
Effect of metal loading sequences in CO2 methanation activity on samarium-doped ceria supported bimetallic catalysts†
Andrew C.
Chien
 *ab and
Corinna C.
Chi
a
*ab and
Corinna C.
Chi
a
aDepartment of Chemical Engineering, Feng Chia University, Taichung 40724, Taiwan. E-mail: andrew@mail.fcu.edu.tw; Fax: +886 424510890; Tel: +886 424517250 ext. 3691
bGreen Energy Development Center, Feng Chia University, Taichung 40724, Taiwan
First published on 21st October 2024
Abstract
A bimetal-supported catalyst, lanthanum and nickel (La and Ni) on samarium-doped ceria (SDC), was synthesized by a sequential microwave heating process for the CO2 methanation reaction. The effect of altering orders of metal addition to SDC on the methanation activity was investigated. The La/Ni/SDC catalyst obtained in co-microwave synthesis (all precursors were added at the same time) had the best methanation activity with a CO2 conversion of approximately 60% at 353 °C, compared to the catalyst that either La or Ni is loaded first on SDC. The better activity of the co-microwave catalyst was attributed to a synergistic function of the carrier and metals due to intimate contact, thus provoking a unique metal–support interaction and optimal CO2 affinity. This was evidenced by surface characterisation including XRD, TEM and XPS analysis, revealing that La/Ni/SDC exhibits low crystallinity in nanoscale size and contains more active oxygen species, as well as Ce3+/Ce4+ redox couples. Besides, working mechanisms of the La/Ni/SDC catalyst were postulated according to in situ infrared studies. The result demonstrated that sequences of metal loading in the synthesis of the catalyst are critical to the metal–support interaction for the methanation activity. Accordingly, this will allow the design of effective bimetallic catalysts capable of conducting potential reactions with optimised synthesis conditions.
1. Introduction
Methanation of carbon dioxide (CO2) is a promising process receiving considerable attention. CO2 methanation produces synthetic methane (CH4) through hydrogenation, giving advantages of direct CO2 mitigation and reduction of energy dependency on fossil fuels.1,2 When the methanation reactant, hydrogen, is supplied from electrolysis of water by renewable energy sources such as solar or wind energy, the whole process becomes potentially sustainable and more attractive. However, CO2 methanation is a mildly exothermic reaction, which is intrinsically favoured at low temperatures.3 Accordingly, activation of the C–O bond at target temperatures and fast enough reaction kinetics are challenges. Designing highly active yet stable catalysts for selective CH4 production would provide a solution to the underlying problem.The development of CO2 methanation catalysts has been largely addressed by previous studies on hydrogenation reactions.4,5 From a mechanistic perspective, reduction of CO2 to CH4 by hydrogen proceeds through an eight-electron reduction process with high-energy barriers. In the past, noble metal-supported materials have been applied and shown to possess good hydrogenation activity. Several group VIII B transition metal catalysts,6–9i.e., iron (Fe), cobalt (Co) and nickel (Ni), are also exploited due to the optimal balance of material cost and reaction activity. In particular, Ni-based catalysts usually have the best activity-to-cost ratio; however, the catalysts require operation at higher temperatures (>350 °C) compared to platinum-group ones. Therefore, the development of highly effective Ni-based catalysts for low temperature CO2 methanation is critical to the advancement of this field.10
On the other hand, supporting materials for metal catalysts also play a significant role in superior methanation activity. Various oxides, such as Al2O3, MgO, ZrO2, TiO2, and CeO2, and carbon (C) are employed.3,11–13 Some oxides are found to increase hydrogenation activity by weakening C–O bonds of CO2 molecules, while others show high CO2 methanation performance due to surface oxygen vacancies. Among these materials, ceria (CeO2) is considered a promising substrate not only because of unique metal–support interactions, but also due to adjustable oxygen vacancies through doping of a trivalent cation such as samarium.14,15 Moderate support–metal interactions allow high dispersion of metal nanoparticles over the support. Oxygen vacancies induce the activation of CO2 molecules on the surface and subsequent dissociation, thus increasing the rate of methanation.16,17
An interesting approach to develop a good methanation catalyst is employing a facile synthesis to make a synergistic combination of an excellent hydrogenation Ni metal and a redox ceria support. Previous studies involving the Ni catalyst demonstrate that addition of ceria effectively activates CO2 to improve the performance of methanation.18–21 The improvement is attributed to surface basicity and partial reduction of the support, which increase surface coverage of acidic CO2 species at low temperatures. Furthermore, it has been reported that incorporation of alkaline earth metals into the Ni/CeO2 catalyst increases alkalinity and generates more oxygen vacancies with higher CO2 methanation performance.13,22–24 However, the activity of the promoted catalyst and the interaction of the promoter, Ni species, and ceria in the methanation reaction need further studies. Moreover, conventional preparation methods used for the synthesis of doped ceria and subsequent metal-supported catalysts need to be optimized. The resulting material could contain a large size distribution of particles and lack reproducibility due to multistep procedures.
