
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBis-thiourea and macrocyclic polyamines as binary organocatalysts for the ROP of lactide†
Assunta
D'Amato‡
 a,
Maria
Voccia‡
b,
Filippo
Bruno
a,
Sara
D'Aniello
a,
Lucia
Caporaso
*a,
Francesco
De Riccardis
a,
Irene
Izzo
a,
Giorgio
Della Sala
*a and
Mina
Mazzeo
*a
a,
Maria
Voccia‡
b,
Filippo
Bruno
a,
Sara
D'Aniello
a,
Lucia
Caporaso
*a,
Francesco
De Riccardis
a,
Irene
Izzo
a,
Giorgio
Della Sala
*a and
Mina
Mazzeo
*a
aDipartimento di Chimica e Biologia “Adolfo Zambelli”, University di Salerno, via Giovanni Paolo II, 132, 84084 Fisciano, SA, Italy. E-mail: lcaporaso@unisa.it; gdsala@unisa.it
bDipartimento di Scienza dei Materiali, Università di Milano-Bicocca, via R. Cozzi, 55, I-20125 Milano, Italy
First published on 27th September 2024
Abstract
New binary catalysts formed by 1,1′-(propane-1,3-diyl)bis(3-(3,5-bis(trifluoromethyl)phenyl)thiourea) and a series of specifically designed flexible polyaza-macrocycles, prepared by a convenient iterative solid-phase synthesis and reduction, have been investigated for the ring opening polymerization (ROP) of L-lactide. These systems, in the presence of benzyl alcohol as initiator, showed high activity, delivering polylactides with high chain-end fidelity and controlled molecular weights with narrow dispersities. A detailed study of the structure–activity relationship for various polyaza-macrocycles, with different sizes and N-side chains, was performed. The best results were exhibited by cyclen derivatives, improving the performances achieved by the well-established triazacyclononane (TACN) co-catalyst. DFT (density functional theory) calculations, performed on each putative polyazamacrocycle/benzyl alcohol complex, assessed both the ring size and the N-alkyl steric hindrance roles on the initiator activation, providing a rationale for the activity scale experimentally observed for macrocyclic polyamines.
Introduction
In the last two decades, organocatalytic ring-opening polymerization (ROP) of various monomers, such as cyclic esters,1–3 epoxides in combination with CO2 (ref. 4–6) or anhydrides,7–11 and N-carboxy anhydrides,12 has emerged as an attractive route for the synthesis of functional, biocompatible, and biodegradable materials.13–16The high functional group tolerance of many organic catalysts and their ability to efficiently control macromolecular parameters, such as the structure of chain end groups as well as the molecular masses and their distribution, have provided new opportunities for macromolecular synthesis and design.11,17–20
Moreover, organic catalysts offer the additional advantage of lower toxicity and ease of removal in comparison to many metal-based catalysts, representing valuable tools to produce polymeric materials for biomedical and food packaging applications.
Different families of organocatalysts have been effectively employed in ROP, including Brønsted/Lewis acids and bases such as amines,21–24 ureas,25–27 phosphoric acids,28 phosphazenes,2,10,29 and N-heterocyclic carbenes (NHCs).30–32
In particular, bicomponent catalysts formed by an H-bond donating (thio)urea in combination with an H-bond accepting organic base are among the most efficient organocatalysts for the ROP of cyclic esters and carbonates.16,26,27,33
In the presence of alcohol initiators, they generate bifunctional cooperative systems where the amine base was proposed to serve as the H-bond acceptor, activating the alcohol in the initiation step (and the alcoholic growing chain during propagation), while the Lewis acidic thiourea activates the monomer.26
Specifically, bis-(thio)ureas, as H-bond donating co-catalysts, revealed enhanced catalytic activity in comparison to the related mono-(thio)ureas in the ROP of L-lactide, thanks to the autoactivation phenomena between the (thio)urea pockets (Fig. 1).34
 | ||
| Fig. 1 Possible mode of bis-thiourea activation. | ||
The cooperative mechanism reaches its maximum efficiency when the pKa of the base and (thio)urea are closely matched.21 A strategy adopted for the amplification of catalytic activity is to expand the number of basic amine sites.
In their recent studies, Coady and Hedrick35 demonstrated that polyamines characterized by a N–N interatomic distance close to 3.0 Å, and an orientation angle of their respective lone pairs close to 50°, are the most efficient bases to promote this process and at the same time suppressing deleterious transesterification reactions. Such structural features enable chelative association of the nitrogen atoms with the hydroxyl proton of the alcohol initiator as well as of the growing chain, enhancing OH nucleophilicity toward the incoming monomer. Molecules that were found to combine these principles were sparteine and 1,4,7-trimethyl-1,4,7-triazacyclononane (Me3TACN), in which the reduced conformational freedom properly pre-orients the nitrogen atom lone pairs for the activation of the hydroxyl group. Despite the high potentials of cyclic polyamines as the Lewis base of bicomponent organocatalysts, a systematic study of the effect of their structural modifications on the catalytic activity has never been carried out.
