 3020–3029 CrossRef CAS PubMed.
- P.-T. Chiang, J. Mielke, J. Godoy, J. M. Guerrero, L. B. Alemany, C. J. Villagómez, A. Saywell, L. Grill and J. M. Tour, ACS Nano, 2012, 6, 592–597 CrossRef CAS PubMed.
- N. Koumura, R. W. J. Zijistra, R. A. Van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed.
- D. Roke, M. Sen, W. Danowski, S. J. Wezenberg and B. L. Feringa, J. Am. Chem. Soc., 2019, 141, 7622–7627 CrossRef CAS PubMed.
- D. R. S. Pooler, A. S. Lubbe, S. Crespi and B. L. Feringa, Chem. Sci., 2021, 12, 14964–14986 RSC.
- B. Oruganti, C. Fang and B. Durbeej, Phys. Chem. Chem. Phys., 2015, 17, 21740–21751 RSC.
- L. Greb and J.-M. Lehn, J. Am. Chem. Soc., 2014, 136, 13114–13117 CrossRef CAS PubMed.
- J. Lehn, Chem.–Eur. J., 2006, 12, 5910–5915 CrossRef CAS PubMed.
- L. Greb, A. Eichhöfer and J. Lehn, Angew. Chem., 2015, 127, 14553–14556 Search PubMed.
- Y. Wang, T. Zhou, S. Ruan, H. Feng, W. Bi, J. Hu, T. Chen, H. Liu, B. Yuan and N. Zhang, Nano Lett., 2022, 22, 6622–6630 CrossRef CAS PubMed.
- W. A. Sokalski, R. W. Góra, W. Bartkowiak, P. Kobyliński, J. Sworakowski, A. Chyla and J. Leszczyński, J. Chem. Phys., 2001, 114, 5504–5508 CAS.
- X. Liu, W. Zhang, Y. Wang, Z.-X. Zhang, L. Jiao and Q. Liu, J. Am. Chem. Soc., 2018, 140, 6873–6882 CAS.
- A. Houmam, Chem. Rev., 2008, 108, 2180–2237 CAS.
- E. Böhler, J. Warneke and P. Swiderek, Chem. Soc. Rev., 2013, 42, 9219–9231 Search PubMed.
- S. Klaiman and L. S. Cederbaum, Angew. Chem., 2015, 127, 10616–10619 Search PubMed.
- Y. Zhang, H. Kersell, R. Stefak, J. Echeverria, V. Iancu, U. G. E. Perera, Y. Li, A. Deshpande, K.-F. Braun and C. Joachim, Nat. Nanotechnol., 2016, 11, 706–712 CAS.
- M. Alemani, S. Selvanathan, F. Ample, M. V Peters, K.-H. Rieder, F. Moresco, C. Joachim, S. Hecht and L. Grill, J. Phys. Chem. C, 2008, 112, 10509–10514 CAS.
- K. Huang, L. Leung, T. Lim, Z. Ning and J. C. Polanyi, J. Am. Chem. Soc., 2013, 135, 6220–6225 CAS.
- G. D. Harzmann, R. Frisenda, H. S. J. van der Zant and M. Mayor, Angew. Chem., Int. Ed., 2015, 54, 13425–13430 CAS.
- S. Mangel, M. Skripnik, K. Polyudov, C. Dette, T. Wollandt, P. Punke, D. Li, R. Urcuyo, F. Pauly and S. J. Jung, Phys. Chem. Chem. Phys., 2020, 22, 6370–6375 CAS.
- A. Jaros, E. F. Bonab, M. Straka and C. Foroutan-Nejad, J. Am. Chem. Soc., 2019, 141, 19644–19654 CAS.
- Y. Li, W. Wang and F. Liu, Chem.–Eur. J., 2019, 25, 4194–4201 CAS.
- L. Liu, W.-H. Fang and T. J. Martinez, J. Am. Chem. Soc., 2023, 145, 6888–6898 CrossRef CAS PubMed.
- J. Lv, Y. Ding, P. Sun, J. Wang, T. Chen, X. Zhao, C. Fang and L. Wang, Chem. Phys. Lett., 2023, 811, 140245 CrossRef CAS.
- M. Wang, X. He, M. Taylor, W. Lorpaiboon, H. Mun and J. Ho, J. Chem. Theory Comput., 2023, 19, 5036–5046 CrossRef CAS PubMed.
- I. F. Galván, M. G. Delcey, T. B. Pedersen, F. Aquilante and R. Lindh, J. Chem. Theory Comput., 2016, 12, 3636–3653 CrossRef PubMed.
- M. G. Delcey, T. B. Pedersen, F. Aquilante and R. Lindh, J. Chem. Phys., 2015, 143(4) CrossRef CAS PubMed.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- E. I. Alanís-Manzano and A. Ramírez-Solís, Comput. Theor. Chem., 2021, 1200, 113230 Search PubMed.
- J. K. Cooper, C. D. Grant and J. Z. Zhang, Rep. Theor. Chem., 2012, 11–19 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CAS.
- I. V Rostov, R. D. Amos, R. Kobayashi, G. Scalmani and M. J. Frisch, J. Phys. Chem. B, 2010, 114, 5547–5555 Search PubMed.
- M. Walker, A. J. A. Harvey, A. Sen and C. E. H. Dessent, J. Phys. Chem. A, 2013, 117, 12590–12600 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, et al., Gaussian 16, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- A. Kazaryan, J. C. M. Kistemaker, L. V Schafer, W. R. Browne, B. L. Feringa and M. Filatov, J. Phys. Chem. A, 2010, 114, 5058–5067 CAS.
- M. Korivand and M. Zamani, J. Solid State Chem., 2021, 294, 121851 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06792d |
| This journal is © The Royal Society of Chemistry 2025 |