In this work, we made a lanthanum and nickel (La and Ni)-supported samarium-doped ceria (SDC) catalyst via a microwave-assisted method. The effect of the combination of the metals with SDC on the methanation activity was investigated. The advantages of employing microwave irradiation in the synthesis are detailed elsewhere,25,26 including short processing time, rapid volumetric heating, and homogeneous temperature distribution. Our results showed that the co-microwave heating synthesis produced a catalyst, La/Ni/SDC, with the best methanation activity. The high catalytic performance was attributed to the unique support–metal interaction arising from both structure and electronic promotion, as characterised by XRD, TEM, TPD, and XPS analysis. In addition, in situ infrared studies were used to characterise reaction intermediates and to elucidate the reaction pathways. This study provides valuable information on the design of bimetallic catalysts for the methanation reaction and optimisation of the synthesis conditions.
2. Experimental
2.1 Catalyst preparation
The catalyst, nickel (Ni)/lanthanum (La) supported by samarium-doped cerium oxide (SDC), was prepared by a microwave-assisted method.27 Three kinds of catalysts were made according to different sequences of precursor addition: (i) mixing Ni, La, and SDC precursors together for microwave irradiation, (ii) mixing Ni and SDC precursors first for irradiation, then adding La to Ni/SDC for irradiation, and (iii) reversing the order of adding Ni and La in (ii). The three kinds of catalysts were denoted as La/Ni/SDC, La–Ni/SDC, and Ni–La/SDC, respectively. The Ni and La precursors were nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O) and lanthanum(III) nitrate hexahydrate (La(NO3)3·6H2O) with a loading of 20% by weight with respect to the SDC. The SDC precursors were samarium(III) nitrate hexahydrate (Sm(NO3)3·6H2O) and cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O), which were mixed uniformly and dissolved in ethanol as a precursor solution. The amounts of the SDC precursors used were calculated according to the molar ratio of Sm0.2Ce0.8O1.9. The precursor solution was mixed well at room temperature and set at pH = 9 by adding sodium hydroxide (10 M NaOH) dropwise as a precipitant. Microwave-assisted synthesis was conducted in a household microwave reactor with an irradiation intensity of 133 W and an on–off cycle of 30 s (10 s on and 20 s off) for 10 min. After the reaction, the mixture was filtered to collect the precipitates, which were washed with ethanol and distilled water, respectively. The sample was dried at 80 °C overnight and calcined at 600 °C in air for 4 h through crystallisation to obtain a fluorite structure. All catalysts were crushed and ground in a mortar for subsequent reaction and characterisation.2.2 Catalytic measurements
The methanation reaction was carried out in a fixed-bed differential reactor (Inconel stainless steel tube: inner diameter 3 mm, length 510 mm) under atmospheric pressure. The catalyst ∼0.1 g was loaded between quartz wool plugs and placed in the middle of the reactor. To evaluate the methanation activity of the catalyst, the reactor was then heated to 500 °C under ambient conditions at a rate of 5.5 °C min−1 with a reactant gas stream of CO2 (1 vol%) and H2 (4 vol%) in Ar (total flow rate: 80 ml min−1) for the temperature-programmed surface reaction (TPSR). The catalyst was pretreated with H2 reduction before the reaction. The effluent gas species were monitored using an online mass spectrometer (Hiden Analytical, HPR 20). The long-term catalytic test was conducted at a reaction temperature of 300 °C. The mass/electron ratio (m/e) in MS were selected for H2 (2), CH4 (15), H2O (18), CO (28), O2 (32) and CO2 (44). The MS response for H2 and CO was obtained by subtracting the intensity of the CH4 fragment from the total MS response at m/e = 2, and subtracting the intensity of the CO2 fragment from the total MS response at m/e =28, respectively. Temperature programmed desorption (TPD) experiments were carried out by (i) exposing the catalyst to a gas mixture containing 10% vol. CO2 in Ar (total flow rates: 50 ml min−1) for 1 h and (ii) increasing the temperature at a rate of 10 °C min−1 from 25 to 600 °C in Ar to study the desorption behaviours. The eluting gas species from the reactor were monitored using an online mass spectrometer.Diffuse reflectance infrared Fourier transform spectroscopy (DRIFT) was used to characterize surface reactions and identify intermediates involved in the course of the reaction. Adequate amounts of the catalyst mixing with potassium bromide (KBr) were loaded in a high-temperature reaction chamber. Before the elevation of temperature, the chamber was purged with argon for 30 min and the background spectra were collected. The background spectra were collected under ambient conditions in pure Ar flow. IR data were presented in the form of absorbance, which is a logarithmic ratio of a series of spectra taken at different temperatures relative to the background one. The catalyst was then pretreated in situ and heated in the reactant flow (CO2/H2 = 1/4 vol%) from room temperature to 500 °C at a rate of 50 °C per 10 min.