Recently, we introduced a convenient method to prepare polyaza-macrocycles with different ring sizes and side chains through reduction of the corresponding cyclic oligoamides, readily accessible through solid-phase synthesis.36
Inspired by the above considerations, in this work we decided to investigate the behaviour of different size polyamine macrocycles, bearing benzyl or methyl substituents (Fig. 2), in combination with the bis-thiourea bTU as catalysts for the polymerization of L-lactide (L-LA).
 | ||
| Fig. 2 Structures of the bis-thiourea and macrocyclic polyamine catalysts. | ||
The number of nitrogen donor sites, the nature of different substituents and the conformational flexibility of the polyaza-macrocycles are parameters that have been screened to elucidate their effect on the catalytic activity. For our comparative studies, the bis-thiourea bTU was selected as the H-bond donating co-catalyst, since it showed enhanced catalytic activity compared to the related mono-thiourea in the ROP of L-lactide.34,37 DFT studies were also performed to rationalize the effect of the structural modifications of cyclic polyamines on their catalytic behaviour.
Experimental
Materials and methods
Moisture and air-sensitive materials were manipulated under nitrogen using Schlenk techniques or an MBraun Labmaster glovebox. The used glassware was dried in an oven at 120 °C and subsequently subjected to vacuum-nitrogen cycles. Toluene and methanol were refluxed over Na and distilled under nitrogen. Tetrahydrofuran (THF) was refluxed over Na and benzophenone and distilled under nitrogen. Deuterated solvents were purchased from Sigma-Aldrich and dried over activated 3 Å molecular sieves prior to use.All the reagents used for the synthesis of the compounds and 1,3-diaminopropane, 3,5-bis(trifluoromethyl)phenyl isothiocyanate were purchased from Sigma-Aldrich Merck. All the other reagents and solvents were purchased from Sigma-Aldrich Merck and used without further purification.
Instruments and measurements
Synthetic procedures
Me 4 cyclen 50 and Me4cyclam51 were obtained from commercially available cyclen and cyclam respectively, as previously described in the literature.
Bn 3 TACN:361H NMR (400 MHz, CDCl3) δ: 7.40–7.30 (15H, m), 5.54 (3H, d, J 14.4 Hz), 4.58 (3H, d, J 15.5 Hz), 4.21 (3H, d, J 14.4 Hz), 3.73 (3H, d, J 15.5 Hz).
Bn 4 cyclen:361H NMR (600 MHz, CDCl3) δ: 7.32–7.08 (20H, m), 5.56 (2H, d, J 14.4 Hz), 5.41 (2H, d, J 14.9 Hz), 4.45 (2H, d, J 17.2 Hz), 4.38 (2H, d, J 17.2 Hz), 4.34 (2H, d, J 17.2 Hz), 3.74 (2H, d, J 14.4 Hz), 3.50 (2H, d, J 14.9 Hz), 3.49 (2H, d, J 17.2 Hz).
Bn 6 hexacyclen:361H NMR (400 MHz, CDCl3, complex mixture of rotamers) δ: 7.40–7.04 (30H, m), 4.86–3.25 (24H, m).
Me 4 cyclen:521H-NMR (400 MHz, CDCl3) δ: 2.20 (12H, s), 2.49 (16H, s).
Me 4 cyclam:511H NMR (400 MHz, CDCl3) δ: 1.64 (4H, q, J 7 Hz), 2.20 (12H, s), 2.43 (16H, m).
Results and discussion
Polymerization of lactide
Initially, N-benzyl polyaza-macrocycles (n = 3, 4, 6, Fig. 2), in combination with one equivalent of bis-thiourea bTU, were selected to investigate the effect of the size of the macrocycle on the catalytic activity in the polymerization of L-lactide (Scheme 1).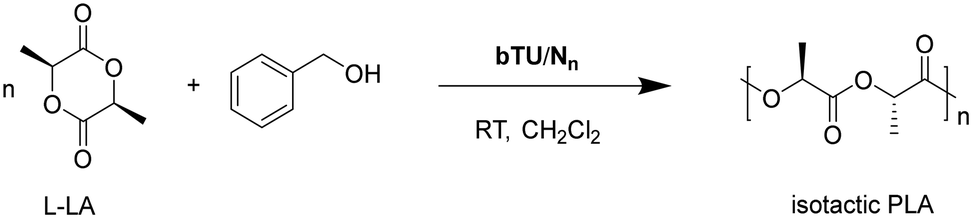 | ||
| Scheme 1 Polymerization of L-lactide (L-LA) by different size polyazamacrocycles (Nn)/bTU. | ||
The polymerization tests were performed in CH2Cl2 (1 M) at 25 °C with 2.5% catalyst loading of macrocyle/bTU, using benzyl alcohol as initiator. The isolated polymers were characterized by 1H NMR, GPC and MALDI-ToF-MS analysis. Representative results are reported in Table 1.
