
Molten salt synthesis of a highly crystalline graphitic carbon nitride homojunction from a deep eutectic solvent for selective photocatalytic oxidation†
Yanglin
Chen
 *a,
Limo
He
a,
Hui-Ru
Tan
b,
Xuanhao
Lin
a,
Stephan
Jaenicke
a and
Gaik-Khuan
Chuah
*a
*a,
Limo
He
a,
Hui-Ru
Tan
b,
Xuanhao
Lin
a,
Stephan
Jaenicke
a and
Gaik-Khuan
Chuah
*a
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Kent Ridge, 117543, Singapore. E-mail: yanglin.chen@ntu.edu.sg; chmcgk@nus.edu.sg
bInstitute of Materials Research and Engineering, A*STAR (Agency for Science, Technology and Research), 3 Research Link, 117602, Singapore
First published on 16th May 2023
Abstract
The construction of homojunctions in graphitic carbon nitride is regarded as an efficient approach to improve the charge separation and photocatalytic activity. However, the enhanced activity is still far from satisfactory due to the low crystalline bulk structure. Herein, a highly crystalline graphitic carbon nitride homojunction is synthesized by calcination of a mixture of NH4SCN and urea, which forms a deep eutectic solvent using NaCl/KCl molten salts as the preparation medium. The homogeneity of the liquid precursor ensures an intimate homojunction interface, facilitating the charge separation, and the highly crystalline bulk structure builds a highway for rapidly transferring charge to the surface. Consequently, the bulk charge recombination is suppressed, and the spatial separation and transfer efficiency of charges are significantly improved. Compared to low crystalline homojunctions, this highly crystalline homojunction showed 15 times higher activity for the selective photocatalytic oxidation of benzyl alcohol to benzaldehyde. This work provides a simple strategy to construct a highly crystalline graphitic carbon nitride homojunction for solar energy conversion.
1. Introduction
Graphitic carbon nitride (CN) is regarded as a promising visible light driven metal-free photocatalyst due to its low cost, suitable energy band position, and high thermal and chemical stability.1 Unfortunately, the intrinsic photocatalytic activity of CN is limited by rapid recombination of photogenerated charges.2 To address this issue, the construction of interface junctions between CN and another semiconductor or conductor has been widely adopted to promote spatial separation of photogenerated charges.3 For example, Zhou and coworkers revealed that the construction of a heterojunction interface between CdS and CN facilitated the enrichment of photogenerated electrons on CN and photogenerated holes on CdS, respectively.4 This spatial charge separation greatly accelerated the photocatalytic CO evolution from biomass. Yang et al. reported that the formation of a Ti3C2/CN Schottky junction inhibited the recombination of photogenerated charges and boosted charge separation.5 However, the lattice mismatch between CN and dissimilar another component resulted in an incoherent heterojunction interface.6 The abundant dangling bonds and defects at this incoherent interface act as charge recombination centers, obstructing the separation of photogenerated charges across the junction interface, and limiting the enhancement of photocatalytic activity. To solve this problem, the construction of a homojunction between two isomorphic CN forms offers an ideal approach to obtain a coherent homogeneous interface free of dangling bonds and interface defects. Since the chemical structure and the crystal phases on both sides of the interface are closely matched, the separation of photogenerated charges can be significantly improved at the homojunction interface.7 For instance, Ye's group found that a p–n CN homojunction interface effectively suppressed charge recombination and enhanced photocatalytic H2 evolution.8 Dong et al. demonstrated that a 2D/3D CN homojunction facilitated the movement of photogenerated charges across the interface leading to improved photoactivity.9Besides interfacial charge separation, charge transfer from the bulk to the exterior surface sites is also essential for photocatalytic activity. Bulk defects can hamper charge transfer to the surface, but this point is rarely considered when discussing CN homojunctions. Hence, it is necessary to consider charge transfer in its entirety. CN prepared by calcination of N-rich precursors is usually rich in bulk defects due to the kinetic hindrance during the solid-state condensation process.10 Therefore, accelerating the mass-transfer of intermediates (such as melem and melon) during the preparation process will be helpful to reduce the density of bulk defects.11 Molten salts have been shown to act as a reactive medium to prepare highly crystalline CN with reduced defect density in the bulk.12 Antonietti and coworkers used LiCl/KCl molten salts to form highly crystalline graphitic carbon nitrides.13 Compared with amorphous CN, the highly crystalline compound had fewer bulk defects, which resulted in higher charge transfer efficiency and enhanced photocatalytic H2 generation. Likewise, Zhang et al. synthesized highly ordered CN by calcination of 5-aminotetrazole with the assistance of NaCl/KCl molten salts.14 The efficient separation of the photogenerated charges in this highly crystalline CN material resulted in an outstanding photocatalytic activity for H2 production from water.
In this work, it is shown that homojunctions in CN can be created using urea and ammonium thiocyanate as precursors. The mixture of urea (melting point 133 °C) and ammonium thiocyanate (melting point 149 °C) forms a deep eutectic solvent (DES) and is a liquid at ambient temperature. The homogeneity of the liquid ensures uniform coupling and intimate contact between the two N-rich precursors.15,16 In conjunction with NaCl/KCl molten salts as the synthesis medium, a highly crystalline graphitic carbon nitride with a coherent and intimate homojunction was prepared. The NaCl/KCl molten salts modulate the crystallinity of CN during the polymerization process. The intimate homojunctions and the high crystallinity of the structure facilitate the separation of photogenerated charges and their diffusion to the surface of the particles. The material was evaluated for its activity in the selective photo-oxidation of alcohols to aldehydes or ketones. Oxidation is an important reaction in organic synthesis and the application of a viable photocatalyst offers a green alternative to traditional toxic oxidants or precious metal-based catalysts.17,18 The highly crystalline graphitic carbon nitride homojunction material described here displayed significantly higher photocatalytic activity than amorphous one. This work outlines an effective strategy to further improve the photocatalytic activity of CN homojunctions.