2.3 Characterisation
The crystallinity and phase composition of the Ni/La catalyst were characterized using a Bruker D8 Discover diffractometer (Cu-Kα) in the angular range of 20–90°. The surface area and pore volume measurements of the synthesised catalyst were determined by nitrogen adsorption/desorption isotherms at 77 K using an automated gas sorption system (Micromeritics ASAP 2020) using the Brunauer–Emmett–Teller (BET) method. All samples were degassed at a temperature of 300 °C for 3 h prior to measurements. The particle size and morphologies of the catalyst were examined using a high resolution transmission electron microscope (JEOL JEM-2100 Plus) equipped with an energy-dispersive X-ray spectrometer (EDX). The measurement of the elemental composition and oxidation states in the sample was performed on an X-ray photoelectron spectrometer (XPS) (ULVAC-PHI, Versa Probe 4) using a monochromated Al Kα X-ray source (energy: 1486.7 eV). In a typical XPS analysis, about 15 mg of the sample is pressed onto the sample holder. To minimise surface contamination possibly from exposure to the atmosphere, in situ calcination and Ar+ etching were conducted before the measurement.3. Results and discussion
3.1 Crystalline structure and morphology
Fig. 1 shows the XRD patterns of the as-synthesized catalysts. The XRD pattern of SDC on all catalysts matched well with that of Sm0.2Ce0.8O2−δ (JCPDS card 75-0158) and indexed to a cubic fluorite structure.14,28 All catalysts showed no sign of dopant samarium oxide and its metal phase, indicating that the dopant ion is fully substituted in the CeO2 lattice to form a solid solution. The XRD pattern of the co-microwaved La/Ni/SDC catalyst reveals the lower crystallinity of SDC, compared to the other two, La–Ni/SDC and Ni–La/SDC. Also, the supported metals, i.e., Ni and La, were barely observed by XRD. This implies that the co-microwave irradiating synthesis gives rise to highly dispersed species in a very small particle size. In contrast, the La–Ni/SDC and Ni–La/SDC catalysts both showed a NiO phase (JCPDS card 71-1179) at 2θ values of 37.1°, 43.3° and 62.9°, which are indexed to the planes (111), (200) and (220), respectively. Additionally, Ni–La/SDC detected the La2O3 phase (JCPDS card 83-1354) at 2θ values of 44.6° and 52.0°, which are indexed to the (110) and (200) planes, respectively. The average size of NiO and La2O3 species was estimated below 5 nm while that of SDC is around 7–10 nm according to the Debye–Scherrer equation. The reason why the La2O3 phase does not appear in La–Ni/SDC is unknown, which may appear in hydroxide form whose patterns overlap with SDC. | ||
| Fig. 1 XRD patterns of the SDC-supported Ni and La catalysts synthesised in different metal-loading sequences. | ||
Fig. 2 presents the MS profiles of the CH4 product and CO2 conversion from the methanation reaction on the three catalysts during heating to 500 °C. The result shows that the co-microwaved La/Ni/SDC catalyst leads to the best methanation activity, which increases with increasing temperature and reaches the highest CH4 intensity at 353 °C. Above 353 °C, the activity decreased with further temperature elevation. The other two catalysts show a similar trend of methanation activity while the CH4 intensity is much lower in conjunction with shifting of the highest production temperature. The Ni–La/SDC one was shifted down to ∼325 °C while the La–Ni/SDC one was shifted up to ∼364 °C. The CO2 conversion was estimated to be about 60%, 50%, and 40% for the three types of catalysts, La/Ni/SDC, Ni–La/SDC, and La–Ni/SDC, respectively, at the highest CH4 production temperature. In addition, it was found that there is a consumption of H2 and CO2 during heating at about 100–200 °C, while no CH4 is produced. It was attributed to the sudden production of water, probably remaining in the line/tube, thus reducing the intensities of the reactants. Since the amount of gas sucked by the MS is constant, any change in the gas flow could cause interference with species detection. On the other hand, the SDC support was also used for the CO2 methanation activity and was compared. The result shows that SDC initiates the formation of methane at 350 °C, which is much higher than that of the SDC-supported Ni/La catalyst (starting from 200 °C). The CO2 conversion of SDC was below 40% even when the temperature reaches 500 °C.
 | ||
| Fig. 2 (a) MS profiles of CH4 intensity and (b) CO2 conversion in the temperature-programmed surface reaction on the three catalysts; gas reactants, CO2/H2, 1/4 vol% in Ar at a total flow rate of 80 ml min−1. | ||
Fig. 3 presents the infrared spectra of the CO2 methanation reaction on the three SDC-supported Ni and La catalysts. At 300 °C, a strong band at 2350 cm−1 assigned to the gaseous CO2 reactant was shown together with a number of weak product bands on all catalysts. The La/Ni/SDC catalyst gave rise to the strongest gaseous methane signals that are bending (1305 cm−1) and C–H stretching H bonding (3020 cm−1) in Fig. 2a. Two bands at 2170 and 2110 cm−1 assigned to gaseous CO were evident on all catalysts (most noticeable for Ni/La–SDC). CO production was speculated to be due to dissociation of CO2. Other bands at 746 (ν4 sym), 857 (ν2 asym), 1089 (ν1 sym), 1447 (ν3 asym), and 1508–1540 (ν3 asym) cm−1 were attributed to carbonate groups while the ones at 1340 and 1560 cm−1 were attributed to formate species (HCOO−).23,29 These oxygen-containing groups were surface adsorbates, and the intensities of which vary with different active sites on the catalyst. As the temperature increases to 350 °C, the absorption of methane and carbon monoxide both increased, while those of carbonates decrease in conjunction with increasing formate species. This manifests the transformation of carbon species through hydrogenation into methane. The downward bands at 1650 and 3450 cm−1 corresponding to the hydroxyl group (OH) were due to the desorption of adsorbed H2O on the surface due to heating. In situ DRIFT measurements characterized these intermediates and allowed the determination of their relationship with catalytic activity and product distribution, thereby gaining a fundamental understanding of the surface reaction.