| Runa | N n | Solvent | t (min) | Convc (%) | M nGPC (kDa) | M thn (kDa) | Đ |
|---|---|---|---|---|---|---|---|
| a All reactions, except run 4 and 5, were conducted at 25 °C with [L-LA] = 1 M, CH2Cl2 = 0.7 mL, Nn = bTU = 17.5 μmol; BnOH 7 μmol. b N n = bTU = 8.8 μmol; BnOH 3.5 μmol. c Conversions of LA were determined by 1H NMR. d Experimental MnGPC (kDa) (corrected using factor of 0.58) and Đ values were determined by GPC analysis using polystyrene standards. e M thn (g mol−1) = MMLA × ([monomer]0/[BnOH]0) × LA conversion. f [rac-LA] = 1 M. g T = 80 °C. | |||||||
| 1 | Bn 3 TACN | CH2Cl2 | 6 | 44 | 5.9 | 6.3 | 1.17 |
| 2 | Bn 4 cyclen | CH2Cl2 | 6 | 95 | 12.2 | 13.7 | 1.14 |
| 3 | Bn 6 hexacyclen | CH2Cl2 | 30 | — | — | — | — |
| 4b | Bn 3 TACN | CH2Cl2 | 30 | 13 | 7.4 | 3.0 | 1.14 |
| 5b | Bn 4 cyclen | CH2Cl2 | 30 | 50 | 13.5 | 14.4 | 1.11 |
| 6b | Bn 4 cyclen | CH2Cl2 | 75 | >99 | 29.7 | 28.8 | 1.13 |
| 7f | Bn 4 cyclen | CH2Cl2 | 75 | 98 | 26.3 | 28.2 | 1.12 |
| 8 | Bn 4 cyclen | THF | 30 | 2 | — | — | — |
| 9g | Bn 4 cyclen | Toluene | 30 | — | — | — | — |
| 10 | Me 4 cyclen | CH2Cl2 | 2 | 94 | 24.5 | 27.1 | 1.07 |
| 11 | Me 4 cyclam | CH2Cl2 | 2 | 40 | 11.5 | 9.8 | 1.07 |
At a monomer-to-initiator ratio of 100 ([L-LA]/[BnOH] = 100), a 44% conversion of lactide was achieved after 6 min (run 1, Table 1). The activity showed by Bn3TACN/bTU was lower than that previously reported with the commercial cyclic amine 1,4,7-trimethyl-1,4,7-triazacyclononane (Me3TACN). Me3TACN gave, under the same reaction conditions, 89% conversion of the monomer.34
The reduced activity of Bn3TACN/bTU was tentatively attributed to the higher steric hindrance created by the benzyl groups on the nitrogen atoms in comparison to the methyl groups of the Me3TACN.
Gratifyingly, under the same reaction conditions, Bn4cyclen/bTU led to quantitative conversion of the monomer after 6 minutes (run 2, Table 1), showing a higher activity than triazacyclononanes.
This could be a consequence of reduced steric encumbrance due to the larger dimension of the macrocycle and/or to a higher basicity of the cyclen structure because of the presence of an additional H-bond acceptor nitrogen atom.
Interestingly, when the larger Bn6hexacyclen was tested under the same reaction conditions, no conversion of L-LA was observed (run 3, Table 1), even after prolonged times (24 hours).
The study proceeded halving the amount of catalyst (L-LA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :polyaza-macrocycles:bTU = 80:1:1) and of the alcohol initiator (in amounts comparable to those used for metal catalysts). Both systems remained highly effective at lower concentrations, and the trend of activity previously observed between the two catalysts was maintained (runs 4 and 5, Table 1), confirming Bn4cyclen/bTU as the best catalyst (with good conversion after only 30 min and full conversion after 90 min). The same activity was observed with the racemic substrate rac-LA (run 7, Table 1).
:polyaza-macrocycles:bTU = 80:1:1) and of the alcohol initiator (in amounts comparable to those used for metal catalysts). Both systems remained highly effective at lower concentrations, and the trend of activity previously observed between the two catalysts was maintained (runs 4 and 5, Table 1), confirming Bn4cyclen/bTU as the best catalyst (with good conversion after only 30 min and full conversion after 90 min). The same activity was observed with the racemic substrate rac-LA (run 7, Table 1).
The solvent optimization runs (entries 6 and 7, Table 1) demonstrated the effect of the medium on the reactions. When the selected solvent changes from dichloromethane to THF or toluene, the catalytic activity of Bn4cyclen/bTU was suppressed, and no conversion was observed even after prolonged reaction times and at temperatures up to 80 °C. These results are not surprising, considering the impact of the solvent on the hydrogen bonding interactions involved in the activation of both the monomer and the alcohol.53,54 In this respect, THF may compete with the monomer in the coordination to the thiourea, whereas the poor conversion achieved in nonpolar toluene is possibly due to its tendency to stabilize an unreactive thiourea-polyamine complex.55,56
More mechanistic information was provided by kinetic studies performed on both the catalytic systems Bn3TACN/bTU and Bn4cyclen/bTU in CD2Cl2 at 25 °C. The conversions of the monomer were monitored by 1H NMR spectroscopy for individual runs at different intervals. For both systems, L-LA polymerizations obeyed first-order kinetics in the monomer with instantaneous initiation (Fig. 3).
 | ||
| Fig. 3 Kinetic plot for ROP of L-LA promoted by Bn3TACN/bTU (N3) and Bn4cyclen/bTU (N4) depicting a reaction order of unity with respect to the monomer concentration. Pseudo first-order rate constants are: kapp = 0.0051 min−1 (R2 = 0.998) for Bn3TACN and kapp = 0.0089 min−1 for Bn4cyclen (R2 = 0.997). | ||
The apparent kinetic constants were 5.1 × 10−3 min−1 and 8.9 × 10−3 min−1 for Bn3TACN and Bn4cyclen respectively, and they well correlated with the activities observed in the polymerization runs reported in Table 1.