2. Experimental section
2.1. Photocatalyst preparation
In a typical procedure, the deep eutectic solvent (DES) containing 1 g of ammonium thiocyanate and 2 g of urea was thoroughly ground with NaCl/KCl (8.4 g/9.6 g) in a mortar to form a slurry (Fig. S1†). This slurry was calcined in a covered crucible in a tube furnace at 550 °C for 4 h with a ramp of 2.3 °C min−1 under an Ar flow. After cooling to room temperature, the solid residue was leached with copious amounts of deionized water to remove the NaCl/KCl salts and dried at 80 °C overnight. The final material was collected as a yellow powder which will be subsequently referred to as CCNA/CCNU, where A and U stand for the ammonium thiocyanate and urea precursors, respectively (Scheme 1). In addition, urea and ammonium thiocyanate were calcined separately in the NaCl/KCl molten salts under otherwise identical conditions to form CCNU and CCNA, respectively. For comparison, a sample, CNA/CNU, was prepared by calcining a mixture of ammonium thiocyanate and urea without NaCl/KCl at 550 °C for 4 h under Ar. | ||
| Scheme 1 Schematic for the preparation of CNA/CNU and CCNA/CCNU. | ||
2.2. Characterization
The X-ray diffraction patterns of photocatalysts were obtained using a Bruker D8 Advance X-ray diffractometer (XRD) with Cu-Kα radiation. The specific surface area and porosity of the photocatalysts were determined from N2 adsorption–desorption isotherms (Micromeritics Tristar 3000). Scanning and transmission electron micrographs were obtained using a JEOL JSM-6071F and a JEOL 3010 microscope respectively. Diffuse reflectance spectra (DRS) were recorded with a Shimadzu UV-2450 UV-vis spectrophotometer using BaSO4 as the reference. A Bruker ALPHA spectrometer with an attenuated total reflection unit was used for infrared spectroscopy. Room-temperature electron paramagnetic resonance (EPR) spectra were recorded by using a Bruker EPR JESFA200 spectrometer. Steady-state photoluminescence (PL) spectroscopy was obtained using a fluorescence spectrophotometer (Hitachi, F-7000). Time-resolved photoluminescence (TR-PL) was performed on a Horiba Fluoro Max-4 Fluorescence spectrometer. X-ray photoelectron spectroscopy (XPS) was carried out with a VG-Scientific ESCALAB Mark 2 spectrometer. The binding energies were corrected for sample changing by referencing to the C 1s signal for adventitious carbon at 284.6 eV. A homebuilt setup was used for oxygen temperature programmed desorption (O2-TPD), where the evolved O2 was detected with a quadrupole mass spectrometer (Balzers QMS 200).2.3. Photoelectrochemical measurements
A CHI660E electrochemical workstation was used for electrochemical and photoelectrochemical measurements. The three-electrode cell comprised a carbon rod counter electrode, an Ag/AgCl reference electrode and indium tin oxide (ITO) glass covered with the photocatalyst as the working electrode. The working electrode was prepared as follows: 4 mg of photocatalyst and 40 μL of Nafion solution were dispersed in 960 μL isopropanol by sonication to form a homogeneous slurry. 40 μL of obtained slurry was deposited onto the ITO glass with an active area of 1 cm2, and air-dried to form the working electrode. The transient photocurrent responses, Mott–Schottky plots, electrochemical impedance spectroscopy (EIS) and linear sweep voltammetry (LSV) curves were recorded in 0.5 mol L−1 Na2SO4 solution.2.4. Procedure for photocatalytic oxidation of alcohols
A Pyrex glass tube was filled with 50 μmol of benzyl alcohol, 3 mL of acetonitrile and 10 mg of the photocatalyst. The suspension was magnetically stirred for 30 min before irradiation with a 9.5 W LED lamp (Megaman). Aliquots of the suspension (0.1 mL) were collected at regular time intervals filtered through a syringe filter and analyzed using a gas chromatograph (Agilent 6890 N, HP-5 column, flame ionization detector). The products were identified using an Agilent/HP 6890 gas chromatograph with a 5973 mass selective detector (GC-MS). For reusability testing, the used catalyst was removed from the reaction mixture by centrifugation and washed with acetonitrile. After drying at 80 °C, the washed catalyst could be reused.2.5. Quenching experiment for active species
To investigate the active species in photo-oxidation of benzyl alcohol, a quenching experiment was carried out. A series of radical scavengers (10 μmol), such as butyl alcohol (BuOH), potassium oxalate (K2C2O4), 1,4-benzoquinone (p-BQ) and furfuryl alcohol (FFA), were added into the above photocatalytic experiment for quenching hydroxide radicals (˙OH), photogenerated holes (h+), and superoxide radical (˙O2−) and singlet oxygen (1O2) species, respectively.2.6. Electron spin resonance (ESR) analysis for active species
The presence of photogenerated superoxide radicals (˙O2−) was tested by trapping with 5,5-dimethyl-L-pyrroline N-oxide (DMPO). About 5 mg of photocatalyst in 4 mL of methanol solution containing 0.2 mmol of DMPO was used. After irradiation for 1 h using a 9.5 W LED lamp, the mixture was filtered with a 0.22 μm microporous membrane and analyzed for the superoxide spin adduct using a Bruker ESR JESFA200 spectrometer. Photogenerated h+ and 1O2 were trapped using 2,2,6,6-tetramethyl-L-piperidine-N-oxyl (TEMPO, 0.2 mmol) in acetonitrile and 2,2,6,6-tetramethylpiperidine (TEMP, 0.2 mmol) aqueous solution, respectively.2.7. DFT calculations
The Dmol3 module of Materials studio was used for density functional theory (DFT) calculations of adsorption energy, electron density and population analysis. The exchange–correlation interaction was calculated by Generalized Gradient Approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) function. The DFT-D method by Grimme was used to describe the van der Waals interaction. The Double Numerical Plus Polarization (DNP) and all electron relativistic (AER) were selected as basis set and core treatment, respectively. A 20 Å vacuum layer was added to avoid the periodic interactions. The Brillouin zone was sampling by using Monkhorst–Pack grids, and the K point was set to 4 × 4 × 1. The convergence thresholds of energy, maximum force and maximum displacement were 1.0 × 10−5 Ha, 2 × 10−3 Ha Å−1 and 5 × 10−3 Å, respectively. The adsorption energy (Eads) was calculated by using the following equation.| Eads = Eadsorbate/slab − (Eadsorbate + Eslab) |
3. Results and discussion
3.1. Characterization of photocatalysts
The morphologies of the prepared photocatalysts were characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). CNA/CNU, synthesized in the absence of NaCl/KCl molten salts, appeared to form a stacked lamellar structure (Fig. 1a and S2†). However, the absence of lattice fringes in the transmission electron micrographs revealed the low crystallinity of the sample (Fig. 1b). In contrast, CCNA/CCNU exhibited a different morphology with dense and aggregated nanoparticles (Fig. 1c and d). Additionally, distinct lattice fringes can be observed in the TEM images (Fig. 1e and f), indicating its crystalline nature. The measured lattice spacings were 1.10 and 0.32 nm, corresponding to the (100) and (002) planes. The results show that using NaCl/KCl molten salts in the synthesis affected the morphology and increased the crystallinity of graphitic carbon nitride. Indeed, the increased crystallinity is also found in CCNA and CCNU prepared from only urea or ammonium thiocyanate, where lattice fringes could be clearly observed (Fig. S3 and S5†). According to the EDX elemental maps, C, N, S, Na and K were present in CCNA/CCNU (Fig. 1g and S4†). The distribution of C, N, Na and K was uniform, whereas the S map showed an uneven distribution with areas void of the element (indicated by red dotted circles). As expected, the crystalline CCNA showed a more uniform distribution of S besides C, N, Na and K (Fig. S3†), whereas the crystalline CCNU had uniformly distributed C, N, Na and K but no S (Fig. S5†). These results confirm the integration of separate CCNA and CCNU domains in CCNA/CCNU. To further investigate the density of local structure defects, room-temperature electron paramagnetic resonance (EPR) was employed. As shown in Fig. 1h, both CNA/CNU and CCNA/CCNU displayed a single Lorentzian line at g = 2.0038, which is caused by the unpaired electrons on the heptazine rings.19 Notably, the EPR intensity of CCNA/CCNU was significantly weaker than that of CNA/CNU, indicating a decreased concentration of unpaired electrons due to lower density of defects in the higher crystalline bulk structure.20 Organic element analysis has been used to identify the atomic ratio of C and N in our samples, and the results are summarized in Table S1.† The C/N atomic ratio was 0.69 for CNA/CNU whereas the melt-formed CCNA, CCNU and CCNA/CCNU have a higher C/N ratio of 0.72, which is close to the value of stoichiometric g-C3N4 (0.75). These results indicate that the NaCl/KCl molten salt medium promotes the polymerization and reduce the structural defects, as also confirmed by the EPR results (Fig. 1h). | ||
| Fig. 1 (a) SEM and (b) HR-TEM images of CNA/CNU; (c) SEM, (d) TEM and (e and f) HR-TEM images of CCNA/CCNU and (g) the corresponding EDX mapping images of C, N, S, Na and K elements; (h) EPR spectra of CNA/CNU and CCNA/CCNU. | ||
The structure of the as-synthesized photocatalysts was characterized by XRD and FT-IR. As shown in Fig. 2a, the XRD pattern of CNA/CNU displayed two characteristic peaks, at 2θ ∼ 12.8° and 27.3° assigned to (100) and (002) planes.21,22 The (100) peak arises from the separation between in-plane packing motifs where neighboring heptazine units are about 0.69 nm apart. The (002) peak shows that the interlayer spacing due to stacking of layers has a periodicity of 0.33 nm. Interestingly, the (100) and (002) peaks of the melt-formed CCNA, CCNU and CCNA/CCNU were located at 2θ ∼ 8° and 27.7°, indicating a very significant increase of the in-plane separation of the heptazine packing to 1.10 nm and a decrease of the interplanar spacing to 0.32 nm. The expansion between the interplanar heptazine packing and contraction of the π–π interlayer stacking distance is attributed to the intercalation of alkali metals into the framework of graphitic carbon nitride.13,23 These observations are consistent with the lattice fringes observed in the TEM images (Fig. 1e and f).