 | ||
| Fig. 3 DRIFT spectra of the CO2 methanation reaction at different temperatures: (a) 300 and (b) 350 °C on the three SDC-supported Ni and La catalysts (CO2/H2, 1/4 vol% in Ar at a total flow rate of 80 ml min−1). | ||
Fig. 4 presents the CO2 temperature-programmed desorption profiles (CO2-TPD) on the three catalysts. The CO2-TPD profiles were divided into three temperature zones: low (<200 °C), medium (250–450 °C) and high (>450 °C). The low temperature peak was attributed to desorption from physically adsorbed species and CO2 at weak basic sites, such as the hydroxyl group (–OH) on the surface. The desorption at medium temperatures was mainly resulted from decomposition of monodentate and polydentate carbonates, which are the precursors of formates and methane. The higher temperature desorption was ascribed to desorption from complex carbonates, for example, La2O2CO3, or strongly bonded carbon species at strong basic sites. The results showed that the La/Ni/SDC catalyst desorbs CO2 with three desorption peaks located at 233, 291, and 368 °C in the medium zone. CO2 desorption on La–Ni/SDC occurred in the low to medium zone (left shift), i.e., 317 °C, whereas the desorption with a right shift to high temperature, i.e., 445 °C, was observed for Ni–La/SDC. The desorption amount was evaluated by calculating the area under the TPD profile, revealing that La/Ni/SDC and Ni–La/SDC give roughly a similar area, whereas La–Ni/SDC is the lowest. Nevertheless, the desorption temperature for La/Ni/SDC at the medium zone is lower than that for Ni–La/SDC, which accounts for a better methanation activity on the co-microwave catalyst.
 | ||
| Fig. 4 CO2 temperature programmed desorption (CO2-TPD) profiles on the three SDC-supported bimetallic catalysts. | ||
Surface analyses of the metal and oxygen species present on the three catalysts were performed by X-ray photoelectron spectroscopy (XPS). Fig. 5 shows the core-level Ce 3d, O 1s, Ni 2p, and La 3d XPS spectra of the three fresh catalysts. The composition percentages from the fitted spectra are summarized in Table 1. The result shows that cerium exists mainly as Ce4+ in all the catalysts. The spectra from the Ce4+ chemical state are characterized by the spin orbit photoelectron peaks Ce 3d5/2 and Ce 3d3/2 with multiple components, labeled as v0, v, v′′, and v′′′ and u, u′, u′′, and u′′′, respectively.30,31 Additionally, the components v0, v′ and u′ are referred to the chemical states of Ce3+. The O 1s spectra show two main peaks around 528.6 and 530.5 eV, which were deconvoluted to three species (OI, OII, and OIII). They were attributed to OI lattice oxygen (O2−: 528.5 eV), OII chemisorbed oxygen (O−: 530.1 eV and O2−: 531.1 eV) and OIII carbonate groups (>532.5 eV), respectively. It was observed that La/Ni/SDC generates more OII and OIII species, which are ascribed to active sites responsible for CO2 methanation. The fitted Ni 2p3/2 XPS spectra show two peaks with binding energies at 