The results obtained suggest that the catalytic activity is strongly dependent on the size of the macrocycle and the type of N-alkyl groups.
As regards to the role of alkyl substituents, their steric hindrance seems to compromise the performance of the base.
To confirm this observation, we purposely synthetized the Me4cyclen macrocycle in which the benzyl groups were substituted by the less sterically encumbered methyl groups.
To our pleasure, under the same polymerization conditions described in run 5, the catalytic system Me4cyclen/bTU was significantly more active than Bn4cyclen/bTU (compare run 8 and run 5, Table 1), as the quantitative conversion of the monomer was achieved after only 2 min, preserving the high ability in controlling the molecular masses of the produced polymer.
Finally, a larger cyclic polyamine, namely 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane (Me4cyclam), was synthetized. In this case the activity of Me4cyclam/bTU was slightly lower than that obtained with Bn4cyclen/bTU (see runs 9 and 5, Table 1).
Probably the longer propylene bridge between adjacent nitrogen atoms and the consequent higher macrocyclic conformational freedom less effectively meets the structural requirements for chelation of the hydroxyl protons of the propagating species.
All the polymerizations described exhibited an efficient control of the molecular masses: the experimental values were close to the theoretical ones with dispersities (Đ) lower than 1.14 (Fig. S10 and S11†). The characteristics of a living polymerization were also evidenced by the linear correlation between Mn (measured by 1H NMR) and monomer conversion (see Table S1†).
The initiation efficiency was investigated by 1H NMR analysis of the end-groups of a low molecular weight PLA initiated by benzyl alcohol (run 2, Table 1). The only end-groups observed were the benzyl ester from the initiating alcohol and the ω-hydroxyl chain-ends, which is indicative of one molecule of initiator per polymer chain (Fig. S7†). In fact, the Mthn evaluated by NMR was coherent with the value experimentally measured (10.5 kDa).
Analogously, the matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-ToF MS) analysis showed a symmetric distribution of peaks associated to a single population of polymeric chains (δ m/z = 144) initiated by benzyl alcohol (Fig. 4). The absence of peak multiples of 72 m/z suggests that no post-polymerization transesterification is occurring.
 | ||
| Fig. 4 MALDI-ToF-MS spectrum of PLA obtained with 20 equivalents of L-LA under reaction conditions described in run 1. | ||
The selectivity of these catalytic systems was also evident in the lack of any observable epimerization of the polylactides. Homonuclear decoupled 1H NMR (Fig. S8†) and 13C NMR spectra of PLA samples show only one resonance in the methine region, suggesting that the configuration of the monomer is retained in the polymerization. Thermal analysis performed by DSC on the PLAs produced in runs 1–4 revealed them to be highly isotactic with a melting point (Tm) of 170 °C (Fig. S5a and S6†).
The analysis of the sample obtained for racemic lactide by homonuclear decoupled 1H NMR spectroscopy revealed an atactic microstructure Fig. S9.†
DFT calculations
For the described systems, a bimolecular hydrogen bond activation of both the monomer and the initiating/propagating alcohol was supposed to be active, in analogy with the previously reported studies.46With the aim to rationalize the experimentally observed relative activity, the interaction between the benzyl alcohol and the polyaza-macrocycles was studied by DFT calculations.26 The species investigated, TACN, cyclen and hexacyclen, bear benzyl or methyl substituents, as described in Fig. 5.
 | ||
| Fig. 5 Schematic structures of the different polyaza-macrocycles investigated. | ||
As reported in the literature,35 the activation of initiating/propagating alcohol by amine compounds originates from a chelative association of the hydroxyl proton of the alcohol with the amine nitrogen atoms of the macrocycle. The cooperation between the nitrogen atoms in the coordination is known to play a crucial role, making the oxygen of the alcohol more nucleophilic and, consequently, more reactive.21
This cooperation takes place when the rigid macrocycle structure adopts a stable conformation showing N–N distances of about 3 Å with lone pairs oriented around 44° above the N–N plane.35
Looking to triazamacrocycles, we performed calculations on Me3TACN/Alcohol and Bn3TACN/Alcohol complexes starting from the corresponding macrocycle and benzyl alcohol at infinite distance.