 | ||
| Fig. 2 (a) XRD patterns, (b) FT-IR spectra, (c) the magnified FT-IR spectra range from 900–700 cm−1, and XPS spectra of (d) C 1s and K 2p, (e) Na 1s, (f) N 1s and (g) S 2p regions of the as-prepared photocatalysts. | ||
The FT-IR spectra for all the photocatalysts exhibited the characteristic peaks of graphitic carbon nitride (Fig. 2). The broad absorption at 3000–3600 cm−1 was assigned to the stretching vibration of –NHx or –OH of adsorbed water.24 The peaks at 1200–1700 cm−1 correspond to the stretching modes of the aromatic C–N heterocycles.25 The peaks at ca. 800 cm−1 were related to the out-of-plane bending mode of heptazine units.26 Three additional peaks were observed for the melt-formed CCNA, CCNU and CCNA/CCNU. The peaks at 990 and 1160 cm−1 were attributed to the symmetric and asymmetric vibrations of the metal–NC2 groups (M–NC2),27 reflecting the incorporation of K and Na into the graphitic carbon nitride structure. The peak at 2185 cm−1 can be assigned to terminal cyano groups (–CN) derived from the transformation of surface –NHx.28 This new band correlated with a reduction in the intensity of the –NHx vibration band (highlighted by a light green rectangle in Fig. 2b). In CCNU, CCNA and CCNA/CCNU, the heptazine band appeared at 803 cm−1. This red-shift from 812 cm−1 in CNA/CNU (Fig. 2c) is caused by the ion–dipole interaction between heptazine and the alkali ions in the form of N–K or N–Na bonds.29
The presence of K and Na in CCNU, CCNA and CCNA/CCNU is further supported by the XPS results (Fig. 2d and e). The binding energies of these elements indicate that they are present as positively charged ions with Na 1s, K 2p3/2 and 2p1/2 peaks at 1071.7, 293.1 and 295.9 eV, respectively.30,31 Even though alkali metals were present, no Cl can be detected (Fig. S6b†). In contrast, no K or Na was detected for CNA/CNU (Fig. S6a†). The C 1s XPS spectrum of the sample showed two peaks, one at 284.6 eV which is due to graphitic carbon (C–C) and another at 288.1 eV assigned to sp2-hybridized carbon (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) in the aromatic heterocycles.32 The C 1s spectra for CCNU, CCNA and CCNA/CCNU are similar to those of CNA/CNU but the peak of the N–CN species is shifted to a higher binding energy (288.4 eV) due to the Na–N and K–N interactions. In addition, a weak peak at a lower binding energy of 286.4 eV can be discerned. This is assigned to the carbon in the cyano group (–C
N) in the aromatic heterocycles.32 The C 1s spectra for CCNU, CCNA and CCNA/CCNU are similar to those of CNA/CNU but the peak of the N–CN species is shifted to a higher binding energy (288.4 eV) due to the Na–N and K–N interactions. In addition, a weak peak at a lower binding energy of 286.4 eV can be discerned. This is assigned to the carbon in the cyano group (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), whose presence in the melt-formed samples had been confirmed by FT-IR.33 The N 1s signal for CNA/CNU can be deconvoluted into three peaks (398.7, 399.8 and 401.2 eV), which can be assigned to N atoms originating from C–NC, N–(C)3 and –NHx species, respectively (Fig. 2f).34 In the CCNA, CCNU and CCNA/CCNU samples, the binding energy for N of the C–NC species is slightly higher (by ∼0.1 eV) owing to the K–N and Na–N interactions. XPS also confirmed the presence of S in CNA/CNU, CCNA and CCNA/CCNU, which contained the C–S–C bond formed by replacing some N atoms in the heptazine rings with S.35 As CCNU was synthesized from only urea, no S could be detected (Fig. S6a†). The O 1s XPS of the as-prepared photocatalysts exhibited one peak at 531.6 eV, which was attributed to the adsorbed water on the surface (Fig. S6c†).36
N), whose presence in the melt-formed samples had been confirmed by FT-IR.33 The N 1s signal for CNA/CNU can be deconvoluted into three peaks (398.7, 399.8 and 401.2 eV), which can be assigned to N atoms originating from C–NC, N–(C)3 and –NHx species, respectively (Fig. 2f).34 In the CCNA, CCNU and CCNA/CCNU samples, the binding energy for N of the C–NC species is slightly higher (by ∼0.1 eV) owing to the K–N and Na–N interactions. XPS also confirmed the presence of S in CNA/CNU, CCNA and CCNA/CCNU, which contained the C–S–C bond formed by replacing some N atoms in the heptazine rings with S.35 As CCNU was synthesized from only urea, no S could be detected (Fig. S6a†). The O 1s XPS of the as-prepared photocatalysts exhibited one peak at 531.6 eV, which was attributed to the adsorbed water on the surface (Fig. S6c†).36
3.2. Optical and photoelectrochemical properties
The optical absorption properties of the as-prepared photocatalysts were characterized by using UV-vis diffuse reflectance spectra (DRS). CCNU, CCNA and CCNA/CCNU possessed wider and stronger visible light absorption than CNA/CNU (Fig. 3a). The absorption band edge of CCNA/CCNU at 466 nm was in between that for CCNU (462 nm) and CCNA (473 nm). This result indirectly verified the construction of the CCNA and CCNU homojunction in CCNA/CCNU. Based on the Tauc plots (Fig. S7†), the band gap energies (Eg) of CCNU and CCNA are calculated to be 2.58 and 2.52 eV, respectively. The flat-band potentials of CCNA and CCNU were obtained from Mott–Schottky plots (Fig. 3b and c). The positive slopes of the Mott–Schottky plots indicate that CCNA and CCNU are n-type semiconductors.37 The flat band potentials of CCNA and CCNU were estimated to be −0.78 and −0.85 V vs. Ag/AgCl (pH = 5.72) from the intercept of the tangent with the x-axis. According to the formula: E(NHE) = E(Ag/AgCl) − Eθ(Ag/AgCl) + 0.059pH, where Eθ(Ag/AgCl) = 0.197 V,38 the flat band potentials of CCNA and CCNU are ca. −0.64 and −0.71 V vs. NHE (pH = 5.72). In general, the surface Fermi level (Ef) of a semiconductor is close to the flat band potential in the electrolytic solution.39 Thus, the Ef of CCNA and CCNU can be taken to be ca. −0.64 and −0.71 V, respectively. To further investigate the band alignments of CCNA and CCNU in the CCNA/CCNU homojunction, the valence band XPS (VB-XPS) spectra were recorded (Fig. 3d). For a semiconductor, the VB-XPS spectrum gives the energy difference between the Ef and the valence band position (Ev).40 Based on the energy gaps between the Ef and Ev of 1.75 and 1.95 eV, the Ev was estimated to be 1.11 and 1.24 eV for CCNA and CCNU, respectively. The conduction band positions (Ec) of CCNA and CCNU were calculated to be −1.41 and −1.34 V from Ec = Ev + Eg.41 The energy band offsets of CCNA and CCNU are summarized in Fig. 3e. In CCNA/CCNU, the higher Ef of CCNU induced the spontaneous flow of electrons from CCNU to CCNA until their Ef equalizes in the dark.42 As a result, accumulation and depletion layers of electrons are generated in the CCNA and CCNU interface regions, respectively. Therefore, the CCNA is negatively charged and its band edge bends downward. On the other hand, the CCNU is positively charged and its band edge bends upward. This results in an internal electric field (IEF) from CCNU to CCNA at the CCNA and CCNU interface (Fig. 3f). Under irradiation, both CCNA and CCNU can be excited to generate electron and hole pairs. Driven by the IEF, the photogenerated electrons migrate from the CB of CCNA to the CB of CCNU, whereas the holes move from the VB of CCNU to the VB of CCNA. The accumulation of electrons in the CB of CCNU and holes in the VB of CCNA leads to the effective separation of charges in the CCNA/CCNU homojunction (Fig. 3g). To further confirm the migration of the photogenerated charge between CCNA and CCNU, Pt (0.3 wt%) was photodeposited on the CCNA/CCNU surface. The EDX maps (Fig. S8†) show that the Pt nanoparticles were mainly distributed in the S-free areas, indicating the reduction of Pt4+ to Pt nanoparticles on the CCNU surface. This result directly reveals the electron transfer from CCNA to CCNU in the CCNA/CCNU homojunction during the photocatalytic process (Fig. 3g). | ||
| Fig. 3 (a) UV-vis DRS of the as-prepared photocatalysts; Mott–Schottky plots of (b) CCNA and (c) CCNU; (d) VB-XPS spectra of CCNA and CCNU; (e–g) schematic illustration of energy band offsets and directional charge transfer on CCNA/CCNU with the impact of the internal electric field (IEF). | ||
The formation of a highly crystalline CCNA/CCNU homojunction is proposed to suppress the recombination of electrons and holes and improve the utilization of photogenerated charge carriers. To assess the radiative charge recombination, steady-state photoluminescence (PL) was employed using an excitation wavelength of 380 nm. A pronounced PL peak centered at 446 nm in the wavelength range of 410–700 nm was observed for CNA/CNU (Fig. 4a). In comparison to CNA/CNU, CCNA and CCNU displayed much weaker PL emission signals and the peak maximum was shifted to a longer wavelength. In general, the PL peak is associated with the band to band transition of electrons from the CB to the VB.43 Hence, the shift in the peak maximum is due to their narrowed band gap (Fig. 3a and S7†).44 The attenuated PL emission signifies a lower rate of radiative recombination of electron–hole pairs with faster transfer of the photo-excited charges due to the high crystallinity of the bulk structure. Notably, the weakest PL signal was obtained with CCNA/CCNU, clearly showing that here, the radiative recombination of electrons and holes was further quenched by the rapid charge separation through the CCNA/CCNU homojunction.
 | ||
| Fig. 4 (a) Steady-state PL spectra, (b) TR-PL spectra, (c) photocurrent transient response and (d) EIS Nyquist plots of the as-prepared photocatalysts. | ||
To gain further insights, time-resolved transient photoluminescence (TR-PL) decay measurements were performed at the PL emission wavelength of each photocatalyst. As shown in Fig. 4b, the TR-PL decay curves can be fitted with a bi-exponential function: I(t) = A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2), where τ1 and τ2 refer to the recombination lifetimes during decay processes, and A1 and A2 refer to the corresponding amplitudes (Fig. 4b).45 The fast decay component (τ1 and A1) is ascribed to the non-radiative recombination of charge carriers at surface defects, whereas the slow decay component (τ2 and A2) is attributed to radiative charge recombination of free excitons in the bulk.46 The detailed fitting parameters for the photocatalysts are listed in Table 1. Despite the presence of a homojunction interface in CNA/CNU (Fig. S2†), the slow decay lifetime (τ2) of CNA/CNU is ca. 3.5 times longer than in the melt-formed CCNA and CCNU, indicating that the photoexcited charges in the melt-formed photocatalysts are able to much more rapidly transfer from the bulk to the surface. This phenomenon strongly suggests that the highly crystalline bulk structure indeed inhibits the bulk recombination of electrons and holes. Compared to CCNU or CCNA, CCNA/CCNU possesses a shorter τ2 (0.81 ns), implying that the bulk separation of photogenerated charges is further improved by the formation of a homojunction. The reduced surface charge recombination in CCNA/CCNU is confirmed by the shortest fast decay lifetime τ1 (0.13 ns).47 According to the bi-exponential function, the average recombination lifetimes (τa) were calculated to be 2.88, 0.68, 0.63 and 0.50 ns for CNA/CNU, CCNA, CCNU and CCNA/CCNU, respectively. The decreased decay lifetime and the quenched photoluminescence intensity suggest that faster exciton separation and transfer occur in CCNA, CCNU and especially CCNA/CCNU; otherwise faster charge recombination will result in enhanced photoluminescence.48,49
exp(−t/τ1) + A2exp(−t/τ2), where τ1 and τ2 refer to the recombination lifetimes during decay processes, and A1 and A2 refer to the corresponding amplitudes (Fig. 4b).45 The fast decay component (τ1 and A1) is ascribed to the non-radiative recombination of charge carriers at surface defects, whereas the slow decay component (τ2 and A2) is attributed to radiative charge recombination of free excitons in the bulk.46 The detailed fitting parameters for the photocatalysts are listed in Table 1. Despite the presence of a homojunction interface in CNA/CNU (Fig. S2†), the slow decay lifetime (τ2) of CNA/CNU is ca. 3.5 times longer than in the melt-formed CCNA and CCNU, indicating that the photoexcited charges in the melt-formed photocatalysts are able to much more rapidly transfer from the bulk to the surface. This phenomenon strongly suggests that the highly crystalline bulk structure indeed inhibits the bulk recombination of electrons and holes. Compared to CCNU or CCNA, CCNA/CCNU possesses a shorter τ2 (0.81 ns), implying that the bulk separation of photogenerated charges is further improved by the formation of a homojunction. The reduced surface charge recombination in CCNA/CCNU is confirmed by the shortest fast decay lifetime τ1 (0.13 ns).47 According to the bi-exponential function, the average recombination lifetimes (τa) were calculated to be 2.88, 0.68, 0.63 and 0.50 ns for CNA/CNU, CCNA, CCNU and CCNA/CCNU, respectively. The decreased decay lifetime and the quenched photoluminescence intensity suggest that faster exciton separation and transfer occur in CCNA, CCNU and especially CCNA/CCNU; otherwise faster charge recombination will result in enhanced photoluminescence.48,49
| Sample | τ 1 (ns) | τ 2 (ns) | A 1 (%) | A 2 (%) | τ a (ns) |
|---|---|---|---|---|---|
| a The average recombination lifetimes (τa) were calculated by using the equation τa = (A1τ12 + A2τ22)/(A1τ1 + A2τ2). | |||||
| CNA/CNU | 0.73 | 3.29 | 46.24 | 53.76 | 2.88 |
| CCNA | 0.16 | 0.96 | 76.78 | 23.22 | 0.68 |
| CCNU | 0.15 | 0.93 | 79.45 | 20.55 | 0.63 |
| CCNA/CCNU | 0.13 | 0.81 | 83.60 | 16.40 | 0.50 |
To further verify the enhancement in charge separation and transfer efficiency, photoelectrochemical measurements were conducted. The photoelectrodes showed rapid and reproducible photocurrent transient responses, indicating their good stability (Fig. 4c). The photocurrent densities of CCNA and CCNU were higher than that of CNA/CNU, attributed to a higher separation and transfer rate of photogenerated charges in the highly crystalline graphitic carbon nitride.50 In addition, the stronger photocurrent response in CCNA/CCNU than CCNA or CCNU suggests that the construction of a highly crystalline homojunction can further inhibit charge recombination. Electrochemical impedance spectroscopy (EIS) was employed to study the charge transfer resistance.51 The impedance arc diameter of CCNA/CCNU was the smallest among the samples, indicating that its electrical resistance was significantly reduced. Hence, it is anticipated that the less electrical resistance and more efficient charge separation and transfer in CCNA/CCNU should be beneficial to its photocatalytic activity.
3.3. Photocatalytic performance
The photocatalytic activity of the materials prepared for this study were evaluated by photo-oxidation of benzyl alcohol using acetonitrile as the solvent, air as the oxidant and a cost-effective 9.5 W LED as the light source. No benzaldehyde was obtained in the dark or without photocatalysts, indicating that both the photocatalyst and light are essential to the photo-oxidation of benzyl alcohol (Fig. 5a). Under light irradiation at a controlled temperature of 28 °C, all the photocatalysts promoted the selective photocatalytic oxidation of benzyl alcohol to benzaldehyde with 100% selectivity. However, they differed greatly in their photocatalytic activity with CNA/CNU being the least active photocatalyst. Higher benzaldehyde yields were obtained for CCNA, CCNU and CCNA/CCNU. Especially, the yield of 85% over CCNA/CCNU was 17 and 2.2 times higher than that of CNA/CNU (5%) and a physical mixture of CCNA and CCNU (38%), respectively. This significant enhancement in photo-oxidation of benzyl alcohol supports the hypothesis that the construction of highly crystalline homojunctions in CCNA/CCNU improves the separation and transfer of photogenerated electrons and holes. The performance of the CCNA/CCNU photocatalyst is superior to that of previously reported graphitic carbon nitride-based catalysts (Table S2†). The reusability of CCNA/CCNU was investigated via multiple cycles of benzyl alcohol photo-oxidation (Fig. 5b). CCNA/CCNU maintained its photoactivity with negligible deactivation for five cycles. The XRD patterns (Fig. 5c) and FT-IR spectra (Fig. 5d) of the used CCNA/CCNU were similar to those of the fresh sample, demonstrating the excellent stability of CCNA/CCNU. The high photoactivity, reusability and structural stability make CCNA/CCNU a promising photocatalyst for benzyl alcohol oxidation. | ||
| Fig. 5 (a) Photocatalytic activity for selective oxidation of benzyl alcohol after 5 h; (b) multiple cycles of benzyl alcohol photo-oxidation over CCNA/CCNU; (c) XRD patterns and (d) FT-IR spectra of fresh and used CCNA/CCNU. | ||
The substrate scope for photo-oxidation of various alcohols over CCNA/CCNU was studied under identical reaction conditions. As summarized in Table 2, all alcohols could be converted to the corresponding carbonyl products with 100% selectivity. The conversion of 4-methoxybenzyl alcohol was higher than that of benzyl alcohol (Table 2, entries 1 and 3). On the other hand, lower conversion was observed for 4-chlorobenzyl alcohol and 4-nitrobenzyl alcohol (Table 2, entries 4 and 5). Based on these results, it appears that an electron-donating group in the para-position improves the conversion, whereas an electron-withdrawing group in this position had the opposite effect.52 The methyl substituent should also act as an electron donating group, but the conversion of 4-methyl benzyl alcohol was slightly lower than that of benzyl alcohol, probably because of its lower dipole moment (Table 2, entry 2).53 The conversion of 1-phenylethanol to acetophenone was lower than for benzyl alcohol due to the higher steric hindrance (Table 2, entry 6). However, para-substitution with the electron-donating 4-methoxy group increased the conversion of 1-(4-methoxyphenyl)ethanol (Table 2, entry 7). Changing the substrate to an alicyclic alcohol, cyclohexanol, gave a lower conversion (52%) (Table 2, entry 8). This is caused by the lower stability of the secondary alcohols. The results show that the highly crystalline CCNA/CCNU homojunction has wide applicability for the photo-oxidation of alcohols to carbonyls without over-oxidation to acids.
| Entry | Substrate | Product | Conv.b (%) | Sel.b (%) |
|---|---|---|---|---|
| a Reaction conditions: 50 μmol substrate, 10 mg CCNA/CCNU photocatalyst, 3 mL acetonitrile, reaction time = 5 h, air (1 bar), 28 °C and 9.5 W LED lamp. b The conversion of alcohols and yield of the corresponding product were determined by GC-FID and GC-MS. | ||||
| 1 |
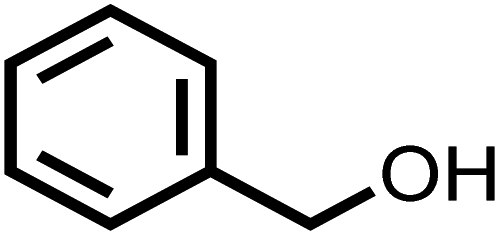
|
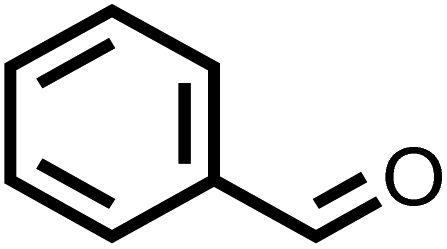
|
85 | 100 |
| 2 |
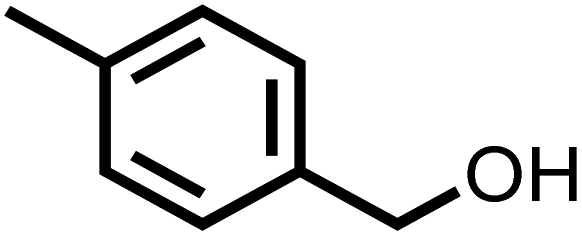
|

|
77 | 100 |
| 3 |
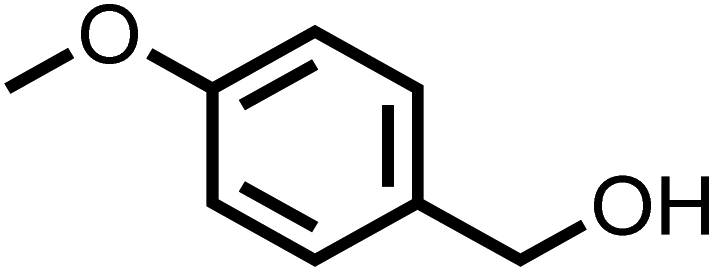
|

|
100 | 100 |
| 4 |
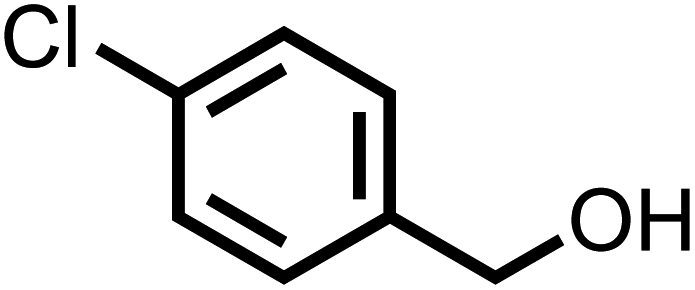
|

|
57 | 100 |
| 5 |

|
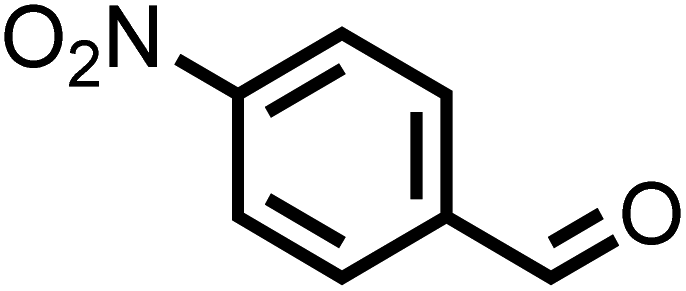
|
40 | 100 |
| 6 |

|

|
71 | 100 |
| 7 |

|

|
98 | 100 |
| 8 |

|

|
52 | 100 |
3.4. Photo-oxidation mechanism
To probe the reactive species during the photocatalytic oxidation of benzyl alcohol to benzaldehyde, free radical quenching experiments were conducted using different scavengers. Butyl alcohol (BuOH), potassium oxalate (K2C2O4), 1,4-benzoquinone (p-BQ) and furfuryl alcohol (FFA) were used as scavengers for hydroxide radicals (˙OH), photogenerated holes (h+), superoxide radicals (˙O2−) and singlet oxygen (1O2), respectively (Fig. 6a).54,55 When BuOH was added into the photocatalytic system, the yield of benzaldehyde was essentially unchanged compared to the control experiment without the scavenger, showing that ˙OH was not involved in the photo-oxidation process. However, the photo-activity was affected by the addition of K2C2O4 and especially of p-BQ and FFA where the benzaldehyde yield fell by 71 and 75%, respectively. From these results, it can be inferred that the main active species for the photo-oxidation of benzyl alcohol are ˙O2− and 1O2 with a minor participation from h+. This is supported by the significant decrease in the benzaldehyde yield when the reaction mixture was bubbled with N2 to remove dissolved oxygen. | ||
| Fig. 6 (a) Photocatalytic activity for selective oxidation of benzyl alcohol over CCNA/CCNU with scavengers or N2 atmospheres; ESR spectra of (b) TEMPO-h+, (c) DMPO-˙O2− and (d) TEMP-1O2 for CNA/CNU and CCNA/CCNU; (e) O2-TPD curves of CNA/CNU and CCNA/CCNU; (f) LSV curves of CNA/CNU and CCNA/CCNU electrodes recorded in O2-saturated Na2SO4 solution. | ||
To further identify the reactive species generated by CCNA/CCNU during the photo-oxidation, electron spin resonance (ESR) experiments were carried out. 2,2,6,6-Tetramethyl-L-piperidine-N-oxyl (TEMPO), 5,5-dimethyl-L-pyrroline N-oxide (DMPO) and 2,2,6,6-tetramethylpiperidine (TEMP) were selected as the trapping agents to probe the generation of h+, ˙O2− and 1O2.56,57 The ESR spectrum of TEMPO displayed a typical triplet signal with an intensity ratio of 1:1:1 (Fig. 6a). After irradiation for 1 h, the intensity of the ESR signal decreased in the order: blank > CNA/CNU > CCNA/CCNU. As TEMPO can be oxidized to the ESR-silent TEMPO+ by photogenerated h+,58 the weak ESR signal indicates more efficient generation of h+ by CCNA/CCNU upon irradiation. Both DMPO and TEMP are ESR inactive (Fig. 6c and d). However, upon irradiation following the addition of CNA/CNU and CCNA/CCNU, the typical quartet ESR signals of DMPO-˙O2− with an intensity ratio of 1:1:1:1 appeared, which implied the formation of ˙O2−.59 The TEMP-1O2 adduct showed a weak characteristic triplet signal with an intensity ratio of 1:1:1.60 The signal intensities were stronger for CCNA/CCNU than for CNA/CNU, showing that under the reaction conditions, more ˙O2− and 1O2 were generated over the former. The surface properties of CCNA/CCNU can affect the formation of these reactive oxygen species (˙O2− and 1O2). Oxygen temperature-programmed desorption measurements showed that despite a 5-fold lower specific surface area (Fig. S9†), more O2 desorbed from CCNA/CCNU than from CNA/CNU (per g) (Fig. 6d). Furthermore, the desorption occurred at higher temperatures than for CNA/CNU, implying a higher affinity of O2 for CCNA/CCNU. Linear sweep voltammetry (LSV) was applied to investigate the O2 reduction ability of CNA/CNU and CCNA/CCNU in O2-saturated Na2SO4 solution. The cathodic current of CCNA/CCNU was greatly enhanced over that observed with CNA/CNU (Fig. 6f). The easier reduction which is instrumental to the formation of the reactive oxygen species can be attributed to the presence of alkali ions in CCNA/CCNU and surface cyano groups, which facilitate the transfer of photogenerated e− to the adsorbed O2.
The influence of alkali ions and surface cyano groups on the activation of O2 and benzyl alcohol was therefore investigated by density functional theory (DFT) calculations. To simplify the DFT calculations, K-doped graphitic carbon nitride with cyano groups is used as a model to represent alkali ion doped graphitic carbon nitride with cyano groups. The optimized adsorption configurations of O2 and benzyl alcohol on pure graphitic carbon nitride and K-doped graphitic carbon nitride with cyano groups are illustrated in Fig. 7. In comparison with graphitic carbon nitride (0.17 eV), a lower adsorption energy (Eads) of O2 (−0.09 and −0.07 eV) was observed for the optimized structure of K-doped graphitic carbon nitride with cyano groups (Fig. 7avs.7b and c). In addition, the optimized distances between O2 and the melon plane (2.83 and 2.85 Å) are shorter than on pure graphitic carbon nitride (2.91 Å). From the significant charge redistribution with electron accumulation on the adsorbed O2 (blue isosurfaces) and a strong electron depletion region on the melon plane (yellow isosurfaces), it can be deduced that the electrons on the melon plane can transfer to the adsorbed O2. Notably, the introduction of K and cyano groups into the graphitic carbon nitride structure results in higher electron density (−0.31 |e| and −0.19 |e|) on the adsorbed O2, which favors the generation of reactive oxygen species. Similarly, the smaller Eads and shorter distance between benzyl alcohol and melon plane are recorded due to the presence of K and cyano groups (Fig. 7dvs.7e). Interestingly, the charge density difference reveals that benzyl alcohol receives electrons from the melon plane in pure graphitic carbon nitride, whereas the benzyl alcohol donates electrons to the melon plane in K-doped graphitic carbon nitride with cyano groups. This implies that the presence of K and cyano groups facilitates the oxidation of benzyl alcohol. As a consequence, the incorporation of K and cyano groups into the graphitic carbon nitride structure not only improves the affinity of substrates (O2 and benzyl alcohol) on the melon plane, but also enhances the charges transfer between substrates and the melon plane.
 | ||
| Fig. 7 Top and side views of the structure with different charge density distributions for O2 and benzyl alcohol adsorption on (a and d) graphitic carbon nitride and (b, c, and e) K-doped graphitic carbon nitride with cyano groups. Eads is the adsorption energy and d is the distance between the substrate and photocatalysts. qO2 and qB represent the accumulated electron charge on O2 and benzyl alcohol, respectively. The blue and yellow isosurfaces represent electron accumulation and depletion with 0.003 e Å−3. The brown, gray, red, green and pink balls represent carbon, nitrogen, oxygen, potassium and hydrogen, respectively; (f) schematic illustration of the proposed photocatalytic mechanism for selective oxidation of alcohols to aldehydes or ketones over CCNA/CCNU. | ||
Based on the above investigation of band alignments, reactive species and DFT calculations, the photocatalytic mechanism of alcohol oxidation over CCNA/CCNU is proposed (Fig. 7f). Under irradiation, driven by the IEF between CCNA and CCNU, the photogenerated e− accumulate in the CB of CCNU, while the photogenerated h+ accumulate in the VB of CCNA. Thus, highly efficient charge separation and transfer of photogenerated e− and h+ were achieved. The h+ in the VB of CCNA participate in the oxidation of the alcohol, converting it to an aldehyde or ketone. At the same time, the e− from the CB of CCNU can be accepted by adsorbed O2 to generate ˙O2− species. The ˙O2− species can either act as an oxidant to directly convert the alcohol to the carbonyl compound or it can be transformed into 1O2 species by reacting with h+. The 1O2 species can then act as the active species for alcohol oxidation.
4. Conclusions
Highly crystalline graphitic carbon nitride homojunctions were synthesized by calcination of a mixture of NH4SCN and urea, which form a low melting eutectic in NaCl/KCl molten salts as the preparation medium. The coherent homojunction interface improved the spatial separation of photogenerated charges by the internal electric field. The highly crystalline structure inhibited the bulk recombination of photogenerated charges and accelerated the charge transfer from the bulk to the surface. Moreover, the introduction of alkali ions and surface cyano groups into the crystalline graphitic carbon nitride structure improves the affinity of substrates and enhances the charge transfer between the adsorbed substrates and photocatalyst. The highly crystalline graphitic carbon nitride obtained by this method proved to be an efficient photocatalyst for the selective oxidation of alcohols. In the oxidation of benzyl alcohol, the benzaldehyde yield of 85% was 17 times higher than for a low crystalline graphitic carbon nitride homojunction material. Its applicability for the selective photo-oxidation of other aromatic and alicyclic alcohols makes the material a very useful photocatalyst.Author contributions
Y. L. C. conceived the idea, prepared the photocatalysts and wrote the original draft. L. M. H. and X. H. L. conducted DFT calculations. H. R. T. carried out microscopy characterisation. S. J. reviewed and edited the manuscript. G. K. C. supervised the project, and reviewed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by FOS ARC Tier 1 grant A-0004107-00-00. And Yanglin Chen wants to thank his beautiful wife Ye Qu for her care and support over the years.Notes and references
- M. Z. Rahman, M. G. Kibria and C. B. Mullins, Chem. Soc. Rev., 2020, 49, 1887–1931 RSC.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- H. Zhou, M. Wang and F. Wang, Chem, 2022, 8, 465–479 CAS.
- Y. Yang, Z. Zeng, G. Zeng, D. Huang, R. Xiao, C. Zhang, C. Zhou, W. Xiong, W. Wang, M. Cheng, W. Xue, H. Guo, X. Tang and D. He, Appl. Catal., B, 2019, 258, 117956 Search PubMed.
- S. Xue, W. Huang, W. Lin, W. Xing, M. Shen, X. Ye, X. Liang, C. Yang, Y. Hou, Z. Yu and X. Wang, Chem Catal., 2022, 2, 125–139 CrossRef CAS.
- S. Wu, X. Liu, X. Lian, G. Tian, C. Janiak, Y. Zhang, Y. Lu, H. Yu, J. Hu, H. Wei, H. Zhao, G. Chang, G. Van Tendeloo, L. Wang, X. Yang and B. Su, Adv. Mater., 2018, 30, 1802173 CrossRef PubMed.
- G. Liu, G. Zhao, W. Zhou, Y. Liu, H. Pang, H. Zhang, D. Hao, X. Meng, P. Li, T. Kako and J. Ye, Adv. Funct. Mater., 2016, 26, 6822–6829 Search PubMed.
- H. Dong, X. Zhang, J. Li, P. Zhou, S. Yu, N. Song, C. Liu, G. Che and C. Li, Appl. Catal., B, 2020, 263, 118270 CrossRef CAS.
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6, 3921–3931 Search PubMed.
- L. Lin, Z. Yu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 6164–6175 CrossRef CAS PubMed.
- Y. Li, B. H. Li, D. Zhang, L. Cheng and Q. Xiang, ACS Nano, 2020, 14, 10552–10561 CrossRef CAS.
- G. Zhang, G. Li, Z. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2017, 56, 13445–13449 CrossRef CAS PubMed.
- G. Zhang, G. Li, T. Heil, S. Zafeiratos, F. Lai, A. Savateev, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 3433–3437 Search PubMed.
- H. Mou, J. Wang, D. Zhang, D. Yu, W. Chen, D. Wang and T. Mu, J. Mater. Chem. A, 2019, 7, 5719–5725 RSC.
- H. Mou, J. Wang, D. Yu, D. Zhang, W. Chen, Y. Wang, D. Wang and T. Mu, ACS Appl. Mater. Interfaces, 2019, 11, 44360–44365 Search PubMed.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17–45 RSC.
- W. Huang, B. C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. A. Zhang, ACS Catal., 2017, 7, 5438–5442 CrossRef CAS.
- M. Liu, C. Wei, H. Zhuzhang, J. Zhou, Z. Pan, W. Lin, Z. Yu, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2022, 61, e202113389 CAS.
- Q. Liang, Z. Li, Z. H. Huang, F. Kang and Q. H. Yang, Adv. Funct. Mater., 2015, 25, 6885–6892 CrossRef CAS.
- Y. Chen, X. Liu, L. Hou, X. Guo, R. Fu and J. Sun, Chem. Eng. J., 2020, 383, 123132 CrossRef CAS.
- J. Sun, W. Zhen and C. Xue, Appl. Surf. Sci., 2023, 623, 157131 CrossRef CAS.
- H. Song, X. Liu, Y. Wang, L. Chen, J. Zhang, C. Zhao, F. He, P. Dong, B. Li, S. Wang, S. Wang and H. Sun, J. Colloid Interface Sci., 2022, 607, 1603–1612 CrossRef CAS.
- R. Fang, Z. Yang, Z. Wang, J. Ran, Y. Yan and L. Zhang, Energy, 2022, 244, 123187 CrossRef CAS.
- N. Meng, J. Ren, Y. Liu, Y. Huang, T. Petit and B. Zhang, Energy Environ. Sci., 2018, 11, 566–571 RSC.
- S. Guo, Z. Deng, M. Li, B. Jiang, C. Tian, Q. Pan and H. Fu, Angew. Chem., Int. Ed., 2016, 55, 1830–1834 CrossRef CAS.
- J. Cheng, Y. Hou, K. Lian, H. Xiao, S. Lin and X. Wang, ACS Catal., 2022, 12, 1797–1808 CrossRef CAS.
- X. Xu, T. Fan, W. Chen, Z. Chen, Y. Dong, H. Fan, W. Fang and X. Yi, Carbon, 2019, 144, 649–658 CrossRef.
- P. Zhang, Y. Tong, Y. Liu, J. J. M. Vequizo, H. Sun, C. Yang, A. Yamakata, F. Fan, W. Lin, X. Wang and W. Y. Choi, Angew. Chem., Int. Ed., 2020, 59, 16209–16217 CrossRef CAS PubMed.
- Y. Xu, X. He, H. Zhong, D. J. Singh, L. Zhang and R. Wang, Appl. Catal., B, 2019, 246, 349–355 CrossRef CAS.
- X. Liu, J. Wang, D. Wu, Z. Wang, Y. Li, X. Fan, F. Zhang, G. Zhang and W. Peng, Appl. Catal., B, 2022, 310, 121304 CrossRef CAS.
- P. Hu, C. Chen, R. Zeng, J. Xiang, Y. Huang, D. Hou, Q. Li and Y. Huang, Nano Energy, 2018, 50, 376–382 Search PubMed.
- L. Lin, W. Ren, C. Wang, A. M. Asiri, J. Zhang and X. Wang, Appl. Catal., B, 2018, 231, 234–241 CrossRef CAS.
- Z. Chen, A. Savateev, S. Pronkin, V. Papaefthimiou, C. Wolff, M. G. Willinger, E. Willinger, D. Neher, M. Antonietti and D. Dontsova, Adv. Mater., 2017, 29, 1700555 CrossRef PubMed.
- H. Wang, Y. Bian, J. Hu and L. Dai, Appl. Catal., B, 2018, 238, 592–598 CrossRef CAS.
- S. Cao, N. Zhou, F. Gao, H. Chen and F. Jiang, Appl. Catal., B, 2017, 218, 600–610 CrossRef CAS.
- B. Li, S. Liu, C. Lai, G. Zeng, M. Zhang, M. Zhou, D. Huang, L. Qin, X. Liu, Z. Li, N. An, F. Xu, H. Yi, Y. Zhang and L. Chen, Appl. Catal., B, 2020, 266, 118650 CrossRef CAS.
- X. Yu, H. Su, J. Zou, Q. Liu, L. Wang and H. Tang, Chin. J. Catal., 2022, 43, 421–432 CrossRef CAS.
- P. Xia, S. Cao, B. Zhu, M. Liu, M. Shi, J. Yu and Y. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5218–5225 CrossRef CAS.
- N. Tian, Y. Zhang, X. Li, K. Xiao, X. Du, F. Dong, G. I. N. Waterhouse, T. Zhang and H. Huang, Nano Energy, 2017, 38, 72–81 CrossRef CAS.
- R. Das, S. Sarkar, R. Kumar, S. D. Ramarao, A. Cherevotan, M. Jasil, C. P. Vinod, A. K. Singh and S. C. Peter, ACS Catal., 2022, 12, 687–697 CrossRef CAS.
- H. Zeng, Z. Li, G. Li, X. Cui, M. Jin, T. Xie, L. Liu, M. Jiang, X. Zhong, Y. Zhang, H. Zhang, K. Ba, Z. Yan, Y. Wang, S. Song, K. Huang and S. Feng, Adv. Energy Mater., 2022, 12, 2102765 CrossRef CAS.
- Y. Chen, Y. Qu, P. Xu, X. Zhou and J. Sun, J. Colloid Interface Sci., 2021, 601, 326–337 CrossRef CAS PubMed.
- G. Zhang, J. Zhu, Y. Xu, C. Yang, C. He, P. Zhang, Y. Li, X. Ren and H. Mi, ACS Catal., 2022, 12, 4648–4658 CrossRef CAS.
- X. Wang, C. H. Liow, A. Bisht, X. Liu, T. C. Sum, X. Chen and S. Li, Adv. Mater., 2020, 27, 2001385 CrossRef PubMed.
- S. Wang, T. He, P. Chen, A. Du, K. Ostrikov, W. Huang and L. Wang, Adv. Mater., 2020, 32, 2001385 CrossRef CAS.
- X. Lu, J. Xie, X. Chen and X. Li, Appl. Catal., B, 2019, 252, 250–259 CrossRef CAS.
- Z. Zhang, Y. Huang, K. Liu, L. Guo, Q. Yuan and B. Dong, Adv. Mater., 2015, 27, 5906–5914 CrossRef CAS PubMed.
- X. Wang, C. H. Liow, A. Bisht, X. Liu, T. C. Sum, X. Chen and S. Li, Adv. Mater., 2015, 27, 2207–2214 CrossRef CAS PubMed.
- Q. Shen, T. Xu, G. Zhuang, Y. Zhuang, L. Sun, X. Han, X. Wang and W. Zhan, ACS Catal., 2021, 11, 12931–12939 CrossRef CAS.
- P. Zhang, D. Sun, A. Cho, S. Weon, S. Lee, J. Lee, J. W. Han, D.-P. Kim and W. Y. Choi, Nat. Commun., 2019, 10, 940 CrossRef PubMed.
- H. Zhang, X. Li, K. S. Chooi, S. Jaenicke and G. K. Chuah, Catal. Today, 2021, 375, 558–564 CrossRef CAS.
- X. Zhang, X. Ke and H. Zhu, Chem. Eur. J., 2012, 18, 8048–8056 CrossRef CAS PubMed.
- M. Zhou, P. Yang, S. Wang, Z. Luo, C. Huang and X. Wang, ChemSusChem, 2018, 11, 3949–3955 CrossRef CAS PubMed.
- R. Tang, D. Gong, Y. Zhou, Y. Deng, C. Feng, S. Xiong, Y. Huang, G. Peng, L. Li and Z. Zhou, Appl. Catal., B, 2022, 303, 120929 CrossRef CAS.
- H. Wang, S. Jiang, S. Chen, D. Li, X. Zhang, W. Shao, X. Sun, J. Xie, Z. Zhao, Q. Zhang, Y. Tian and Y. Xie, Adv. Mater., 2016, 28, 6940–6945 CrossRef CAS PubMed.
- J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed.
- Y. Lin, Y. Yang, W. Guo, L. Wang, R. Zhang, Y. Liu and Y. Zhai, Appl. Surf. Sci., 2021, 560, 150029 Search PubMed.
- Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2014, 4, 774–780 CrossRef CAS.
- X. Wang, J. Meng, X. Zhang, Y. Liu, M. Ren, Y. Yang and Y. Guo, Adv. Funct. Mater., 2021, 31, 2010763 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01460f |
| This journal is © The Royal Society of Chemistry 2023 |