854.41 and 852.01 eV, confirming the existence of both NiO and metallic Ni on the surfaces of fresh catalysts (Fig. 5c). The La/Ni–SDC one contained the highest content of metallic nickel. With respect to the La 3d5/2 spectra, all the catalysts contained two 3d5/2 peaks, one at ∼834.0 and the other at 838.0 eV. The ΔE values between these two La 3d5/2 peaks were 3.39, 3.97, and 3.72 eV for La/Ni/SDC, La–Ni/SDC and Ni–La/SDC, respectively. This result indicated that there is probably La2O2CO3 existing on La–Ni/SDC while more La(OH)3 are in La/Ni/SDC.22,32
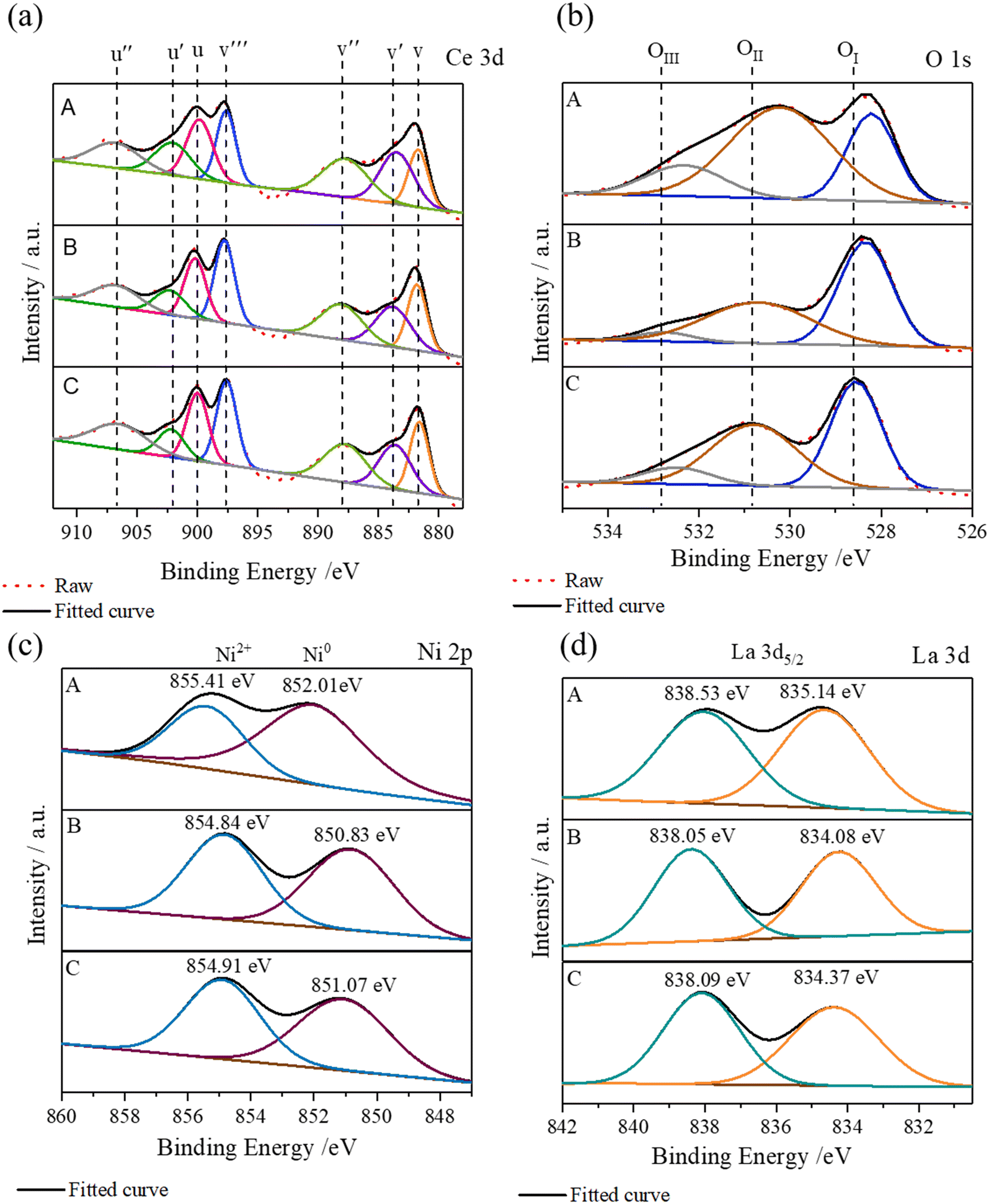 | ||
| Fig. 5 Core level (a) Ce 3d, (b) O 1s, (c) Ni 2p, and (d) La 3d XPS spectra of the three catalysts; A: La/Ni/SDC, B: La–Ni/SDC and C: Ni–La/SDC. | ||
| Sample | Ce3+ (%) | Ce4+ (%) | Ce3+/Ce4+ | OI (%) | OII (%) | OIII (%) | Ni0 (%) | Ni2+ (%) | ΔE (eV) (La3d) |
|---|---|---|---|---|---|---|---|---|---|
| La/Ni/SDC | 29.2 | 70.8 | 0.41 | 26.76 | 58.17 | 15.07 | 67.16 | 32.84 | 3.39 |
| La–Ni/SDC | 24.53 | 85.16 | 0.29 | 53.13 | 41.93 | 4.93 | 51.52 | 48.48 | 3.97 |
| La–Ni/SDC | 22.55 | 77.45 | 0.29 | 45.68 | 44.73 | 9.59 | 51.37 | 48.63 | 3.72 |
The above experimental results demonstrated that the catalytic activity in the CO2 methanation reaction is strongly influenced by the sequences of metal loading on the catalyst and the way the metal and support combined. Present studies show that the SDC-supported bimetallic catalyst gives a substantial increase in activity while the bare SDC is not active. Among the catalysts under investigation, Ni/La/SDC performed much better compared to the other two catalysts, i.e., Ni–La/SDC and La–Ni/SDC. One explanation lies in the difference of the crystalline phase and particle size. The X-ray diffraction (XRD) result showed that the Ni/La/SDC catalyst presents a lower crystalline SDC structure without detection of any metal phase, implying the formation of relatively small crystallites both for the carrier and metal sites. Temperature-programed desorption studies revealed that the Ni/La/SDC catalyst displayed an optimum affinity for CO2. There existed a synergistic effect of the carrier and metal on this catalyst for excellent methanation activity, which could be attributed to the intimate contact between Ni and SDC because of their small size. In contrast, the catalyst prepared in a separate synthesis step produced metals and SDC both in larger sizes according to XRD characterization. The particle size and morphology of the catalyst were analysed by TEM too. Fig. 6 reveals that La/Ni/SDC consists of sphere-shape nanoparticles of NiO and SDC (∼5 nm), which are well-dispersed and distinguished by the (111) crystal plane. In contrast, NiO on Ni–La/SDC agglomerated into a larger cluster. On the other hand, the La species appeared on a big chunk in a polygonal shape, sitting apart from NiO and SDC. The effect of lanthanide on the catalytic behaviour was not evident in terms of structure and morphology.
 | ||
| Fig. 6 TEM images of SDC and the as-synthesised La/Ni/SDC catalyst with particle size distribution (inset). | ||
The other explanation for the activity deviation among the three catalysts was the effect of different active oxygen species and their concentrations. As characterised by the XPS analysis on the surface, the elemental compositions in terms of the main photoelectron lines, Ni 2p3/2, Ce 3d5/2 (V component) and O 1s with molar ratios of different species, are summarized in Table 1. The result shows that La/Ni/SDC contains the electrophilic oxygen species (OII and OIII) up to ∼75% of the total oxygen, compared with ∼50% for the other two catalysts. The OII and OIII species (that are O−, O2− and CO32−) were considered to be those with high oxygen mobility while the OI species are stationary lattice sites. According to the O 1s result, the analysis of the Ce 3d5/2 spectrum indicated that on La/Ni/SDC there exists the highest ratio of Ce3+/Ce4+, mixed-valence redox couples, implying high oxygen lability on the co-microwave sample with plentiful oxygen vacancies. On the other hand, it was found that both metallic Ni and oxide NiO are present on all fresh catalysts, where La/Ni/SDC presents the largest metal phase ∼67% (Ni0/Ni0 + Ni2+). It was speculated that the metal and its oxide phase could both be observed on the XPS analysis for the SDC–Ni catalyst. Shifting to a metal phase may occur with a stronger metal–support interaction as nickel species remain adjacent to the support. The high metallic Ni percentage was favoured for CO2 methanation, since nickel is considered as active sites for hydrogen adsorption and dissociation. On the other hand, there was still no definite evidence to explain the role of La species. However, it was suggested that lanthanum promotes nickel species on the co-microwave bimetallic catalyst and allows the metallic phase to a great extent, thus provoking H2 activation and subsequent hydrogenation.
Based on the results and characterization discussed above, it was interesting to observe that the concentration of intermediates such as monodentate and polydentate carbonates that are formed follows the order La/Ni/SDC, Ni–La/SDC, and La–Ni/SDC (from high to low) at 200 °C. Most of the intermediates in the catalysts were transformed into bicarbonates and formates at 300 °C, while La–Ni/SDC just produced comparable monodentate carbonates then. Thereafter, CO was generated on all three catalysts. As a result, the reaction pathways of CO2 methanation on La/Ni/SDC were proposed as follows: (i) CO2 first adsorbs as monocarbonates and bicarbonates on the surface of the catalyst, (ii) transformation of the carbonates into bidentate and/or monodentate carbonates occurs with increasing temperatures (near 200 °C), (iii) the transformed carbonates are reduced to formates by reacting with H atoms via H2 activation and dissociation at metal sites, followed by spillover and migration to the metal–support interface, and (iv) the formate species are finally converted to CH4. Compared with the findings in the literature, the reaction pathway for CO2 methanation on the La/Ni/SDC catalyst complied with the so-called associative mechanism through carbonate and formate species as the main intermediates.16,24,33 However, the chemisorption of CO2 and direct dissociation were not ruled out to take place as the temperature is elevated. The intimate contact of La/Ni and SDC and the resulting interface is thought to provide a facile transfer of oxygen vacancies and active sites to activate CO2via dissociation.32 Consequently, the consecutive associative and dissociative mechanisms probably explained the excellent methanation activity and selectivity of the co-microwave synthesised La/Ni/SDC catalyst.
Conclusions
The effect of metal loading sequences on CO2 methanation activity was investigated in samarium-doped ceria. The La/Ni/SDC catalyst obtained by co-microwave heating produced the best methanation activity with a CO2 conversion of about 60% at 353 °C. Compared to the other two catalysts, either La or Ni first loaded on SDC (i.e., Ni–La/SDC and La–Ni/SDC), the better catalytic performance of La/Ni/SDC was attributed to a synergistic function of the carrier and metal due to intimate contact. XRD and TEM analysis revealed that the co-microwave heating synthesis produced a material of low crystallinity and small particle size. XPS analysis showed that Ni/La/SDC contains a high ratio of Ni phase, abundant active oxygen sites, and fast oxygen lability due to more redox couples. An optimum CO2 affinity was also evidenced in the TPD measurement. Moreover, in situ infrared studies indicated that La/Ni/SDC catalyses the methanation reaction via an associative mechanism with carbonate and formate species as main intermediates at low temperatures, followed by dissociative pathways to produce CO as the temperature is elevated. All of these characterisation results were correlated with the reaction studies, demonstrating that the co-microwave-synthesised material exhibits unique metal–support interactions. This study provides valuable information to understand the effects of metal loading sequences; in turn, this will allow the design of an effective material capable of conducting methanation reactions and optimising the synthesis conditions of bimetallic catalysts for potential applications.Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its ESI.† Should any raw data files be needed in another format, they are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support from Feng Chia University and the National Science Council of Taiwan under contract 1410-D1-23H00804, MOST 109-2221-E-035-021-MY3, NSTC 113-2221-E-035-004-MY3.References
- A. Tripodi, F. Conte and I. Rossetti, Carbon Dioxide Methanation: Design of a Fully Integrated Plant, Energy Fuels, 2020, 34(6), 7242–7256, DOI:10.1021/acs.energyfuels.0c00580.