Calculations showed that the adduct is in a thermodynamic equilibrium with the reagents for both Me3TACN/Alcohol and Bn3TACN/Alcohol since ΔG is 0.3 kcal mol−1 and 1.5 kcal mol−1, respectively (see Fig. 6).
 | ||
| Fig. 6 Free energies (kcal mol−1) of the adduct formation obtained by the corresponding benzyl alcohol and macrocycle at infinite distance. | ||
Previously reported X-ray analysis of Na+ and H+ complexes of Bn3TACN and Bn4cyclen in the solid state revealed “closed” structures, with the N-benzyl moieties arranged almost perpendicularly to the mean plane of the macrocycle wrapping around the cation.36 In contrast, calculations show that on the respective benzyl alcohol adduct, the benzyl rings move on a parallel plane (“open” geometry) minimizing steric repulsions and allowing the hydroxyl proton to interact with greater extent with the nitrogen atoms of the macrocycle. In fact, for Bn3TACN, this “open” geometry is about 2.0 kcal mol−1 more stable than the “closed” one (see Fig. 7).
 | ||
| Fig. 7 The investigated geometries and relative free energy of Bn3TACN/Alcohol complexes. The calculated distances between the three N atoms along with the alcohol proton are 2.12, 2.77 and 2.40 Å for the open geometry and 1.88, 3.14 and 3.16 Å for the close geometry. | ||
In analogy to what already reported for Me3TACN, showing a N–N distance of 2.96 Å and an orientation angle between H⋯N⋯N atoms of 55° and 42°,35 the calculations show that for Bn3TACN/Alcohol Open, the distance calculated between two adjacent nitrogen atoms (2.98 Å) and the angles between the H atom of alcohol and the two involved N atoms (53° and 45°) were values useful to favour an effective interaction.
Interestingly, although the conformation of macrocycles in the two complexes is similar, the formation of the Bn3TACN/Alcohol is less favoured by 1.2 kcal mol−1 than that of Me3TACN/Alcohol. From a deeper geometry analysis, it is shown that steric repulsion between the N-benzyl substituents and the macrocycle's backbone pushes the –CH2-benzyl side chains closer to the alcohol's –CH2–, slightly destabilizing the complex (compare dihedral angles and –CH2–⋯–CH2– distances of the geometries in Fig. 8).
 | ||
| Fig. 8 Comparison of the Me3TACN/Alcohol, Bn3TACN/Alcohol, Me4cyclen/Alcohol, Bn4cyclen/Alcohol structures. For Me3TACN/Alcohol and Bn3TACN/Alcohol the values of the main dihedral angles (d) are reported. | ||
To reduce this steric interaction, the alcohol moves away from the nitrogen atoms (compare the distances N⋯H–O for Me3TACN/Alcohol and for Bn3TACN/Alcohol reported in Table S2 and Fig. S15†), and consequently the activation of the oxygen of the benzyl alcohol should be less favoured for the Bn3TACN/Alcohol complex compared to the Me3TACN/Alcohol one, with the latter bearing the less encumbered methyl substituents.
The distortion energies (ΔE kcal mol−1)29 calculated for the formation of the adduct show that no important geometric deformations are generated (the ΔE is less than 1 kcal mol−1), confirming that in the case of Bn3TACN the alcohol is less well accommodated within the macrocycle for steric reasons and therefore interacts less effectively with the nitrogen atoms. As confirmation, the Mulliken population analysis performed on these complexes (see Table 2) shows a greater negative charge on the oxygen atom for Me3TACN/Alcohol compared to Bn3TACN/Alcohol, in line with the higher experimental activity of the former.21,27,35
| Species | Charge on O | Charge on H | Charge on N1, N2, N3, N4a |
|---|---|---|---|
| a Values related to Me4cyclen/Alcohol and Bn4cyclen/Alcohol. | |||
| Alcohol | −0.275 | 0.173 | |
| Me 3 TACN/Alcohol | −0.321 | 0.215 | −0.354, −0.319, −0.330 |
| Bn 3 TACN/Alcohol | −0.313 | 0.216 | −0.324, −0.311, −0.341 |
| Me 4 cyclen/Alcohol | −0.337 | 0.199 | −0.353, −0.316, −0.299, −0.312 |
| Bn 4 cyclen/Alcohol | −0.334 | 0.206 | −0.350, −0.306, −0.298, −0.307 |
Moving to cyclen derivatives, we performed calculations on Me4cyclen/Alcohol and Bn4cyclen/Alcohol complexes. As observed for Bn3TACN, the conformation of Bn4cyclen is different depending on whether it hosts a small cation (“closed” conformation)36 or a larger neutral molecule as benzyl alcohol (“open” conformation).
In both the Me4cyclen/Alcohol and Bn4cyclen/Alcohol complexes the calculated geometries are compatible with the aforementioned cooperation of the nitrogen atoms (distances between the nitrogen atoms of 3.1 Å and angle values of 60° and 38°, for both adducts). Notably, the shorter OH⋯N distance in the case of tetrazamacrocycles, compared to the corresponding triazamacrocycles, (see Fig. S15†) agrees with their higher experimental activity.21
By comparing Me4cyclen/Alcohol with Bn4cyclen/Alcohol, calculations show that the methyl-substituted complex is about 1 kcal mol−1 more stable than the benzyl-substituted one for the same steric reasons discussed for the TACN analogues (see Fig. S15†). Consequently, for Me4cyclen/Alcohol the N⋯H–O interaction is more effective (see Tables 2 and S2†), leading a more negative charge on the corresponding alcohol oxygen, in agreement with the greater activity observed experimentally.
Interestingly, by comparing the ΔG of formation of the triaza- and tetraza-macrocyles complexes with benzyl alcohol, it emerges that the Me4cyclen/Alcohol and Bn4cyclen/Alcohol are more favored than Me3TACN/Alcohol by 2.3 and 1.2 kcal mol−1, respectively. Interestingly, the calculated distortion energy is almost zero for both complexes, indicating that no deformation of the macrocycles in the interaction with the alcohol occurs.
Accordingly, the O–H⋯N distance decreases from 2.00 to 1.96 and to 1.92 Å moving from Me3TACN/Alcohol to Bn4cyclen/Alcohol and Me4cyclen/Alcohol, respectively (Table 3), and the Mulliken population analysis also shows a greater negative charge on the oxygen for both Me4cyclen/Alcohol and Bn4cyclen/Alcohol when compared to Me3TACN/Alcohol (see Tables 2 and S2† and Fig. 8). In conclusion, these results agree with the higher experimental activity observed for macrocycles with methyl substituents compared to macrocycles with benzyl substituents, as well as with the higher experimental activity observed for the Me4cyclen/Alcohol and Bn4cyclen/Alcohol compared with that of the analogous N3 co-initiators.
| Adducts | O–H⋯N1 in Å | O–H⋯N2 in Å | O–H⋯N3 in Å | O–H⋯N4 in Å |
|---|---|---|---|---|
| Me 3 TACN/Alcohol | 2.00 | 2.45 | 2.85 | |
| Bn 3 TACN/Alcohol | 2.12 | 2.40 | 2.77 | |
| Me 4 cyclen/Alcohol | 1.92 | 2.73 | 2.99 | 3.93 |
| Bn 4 cyclen/Alcohol | 1.96 | 2.75 | 2.86 | 3.82 |
Since both the calculations and the experimental results suggest that the activation of benzyl alcohol is affected by both the size of the macrocycle and the nature of its substituents, we decided to perform calculations on the six-membered complex Bn6hexacyclen/Alcohol in an attempt to rationalize the unexpected inactivity of Bn6hexacyclen in the L-lactide polymerization.
Interestingly, from the analysis of the optimized geometry of the macrocycle alone, it is shown that the conformation of Bn6hexacyclen is not compatible with the demanded nitrogen atom cooperation for the alcohol activation, since the N–N distances are equal to 3.82 Å and the angle values are 96° and 25° (see Fig. 9). Therefore, to adopt the proper catalytically active conformation, Bn6hexacyclen is forced to an unstable geometry. As a matter of fact, the energy of its complex with benzyl alcohol is approximately 3 kcal mol−1 from the reactants at infinite distance, with a macrocycle distortion energy of 1.2 kcal mol−1.
 | ||
| Fig. 9 Structures of A) the Bn6hexacyclen macrocycle and B) the Bn6hexacyclen/Alcohol complex. | ||
A detailed analysis of the optimized geometry of the Bn6hexacyclen macrocycle reveals that the nitrogen atoms are positioned alternately above and below the macrocycle plane and that the aromatic hydrogens interact with the N atoms stabilizing the structure (see Fig. 9A).
In the formation of the complex with the alcohol, one of these interactions is lost to be replaced by the hydrogen bond interaction with OH, leading to destabilization (see Fig. 9B).
Furthermore, whereas all N-substituents of TACN and cyclen derivatives are oriented out of the plane of the cycle; in the case of the Bn6hexacyclen macrocycle, the substituents are approximately lying in the plane. Consequently, the benzyl substituents approach the nitrogen by interacting with the alcohol, crowding the catalytic space where the reaction with the monomer will take place. This could make the reaction less kinetically favoured for steric reasons (see Fig. 9B), in line with experimental kinetics studies, suggesting the participation of both thiourea and macrocycle in the decisive rate determining transition state.
Moreover, we believe that additional factors may contribute to the reduced catalytic activity in presence of Bn6hexacyclen. Since thiourea can compete with alcohol in forming a host–guest complex with the macrocycles, thus deactivating the reaction, we conducted energy calculations for the formation Bn3TACN/bTU, Bn4cyclen/bTU, and Bn6hexacyclen/bTU (see Fig. 10).
 | ||
| Fig. 10 Gibbs energy of the adduct formation obtained by A) Bn3TACN/bTU, B) Bn4cyclen/bTU, and C) Bn6hexacyclen/bTU macrocycle at infinite distance. | ||
Interestingly, all three macrocycles form thermodynamically more favorable complexes with thiourea than with benzyl alcohol. Notably, the Bn6hexacyclen/bTU complex is approximately 2 kcal mol−1 more stable than the corresponding Bn3TACN/bTU and Bn4cyclen/bTU complexes (compare the free energies of formation for the macrocycle/bis-thiourea complexes reported in Fig. 10 with the corresponding free energies of formation for macrocycle/alcohol complexes reported in Fig. 6, Bn3TACN/Alcohol, Bn4cyclen/Alcohol, and Bn6hexacyclen/Alcohol). This result adds another piece to the overall picture of understanding the experimentally observed inactivity with Bn6hexacyclen.