- H. Jiang, L. Wang, H. Kaneko, R. Gu, G. Su and L. Li, et al., Light-driven CO2 methanation over Au-grafted Ce0.95Ru0.05O2 solid-solution catalysts with activities approaching the thermodynamic limit, Nat. Catal., 2023, 6(6), 519–530 CrossRef CAS , Available from: https://www.nature.com/articles/s41929-023-00970-z.
- H. Muroyama, Y. Tsuda, T. Asakoshi, H. Masitah, T. Okanishi and T. Matsui, et al., Carbon dioxide methanation over Ni catalysts supported on various metal oxides, J. Catal., 2016, 343, 178–184, DOI:10.1016/j.jcat.2016.07.018.
- J. Gao, X. Mo, A. C. Y. Chien, W. Torres and J. G. Goodwin, CO hydrogenation on lanthana and vanadia doubly promoted Rh/SiO2 catalysts, J. Catal., 2009, 262(1), 119–126 CrossRef CAS.
- R. P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong and Y. Wang, et al., CO2 hydrogenation to high-value products via heterogeneous catalysis, Nat. Commun., 2019, 10(1), 5698, DOI:10.1038/s41467-019-13638-9.
- B. Li, F. Wang, K. Li, P. Ning, M. Chen and C. Zhang, CeO2-supported Fe, Co and Ni toward CO2 hydrogenation: Tuning catalytic performance via metal-support interaction, J. Rare Earths, 2023, 41(6), 926–932 CrossRef CAS.
- L. Liang, C. Miao, S. Chen, X. Zheng and J. Ouyang, Effective CO2 methanation at ambient pressure over Lanthanides ( La/Ce/Pr/Sm ) modified cobalt-palygorskite composites, J. CO2 Util., 2022, 63, 102114, DOI:10.1016/j.jcou.2022.102114.
- D. Pandey and G. Deo, Effect of support on the catalytic activity of supported Ni-Fe catalysts for the CO2 methanation reaction, J. Ind. Eng. Chem., 2016, 33, 99–107 CrossRef CAS.
- M. M. Jaffar, M. A. Nahil and P. T. Williams, Parametric Study of CO 2 Methanation for Synthetic Natural Gas Production, Energy Technol., 2019, 7(11), 1900795, DOI:10.1002/ente.201900795.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and A. Ahmad, CO2 methanation over heterogeneous catalysts: recent progress and future prospects, Green Chem., 2015, 17(5), 2647–2663 RSC , Available from: http://xlink.rsc.org/?DOI=C5GC00119F.
- L. P. L. Gonçalves, J. Mielby, O. Salom, G. P. Soares, J. P. S. Sousa and D. Y. Petrovykh, et al., In situ investigation of the CO2 methanation on carbon / ceria-supported Ni catalysts using modulation-excitation DRIFTS, Appl. Catal., B, 2022, 312(5), 121376, DOI:10.1016/j.apcatb.2022.121376.
- J. Chen, X. Shen, Q. Wang, J. Wang, D. Yang and T. Bold, et al., CO2 methanation over γ -Al2O3 nanosheets-stabilized Ni catalysts : Effects of MnOx and MoOx additives on catalytic performance and reaction pathway, J. CO2 Util., 2022, 63, 102113, DOI:10.1016/j.jcou.2022.102113.
- Q. Pan, J. Peng, T. Sun, S. Wang and S. Wang, Insight into the reaction route of CO2 methanation: Promotion effect of medium basic sites, Catal. Commun., 2014, 45, 74–78, DOI:10.1016/j.catcom.2013.10.034.
- R. Tao, J. Xu, H. Zhong, W. Wen, Q. Pan and Y. Liu, et al., Finely Tuned Structure and Catalytic Performance of Cerium Oxides by a Continuous Samarium Doping from 0 to 100%, Inorg. Chem., 2019, 58(19), 13066–13076 CrossRef CAS.
- A. C. Chien, N. J. Ye, C. Huang and I.-H. Tseng, Studies of Nickel/Samarium-Doped Ceria for Catalytic Partial Oxidation of Methane and Effect of Oxygen Vacancy, Catalysts, 2021, 11(6), 731 CrossRef CAS , Available from: https://www.mdpi.com/2073-4344/11/6/731.
- P. Hongmanorom, J. Ashok, P. Chirawatkul and S. Kawi, Interfacial synergistic catalysis over Ni nanoparticles encapsulated in mesoporous ceria for CO2 methanation, Appl. Catal., B, 2021, 297(15), 120454, DOI:10.1016/j.apcatb.2021.120454.
- Y. Du, C. Qin, Y. Xu, D. Xu, J. Bai and G. Ma, et al., Ni nanoparticles dispersed on oxygen vacancies-rich CeO2 nanoplates for enhanced low-temperature CO2 methanation performance, Chem. Eng. J., 2021, 418, 129402 CrossRef CAS.
- N. Ullah, R. Tang and Z. Li, Nanostructured Ceria-zirconia Supported Ni Catalysts for High Performance CO2 Methanation: Phase and morphology effect on activity, J. Taiwan Inst. Chem. Eng., 2022, 134, 104317 CrossRef CAS , Available from: https://linkinghub.elsevier.com/retrieve/pii/S187610702200116X.
- J. Ashok, M. L. Ang and S. Kawi, Enhanced activity of CO2 methanation over Ni/CeO2-ZrO2 catalysts: Influence of preparation methods, Catal. Today, 2017, 281, 304–311 CrossRef CAS.
- G. Zhou, H. Liu, K. Cui, A. Jia, G. Hu and Z. Jiao, et al., Role of surface Ni and Ce species of Ni/CeO 2 catalyst in CO2 methanation, Appl. Surf. Sci., 2016, 383, 248–252 CrossRef CAS.
- G. I. Siakavelas, N. D. Charisiou, A. Alkhoori, S. Alkhoori, V. Sebastian and S. J. Hinder, Highly selective and stable Ni/La-M ( M = Sm , Pr , and Mg ) -CeO2 catalysts for CO2 methanation, J. CO2 Util., 2021, 51, 101618, DOI:10.1016/j.jcou.2021.101618.
- J. A. Onrubia-Calvo, S. López-Rodríguez, I. J. Villar-García, V. Pérez-Dieste, A. Bueno-López and J. R. González-Velasco, Molecular elucidation of CO2 methanation over a highly active, selective and stable LaNiO3/CeO2-derived catalyst by in situ FTIR and NAP-XPS, Appl. Catal., B, 2024, 342, 123367 CrossRef CAS.
- M. González-Castaño, J. González-Arias, L. F. Bobadilla, E. Ruíz-López, J. A. Odriozola and H. Arellano-García, In-situ DRIFTS steady-state study of CO2 and CO methanation over Ni-promoted catalysts, Fuel, 2023, 338, 127241 CrossRef.
- J. B. Branco, P. E. Brito and A. C. Ferreira, Methanation of CO2 over nickel-lanthanide bimetallic oxides supported on silica, Chem. Eng. J., 2020, 380, 122465 CrossRef CAS.
- V. Palma, D. Barba, M. Cortese, M. Martino, S. Renda and E. Meloni, Microwaves and heterogeneous catalysis: A review on selected catalytic processes, Catalysts, 2020, 10(2), 246 CrossRef CAS.
- Y. P. Fu, C. H. Lin and C. S. Hsu, Preparation of ultrafine CeO2 powders by microwave-induced combustion and precipitation, J. Alloys Compd., 2005, 391(1–2), 110–114 CrossRef CAS.
- A. C. Chien and N. J. Ye, Effect of preparation method and particle size of Ni/SDC catalyst on methane oxidation, Catal. Commun., 2021, 154, 106312 CrossRef CAS , Available from: https://linkinghub.elsevier.com/retrieve/pii/S1566736721000352.
- K. Polychronopoulou, A. F. Zedan, M. S. Katsiotis, M. A. Baker, A. A. AlKhoori and S. Y. AlQaradawi, et al., Rapid microwave assisted sol-gel synthesis of CeO2 and CexSm1-xO2 nanoparticle catalysts for CO oxidation, Mol. Catal., 2017, 428, 41–55 CrossRef CAS.
- A. C. Chien, I. Z. Xie and C. Yeh, Reaction pathways of oxidative coupling of methane on lithiated lanthanum oxide, Mol. Catal., 2023, 538, 112974, DOI:10.1016/j.mcat.2023.112974.
- R. K. Singha, A. Shukla, A. Yadav, L. N. Sivakumar Konathala and R. Bal, Effect of metal-support interaction on activity and stability of Ni-CeO2 catalyst for partial oxidation of methane, Appl. Catal., B, 2017, 202, 473–488, DOI:10.1016/j.apcatb.2016.09.060.
- G. I. Siakavelas, N. D. Charisiou, A. AlKhoori, V. Sebastian, S. J. Hinder and M. A. Baker, et al., Cerium oxide catalysts for oxidative coupling of methane reaction: Effect of lithium, samarium and lanthanum dopants, J. Environ. Chem. Eng., 2022, 10(2), 107259 Search PubMed , Available from: https://linkinghub.elsevier.com/retrieve/pii/S2213343722001324.
- H. Yang, X. Wen, S. Yin, Y. Zhang, C. e. Wu and L. Xu, et al., The construction of the Ni/La2O2CO3 nanorods catalysts with enhanced low-temperature CO2 methanation activities, J. Ind. Eng. Chem., 2023, 128, 167–183 CrossRef CAS.
- A. Quindimil, J. A. Onrubia-Calvo, A. Davó-Quiñonero, A. Bermejo-López, E. Bailón-García and B. Pereda-Ayo, et al., Intrinsic kinetics of CO2 methanation on low-loaded Ni/Al2O3 catalyst: Mechanism, model discrimination and parameter estimation, J. CO2 Util., 2022, 57, 101888 CrossRef CAS , Available from: https://linkinghub.elsevier.com/retrieve/pii/S2212982022000075.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy01007h |
| This journal is © The Royal Society of Chemistry 2025 |