To summarize, Bn6hexacyclen shows a conformation not compatible with the activation of the benzyl alcohol involving the cooperation of more nitrogen atoms. In fact, the energy formation of the complex with benzyl alcohol is higher than that observed for the corresponding triaza- and tetraza-derivatives by 2–3 kcal mol−1, due to both the distortion of the macrocycle in the complex and the loss of a favourable interaction between the benzyl ortho-hydrogen and the adjacent nitrogen atom of the macrocycle. Moreover, the presence of the benzyl rings in the plane of the macrocycle suggests that the reaction between the alcohol and monomer could be kinetically unfavoured for steric reasons. Finally, the high stability of the host–guest macrocycle–thiourea complex sequesters both the catalytic components making them hardly available for the interaction with the alcohol and the L-lactide, respectively.
The combination of all these factors could explain the lack of catalytic activity experimentally observed for the Bn6hexacyclen macrocycle.
Conclusions
In this work, we report new metal-free catalytic systems for the ring-opening polymerization of L-lactide, formed by a bis-thiourea and conformationally flexible cyclic polyamines with diverse sizes and N-substituents.The polymerization data showed that the catalytic performance of these binary systems is dramatically affected by the structure of the macrocyclic polyamines. In particular, the tetrameric cyclen derived macrocycles were more active than traditionally employed trimeric ones, while the hexameric cyclic polyamine was completely inactive. In addition, N-methyl substituted polyaza-macrocycles were more efficient than the related N-benzyl substituted ones.
DFT calculations rationalized these results, demonstrating that the steric aspects are predominant for the activation of the alcohol by the macrocyclic polyamines. In fact, sterically bulkier substituents at the nitrogen atoms hamper the accessibility of the macrocycle's coordinative niche to the hydroxyl group of the alcohol/growing chains.
In trimeric and tetrameric polyaza-macrocycles, the proper distance between the nitrogen atoms allows their cooperation in the activation of the alcohol hydroxyl proton, that is significantly more efficient when compared to the larger cyclen derivatives.
Differently, for the hexacyclen macrocycle, the most stable conformation does not allow the cooperation of the nitrogen atoms in the activation of the benzyl alcohol. Moreover, a strong interaction between the macrocycle and the thiourea competes with the activation of the alcohol contributing to the inefficiency of the Bn6hexacyclen cycle.
All the catalytic systems active in the ROP of L-lactide revealed a very good control of the polymerization reactions, as neglectable narrowly dispersed polymers of predictable molecular weights were obtained.
In conclusion, cyclic polyamines proved to be useful model compounds for the comprehension of the architectural characteristic of catalysts enabling cooperative activation of the substrates in the ROP polymerization and analogous organocatalyzed reactions. Considering their facile synthesis and the catalytic efficiency, cyclen derivatives have promise to be convenient organocatalysts, as an alternative to the well-established TACN derivatives, in the preparation of polylactide and other biodegradable polyesters.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: G. D. S. and M. M.; methodology: A. D., M. V., F. B. and S. D.; investigation: F. D. R. and I. I.; visualization: F. D. R. and I. I.; data curation: M. M. and L. C.; writing – review and editing: M. M., G. D. S. and L. C. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Federica Santulli for some synthetic work, Dr. Patrizia Iannece for MALDI-ToF analysis, Dr. Patrizia Oliva for NMR technical assistance and Dr. Mariagrazia Napoli for GPC analysis. Financial support from the University of Salerno (FARB), and from PRIN 2020: “Natural Products-Assisted Organic Synthesis” (2020AEX4TA) is acknowledged.Notes and references
- A. Khalil, S. Cammas-Marion and O. Coulembier, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 657–672 CrossRef CAS.
- S. Liu, C. Ren, N. Zhao, Y. Shen and Z. Li, Macromol. Rapid Commun., 2018, 39, 1800485 CrossRef.
- H. Wang, Z. Yao, Z. Li, Y. Zhu, C. Zhang, Z. Luo, T. Guo, Y. Gao, L. Zhang and K. Guo, Eur. Polym. J., 2020, 127, 109570 Search PubMed.
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed.
- M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651–2684 Search PubMed.
- F. Nederberg, B. G. G. Lohmeijer, F. Leibfarth, R. C. Pratt, J. Choi, A. P. Dove, R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2007, 8, 153–160 CrossRef CAS PubMed.
- B. Han, L. Zhang, B. Liu, X. Dong, I. Kim, Z. Duan and P. Theato, Macromolecules, 2015, 48, 3431–3437 CrossRef CAS.
- L. Lin, J. Liang, Y. Xu, S. Wang, M. Xiao, L. Sun and Y. Meng, Green Chem., 2019, 21, 2469–2477 RSC.
- S. Pappuru and D. Chakraborty, Eur. Polym. J., 2019, 121, 109276 CrossRef CAS.
- J. Zhang, L. Wang, S. Liu and Z. Li, J. Polym. Sci., 2020, 58, 803–810 CrossRef CAS.
- D. Ryzhakov, G. Printz, B. Jacques, S. Messaoudi, F. Dumas, S. Dagorne and F. Le Bideau, Polym. Chem., 2021, 12, 2932–2946 RSC.
- W. Zhao, Y. Gnanou and N. Hadjichristidis, Polym. Chem., 2015, 6, 6193 RSC.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- M. Valle, M. Ximenis, X. Lopez de Pariza, J. M. W. Chan and H. Sardon, Angew. Chem., Int. Ed., 2022, 61, e202203043 CrossRef CAS PubMed.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- M. E. G. Mosquera, M. Palenzuela and M. Fernández-Millán, in Noncovalent Interactions in Catalysis, ed. K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, The Royal Society of Chemistry, 2019, 10.1039/9781788016490-00415.
- F. Nederberg, E. F. Connor, M. Moeller, T. Glauser and J. L. Hedrick, Angew. Chem., Int. Ed., 2001, 40, 2712–2715 CrossRef CAS PubMed.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS.
- S. Hu, J. Zhao, G. Zhang and H. Schlaad, Prog. Polym. Sci., 2017, 74, 34–77 CrossRef CAS.
- P. Olsén, K. Odelius, H. Keul and A.-C. Albertsson, Macromolecules, 2015, 48, 1703–1710 CrossRef PubMed.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- B. Lin and R. M. Waymouth, Macromolecules, 2018, 51, 2932–2938 CrossRef CAS.
- L. Zhang, R. C. Pratt, F. Nederberg, H. W. Horn, J. E. Rice, R. M. Waymouth, C. G. Wade and J. L. Hedrick, Macromolecules, 2010, 43, 1660–1664 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS PubMed.
- B. Lin and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 1645–1652 CrossRef CAS.
- I. Jain and P. Malik, Eur. Polym. J., 2020, 133, 109791 CrossRef CAS.
- N. U. Dharmaratne, J. U. Pothupitiya and M. K. Kiesewetter, Org. Biomol. Chem., 2019, 17, 3305–3313 RSC.
- K. Makiguchi, T. Yamanaka, T. Kakuchi, M. Terada and T. Satoh, Chem. Commun., 2014, 50, 2883–2885 RSC.
- L. Zhang, F. Nederberg, R. C. Pratt, R. M. Waymouth, J. L. Hedrick and C. G. Wade, Macromolecules, 2007, 40, 4154–4158 CrossRef CAS.
- O. Coulembier, A. P. Dove, R. C. Pratt, A. C. Sentman, D. A. Culkin, L. Mespouille, P. Dubois, R. M. Waymouth and J. L. Hedrick, Angew. Chem., Int. Ed., 2005, 44, 4964–4968 CrossRef CAS PubMed.
- M. Fevre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142–2172 RSC.
- E. F. Connor, G. W. Nyce, M. Myers, A. Moeck and J. L. Hedrick, J. Am. Chem. Soc., 2002, 124, 914–915 CrossRef CAS PubMed.
- C. Thomas and B. Bibal, Green Chem., 2014, 16, 1687–1699 RSC.
- S. S. Spink, O. I. Kazakov, E. T. Kiesewetter and M. K. Kiesewetter, Macromolecules, 2015, 48, 6127–6131 Search PubMed.
- D. J. Coady, A. C. Engler, H. W. Horn, K. M. Bajjuri, K. Fukushima, G. O. Jones, A. Nelson, J. E. Rice and J. L. Hedrick, ACS Macro Lett., 2012, 1, 19–22 CrossRef CAS PubMed.
- R. Schettini, A. D'Amato, G. Pierri, C. Tedesco, G. Della Sala, O. Motta, I. Izzo and F. De Riccardis, Org. Lett., 2019, 21, 7365–7369 CrossRef CAS PubMed.
- K. V. Fastnacht, S. S. Spink, N. U. Dharmaratne, J. U. Pothupitiya, P. P. Datta, E. T. Kiesewetter and M. K. Kiesewetter, ACS Macro Lett., 2016, 5, 982–986 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B, 1986, 33, 8822 CrossRef PubMed.
- J. P. Perdew, Phys. Rev. B, 1986, 34, 7406 CrossRef PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. R. Ochterski, L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, D. J. DeFrees, J. A. Pople and M. S. Gordon, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. Schaefer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32(7), 1456–1465 CrossRef CAS.
- R. Schettini, A. D'Amato, A. M. Araszczuk, G. Della Sala, C. Costabile, A. M. D'Ursi, M. Grimaldi, I. Izzo and F. De Riccardis, Org. Biomol. Chem., 2021, 19, 7420–7431 RSC.
- A. M. Araszczuk, G. Pierri, R. Schettini, C. Costabile, G. Della Sala, L. Di Marino, C. Tedesco, F. De Riccardis and I. Izzo, Chem. – Eur. J., 2024, 30, e202400904 CrossRef CAS.
- J. Wallick, C. G. Riordan and G. P. A. Yap, J. Am. Chem. Soc., 2013, 135, 14972–14974 CrossRef CAS PubMed.
- E. Evangelio, N. P. Rath and L. M. Mirica, Dalton Trans., 2012, 41, 8010–8021 RSC.
- X.-Q. Xiong, F. Liang, L. Yang, X.-L. Wang, X. Zhou, C.-Y. Zheng and X.-P. Cao, Chem. Biodiversity, 2007, 4, 2791–2797 CrossRef CAS PubMed.
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 13798–13799 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863–7871 CrossRef CAS.
- G. Tárkányi, P. Király, T. Soós and S. Varga, Chem. – Eur. J., 2012, 18, 1918–1922 CrossRef.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00952e |
| ‡ The authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |