
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic charge collecting mechanisms of cobalt phosphate on hematite photoanodes studied by photoinduced absorption spectroscopy †
Dongfeng
Li
ab,
Ruifang
Wei
ac,
Heng
Yin
a,
Hemin
Zhang
 d,
Xiuli
Wang
*a and
Can
Li
*abc
d,
Xiuli
Wang
*a and
Can
Li
*abc
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian National Laboratory for Clean Energy, Dalian 116023, China. E-mail: canli@dicp.ac.cn; xiuliwang@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
cDepartment of Chemical Physics, University of Science and Technology of China, Hefei 230026, China
dCollege of Materials Science and Engineering, Sichuan University, Engineering Research Center of Alternative Energy Materials and Devices, Ministry of Education, Chengdu 610065, China
First published on 20th January 2023
Abstract
Reaction sites, surface states, and surface loaded electrocatalysts are photoinduced charge storage sites and critical to photoelectrochemical (PEC) performance, however the charge transfer mechanisms involved in the three remain poorly understood. Herein, we studied the charge transfer processes in hematite (Fe2O3) without/with loaded cobalt phosphate (CoPi) by operando photoinduced absorption (PIA) spectroscopy. The loaded CoPi receives trapped holes in surface states at low potential and directly captures holes in the valence band at high potential. Through the dynamic hole storage mechanisms, loaded CoPi on Fe2O3 facilitates spatial charge separation and serves as a charge transfer mediator, instead of serving as a catalyst to change the water oxidation mechanism (constant third-order reaction). The spatial separation of photoinduced charges between Fe2O3 and CoPi results in more long-lived holes on the Fe2O3 surface to improve PEC water oxidation kinetically. The dynamic charge collecting mechanism sheds light on the understanding and designing of electrocatalyst loaded photoanodes.
Introduction
Photoelectrochemical (PEC) water splitting is a promising strategy to convert solar energy into clean hydrogen fuel.1 In PEC water splitting, the water oxidation process by a photoanode is generally regarded as a bottleneck and strongly relies on the surface hole density, which is related to various hole storage sites.2–4 A high-performance photoanode is usually composed of a conductive back contact, light-absorbing semiconductor and surface loaded electrocatalyst. The multiple interfaces (semiconductor|electrocatalyst, semiconductor|solution, and electrocatalyst|solution) and charge storage sites (reaction sites, surface states of the photoanode, and the loaded electrocatalyst) complicate the surface reaction mechanisms.5,6 Therefore, understanding the charge transfer mechanisms among reaction sites, surface states of the photoanode and the loaded electrocatalyst in photoelectrocatalysis is critical.Among various photoanodes, hematite (Fe2O3) has received lots of attention owing to its abundant, nontoxic and scalable characteristics.7,8 There have been many research studies on revealing hole storage sites related to PEC properties in the bare Fe2O3 system, including reaction intermediates,9–11 multi-hole cooperation clusters4,12 and filling surface trap states.13 In order to promote PEC performances, the OER catalysts, such as cobalt phosphate (CoPi)14 and NiFe(OH)x electrocatalysts,15–17 are usually loaded on the Fe2O3 surface as new hole storage sites. The loaded electrocatalyst negatively shifts the onset potential and improves the photocurrent, while it definitely influences or fully changes the properties of hole storage sites resulting in complicated charge transfer processes. In particular, the promoted role of the electrocatalyst on the Fe2O3 surface is a subject of debate, particularly CoPi, a common and effective covering layer.6,14,18–21 There appear two opposite explanations about the role of CoPi including a catalyst and spectator by (photo)electrochemical and spectroscopic techniques.18,22,23 Boettcher claimed that potential-sensing electrochemical atomic force microscopy (AFM) in operando gave the direct evidence of loaded CoPi as a catalyst by tracking surface potential variation of CoPi in photoelectrocatalysis.18–20 Nevertheless, in situ spectroscopy likewise offered direct proof of the CoPi layer as a spectator through similar spectra shapes and charge transfer dynamics regardless of transient or steady state spectroscopy.14,24–26 Furthermore, spectroscopic results also revealed a crucial point that the contribution of the CoPi layer to photocurrent was lower than 5%.22 However, these research studies were mainly devoted to revealing the reaction sites of the water oxidation reaction of Fe2O3 modified with an electrocatalyst and ignore interfacial dynamic processes.
In principle, both the surface states of Fe2O3 and loaded electrocatalyst will compete for photoinduced holes with reaction sites on the photoanode surface. The reduction reaction of photoinduced holes by surface states or the electrocatalyst is dependent on their thermodynamic driving force, and should show distinct dynamics/kinetics. Generally, compared with free holes in the valence band (VB), the trapped holes in surface states possess a lower PEC activity and tend to recombine with photoinduced electrons. If the loaded electrocatalyst plays a leading role in surface catalysis, a surface reaction kinetics similar to electrocatalysis is expected, vice versa. But whether the trapped holes interact with the loaded electrocatalyst and its role in photoelectrocatalysis is unclear. More importantly, it is also not studied whether there is charge transfer between the loaded electrocatalyst and reaction sites of Fe2O3 and its crucial roles if loaded electrocatalysts do not serve as a catalyst. Thus, the dynamic interaction between the electrocatalyst and Fe2O3 and its role in PEC performances are quite important and deserve to be studied deeply.
In this work, the charge transfer processes among surface states, reaction sites and the coated electrocatalyst for Fe2O3 without/with CoPi loading were studied by photoinduced absorption (PIA) spectroscopy. Loaded CoPi on the Fe2O3 surface does not remove the mid-gap surface states. The dynamic transition in collecting holes for loaded CoPi from surface states to the VB following increasing potential is discussed. The roles of the dynamic charge collecting mechanism in PEC water oxidation are revealed. This dynamic mechanism sheds light on the understanding and designing of electrocatalyst loaded photoanodes.
Results and discussion
Determining the distribution of surface states in bare Fe2O3 and CoPi/Fe2O3
Fe2O3 photoanodes were prepared by a combined method of solvothermal and high-temperature calcination.27 A subsequent electrodeposition was applied to obtain CoPi/Fe2O3 photoanodes.14 The cyclic voltammetry (CV) curve of CoPi/Fe2O3 shows negligible current at the applied bias of 0.6–1.6 V vs. RHE in the dark (Fig. S1†), while the CV curve of CoPi displays obvious current for cobalt oxidation or water oxidation. The negligible current of CoPi/Fe2O3 suggests a dense and non-porous Fe2O3 film.16 For the PEC performances, loaded CoPi on the Fe2O3 surface negatively shifts onset potential, improves photocurrent, and enhances transient spikes (Fig. 1a and b), in line with previous reports.14,23,28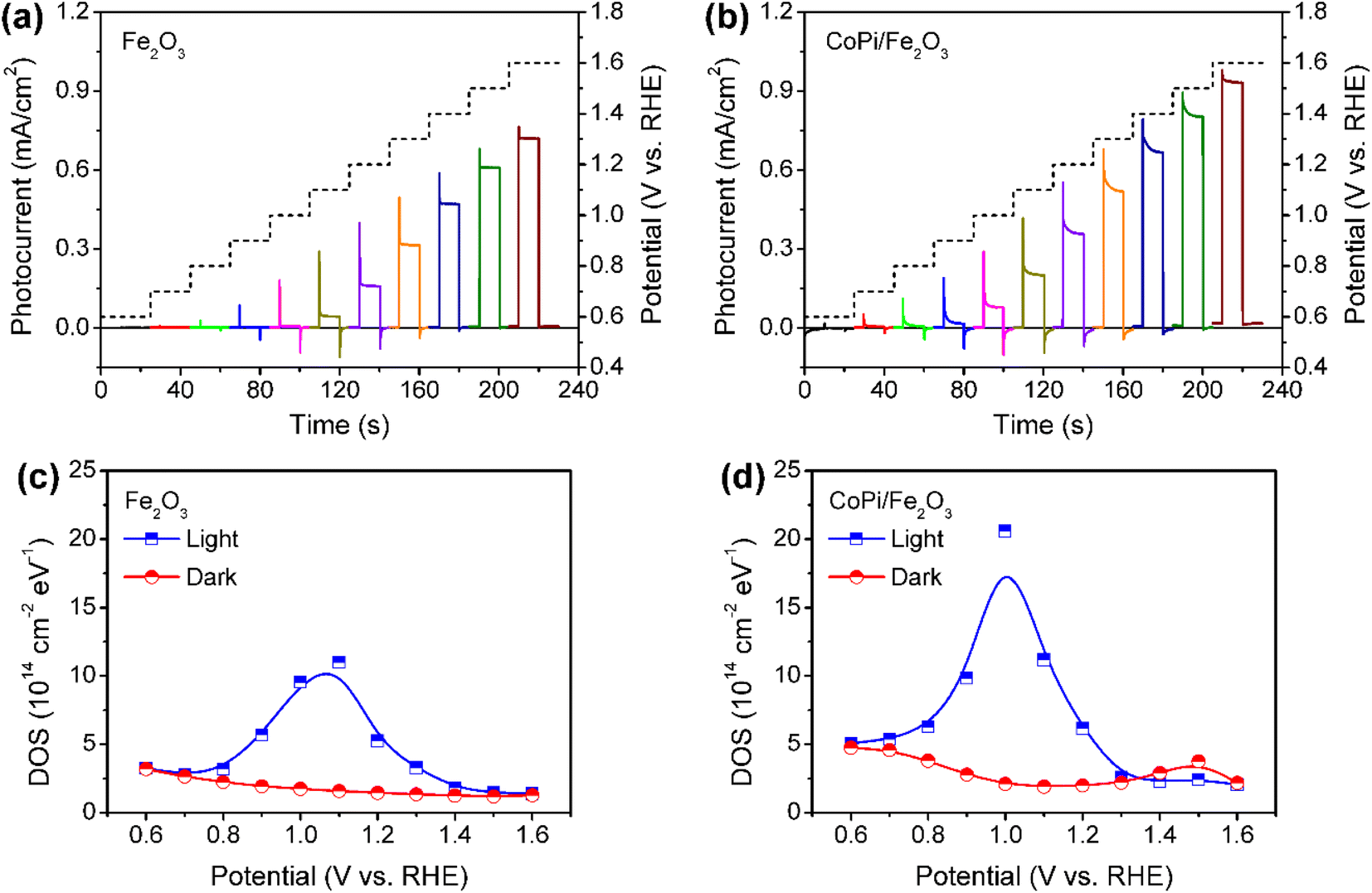 | ||
| Fig. 1 Potential-dependent transient photocurrents for (a) bare Fe2O3 and (b) CoPi/Fe2O3 photoanodes under chopped illumination (AM 1.5 G: 100 mW cm−2). The applied potential is shown by a dashed line in the right axis. DOS of surface states in (c) bare Fe2O3 and (d) CoPi/Fe2O3 as a function of potential under illumination (456 nm LED: 5 mW cm−2) and in the dark. | ||
In order to determine the charge transfer processes among reaction sites, surface states and the surface loaded electrocatalyst, the potential distribution of mid-gap or surface states is first investigated by electrochemical impedance spectroscopy (EIS) under illumination and in the dark. The EIS results (Fig. 1c and d) fitted with the equivalent circuit (Fig. S2†) show that bare Fe2O3 and CoPi/Fe2O3 photoanodes exhibit a similar distribution of density of states (DOS = C/e, where C is the capacitance and e is the elementary charge.) in the 0.7–1.4 VRHE range. Loading CoPi on the Fe2O3 surface shifts the distribution of surface states about 100 meV towards lower potential. Moreover, CV results in Fig. S3† after PEC water oxidation also display similar reduction waves in the 0.8–1.4 VRHE range for bare Fe2O3 or in the 0.6–1.1 VRHE range for CoPi/Fe2O3. Both EIS and CV results demonstrate that there are similar distributions of surface states in bare Fe2O3 and CoPi/Fe2O3 photoanodes in the 0.8–1.4 VRHE range. Therefore, although loading CoPi eliminates fractional surface states and releases Fermi-level pinning revealed by fast decay in chopped OCP results (Fig. S4†), the surface states in the 0.8–1.4 VRHE range still exist in CoPi/Fe2O3.
The surface states in Fe2O3 always act as hole trap states and have been described as water oxidation intermediates9,11 or recombination sites,29 because Fe2O3 is an n-type semiconductor with a Fermi level close to the conduction band (CB) edge. Thus, the surface states in the 0.8–1.4 VRHE range are tentatively assigned as hole trap states for both bare Fe2O3 and CoPi/Fe2O3 photoanodes. In principle, with the existence of surface states, the photoinduced holes in the VB might be consumed by indirect paths (water oxidation by trapped holes: h+surface states + H2O → O2, or trap-mediated recombination) for bare Fe2O3, besides direct paths (water oxidation by VB holes: h+VB + H2O → O2, or recombination). When CoPi is loaded on Fe2O3, the surface states can bring new charge transfer paths between the photoinduced holes, surface states and the loaded CoPi. Namely, the photoinduced holes in Fe2O3 can move to CoPi via surface states (sequential hole transfer) besides the direct transfer of VB holes to CoPi, which may play a vital role in CoPi/Fe2O3 photoelectrocatalysis.
Charging sites of CoPi/Fe2O3 at low and high potentials
Next, the photoinduced charge transfer processes were studied to reveal the direct or sequential hole transfer processes in CoPi/Fe2O3 with the existence of surface states. Based on the position of the energy levels, the oxidation ability of trapped holes (surface states: 0.8–1.4 VRHE) should be lower than that of free holes (VB: 2.5 VRHE). A logical speculation is that a probe with <1.4 VRHE redox potential can be oxidized by trapped holes, otherwise there will be no oxidation as schematically shown in Fig. 2a. Thus, CoPi (Co2+/3+: ∼1.10 VRHE) and NiOOH (Ni2+/3+: ∼1.38 VRHE) electrocatalysts were chosen as probes and deposited on the Fe2O3 surface (Fig. 2b). The role of surface states in charge transfer was first studied by PIA spectroscopy at low potential (0.8 VRHE). The wavelength-dependent PIA responses of CoPi, bare Fe2O3, CoPi/Fe2O3 and NiOOH/Fe2O3 at 0.8 VRHE are shown in Fig. 2c. Bare Fe2O3 and CoPi display negligible PIA intensity, illustrating that the bulk/surface recombination is too fast to probe. In contrast, CoPi/Fe2O3 presents an appreciable PIA intensity, indicating the effective hole transfer from Fe2O3 to CoPi. Moreover, the consistence of PIA spectra of CoPi/Fe2O3 and spectroelectrochemical absorption (SECA) spectra of CoPi (Co2+/3+, for calculation details, see Fig. S5†) in Fig. 2c indicates that CoPi is the main hole storage site at 0.8 VRHE. Amazingly, no PIA response is observed in NiOOH/Fe2O3 at 0.8 VRHE, corresponding to no hole transfer from Fe2O3 to NiOOH and in line with negligible photocurrent in Fig. S6.† Since free holes in the VB of Fe2O3 can oxidize both CoPi and NiOOH, the distinct PIA response should result from the CoPi oxidation by the long-lived trapped holes in the surface states of Fe2O3. Thus, at low potential, the photoinduced holes in Fe2O3 mainly undergo sequential transfer processes from the VB to surface states then to CoPi. The spatial separation between holes in CoPi and electrons in Fe2O3 prolongs charge lifetimes (Fig. S7†) by suppressing charge recombination at 0.8 VRHE. Moreover, transient absorption spectra of CoPi/Fe2O3 at 100 ms at 0.8 VRHE in Fig. S7e† display a broad absorption which is quite similar to the absorption spectra of oxidized Co3+, demonstrating that the long-lived transient absorption signals in CoPi/Fe2O3 at lower potential should originate from cobalt oxidation of Co2+/3+ rather than passivating surface states or increasing band-bending.14 A combination of negligible photocurrent (Fig. 1b) and appreciable PIA (Fig. 2c) at 0.8 VRHE demonstrates that the oxidized CoPi cannot contribute to the water oxidation processes. Therefore, at low potential, the loaded CoPi on the Fe2O3 surface serves as the charging site for photoinduced holes, and prolongs the lifetime of holes by suppressing recombination, but with no catalysis effect for water oxidation. | ||
| Fig. 2 (a) Schematic of charge transfer between surface states and probes. Red and blue arrows refer to the transfer processes of photoinduced holes and electrons, respectively. ‘×’ denotes no charge transfer. (b) CV and corresponding redox potentials of CoPi and NiOOH. (c) Wavelength-dependent PIA spectra of bare Fe2O3, CoPi, CoPi/Fe2O3 and NiOOH/Fe2O3 at low potential of 0.8 VRHE; left axis is PIA spectra and right axis is the SECA spectrum of CoPi (Co2+/3+: 1.3 VRHE – 1.0 VRHE). (d) Wavelength-dependent PIA spectra of bare Fe2O3, CoPi and CoPi/Fe2O3 at high potential of 1.4 VRHE; left axis is PIA spectra and right axis is the SECA spectrum of CoPi (Co3+/4+: 1.7 VRHE – 1.4 VRHE). Pump wavelength in PIA is 456 nm (5 mW cm−2). | ||
Then, the charge transfer processes were studied by PIA spectroscopy at high potential of 1.4 VRHE to reveal the main charging sites during the OER. In wavelength-dependent PIA spectra (Fig. 2d), bare Fe2O3 and CoPi/Fe2O3 display similar spectral shapes from 500 to 900 nm, while CoPi still shows no photo-response. The PIA spectra shape is in line with the absorption spectra of photoinduced holes in Fe2O3 and different from absorption properties of oxidized cobalt species in CoPi (Co2+/3+ shown in Fig. 2c and Co3+/4+ shown in Fig. 2d).12,29,30 These results suggest that the charging sites of bare Fe2O3 and CoPi/Fe2O3 photoanodes at high potential (1.4 VRHE) may be mainly on the Fe2O3 surface. Moreover, the similar transient absorption results of bare Fe2O3 and CoPi/Fe2O3 photoanodes further confirm the assignment of same charging sites (Fig. S8†).14,24
Besides the spectral shape, an obvious increase of PIA amplitude for CoPi/Fe2O3 is observed relative to that of Fe2O3 in Fig. 2d. In order to uncover the origin of the increased PIA amplitude, a quantitative comparison of ratios of PIA amplitudes (PIACoPi/Fe2O3/PIAFe2O3 ≥1.8) and photocurrents (JCoPi/Fe2O3/JFe2O3 ∼ 1.8) for the Fe2O3 and CoPi/Fe2O3 photoanodes is displayed in Figs. S8a† and S8b. The ratios display the proportional relationship between photocurrent and hole density (J ∝ [h+]1). However, CoPi/Fe2O3 at this potential (1.4 VRHE) and illumination (5 mW cm−2) displays a third-order kinetics (J ∝ [h+]3) which will be further discussed in detail below, suggesting that the increased PIA amplitude should lead to a much higher ratio of photocurrents (1.833 ≈ 5.8). This disagreement indicates that the increased PIA amplitude is not only derived from the increased holes in Fe2O3, but also from the effective hole storage in loaded CoPi. Since the PIA data are obtained from quasi-steady-state PIA spectra measured with a continuous pulsed light on, the initial states of cobalt ions in CoPi/Fe2O3 for PIA at 1.4 VRHE should be the Co3+ state due to the sequential and direct hole transfer processes (Fig. S9c†). The increased PIA amplitude in CoPi/Fe2O3 (Fig. 2d) is derived from the oxidation process of Co3+/4+ by photoinduced holes in the VB of Fe2O3 besides increased holes in Fe2O3, since the trapped holes in surface states cannot oxidize Co3+/4+ (redox potential: 1.47 VRHE). The Co3+/4+ oxidation process via direct hole transfer contributes little to the photocurrent of CoPi/Fe2O3 at 1.4 VRHE, which should be less than ∼1 μA by evaluation from the PIA amplitude in the I–t curve in Fig. S10.† Loaded CoPi on the Fe2O3 surface still exhibits negligible catalysis at high potential. Therefore, at high potential (1.4 VRHE), the main charging sites of photoinduced holes in the CoPi/Fe2O3 photoanode are the reaction sites on the Fe2O3 surface and Co3+/4+ in the CoPi electrocatalyst. Moreover, the same as that at low potential (0.8 VRHE), the spatial separation between holes in CoPi and electrons in Fe2O3 also prolongs charge lifetimes by suppressing charge recombination (Fig. S11†) at high potential (1.4 VRHE).
Potentiodynamic charge storage processes
Wavelength-dependent PIA spectra of bare Fe2O3, CoPi and CoPi/Fe2O3 demonstrate that loaded CoPi displays two distinct hole-storage mechanisms, where loaded CoPi on the Fe2O3 surface receives trapped holes in surface states and free holes in the VB at low and high potentials, respectively. In order to further uncover potential-dependent charge storage mechanisms, the PIA spectra were recorded at a fixed probe wavelength and varying potentials (0.6 VRHE ≤ potential ≤ 1.6 VRHE). Since the probe wavelength of > 650 nm in PIA spectra has been assigned to photoinduced holes on the Fe2O3 surface by the hole scavenger and potential-dependent TAS experiments (Fig. S8†),15,31 the probe wavelength of 694.3 nm was used to monitor photoinduced hole dynamics on the Fe2O3 surface. The reasonability is confirmed by the linear relationship of PIA intensity and photoinduced hole density in Fe2O3 in Fig. S10.† Moreover, the linear relationship between absorbance and high-valent cobalt species (holes) in Fig. S9† demonstrates that the probe wavelength of 694.3 nm is also suitable for monitoring holes in CoPi.Fig. 3a–c show the potential-dependent PIA spectra of bare Fe2O3, CoPi and CoPi/Fe2O3 photoanodes. There are three regions for a PIA spectrum, including baseline in the dark (0–0.5 s), photoinduced species accumulation (0.5–6 s) and species consumption after turning the light off (6–10 s). The variation trend of PIA amplitudes with applied potential is extracted and shown in Fig. 3d. CoPi shows no PIA response at 0.6–1.6 VRHE, suggesting that PEC charging of CoPi is only through collecting extraneous charges from Fe2O3. For the bare Fe2O3 photoanode, the PIA amplitudes increase monotonically to a saturation intensity. The invariable PIA intensity above 1.2 VRHE can originate from the signal overlap of inactive holes (surface states) and active holes (VB), which is a common phenomenon for the Fe2O3 photoanode by the solvothermal method.29 Increasing the external potential results in higher percentage of active holes, but doesn't influence PIA intensity. However, potential-dependent PIA amplitudes of CoPi/Fe2O3 display a complicated trend, first increasing (0.7–1.0 VRHE) and then decreasing (1.0–1.6 VRHE) resulting in a peak at 1.0 VRHE. Moreover, at the same potential, the PIA amplitudes of CoPi/Fe2O3 are greater than those of bare Fe2O3. Because the extinction coefficients of Co2+/Co3+ (ε1 = 7332 M−1 cm−1) and Co3+/Co4+ (ε2 = 1833 M−1 cm−1) in CoPi are larger than the extinction coefficient of photoinduced holes (ε = 637 M−1 cm−1) in bare Fe2O3 (Fig. S10†), the increased PIA amplitude in the whole applied potential range (0.6–1.6 VRHE) should originate from hole transfer from the Fe2O3 surface to CoPi. Combined with all PIA results in Fig. 2 and 3, the complicated variation of PIA amplitudes in CoPi/Fe2O3 can be attributed to the transition from oxidation of Co2+/Co3+ by trapped holes in surface states at low potentials to oxidation of Co3+/Co4+ by free holes in the VB at high potentials. That's to say, the difference of extinction coefficients should be the origin of the complicated trend in potential-dependent PIA amplitudes for CoPi/Fe2O3. Furthermore, the potential-dependent PIA spectra of CoPi/Fe2O3–air and CoPi/Fe2O3–Ar (Fig. S12†) with modified surface states display a similar change trend, suggesting that the potential-dependent PIA variation in electrocatalyst-coated Fe2O3 can be related to the distribution of surface states, free holes in the VB and oxidation states of the electrocatalyst.
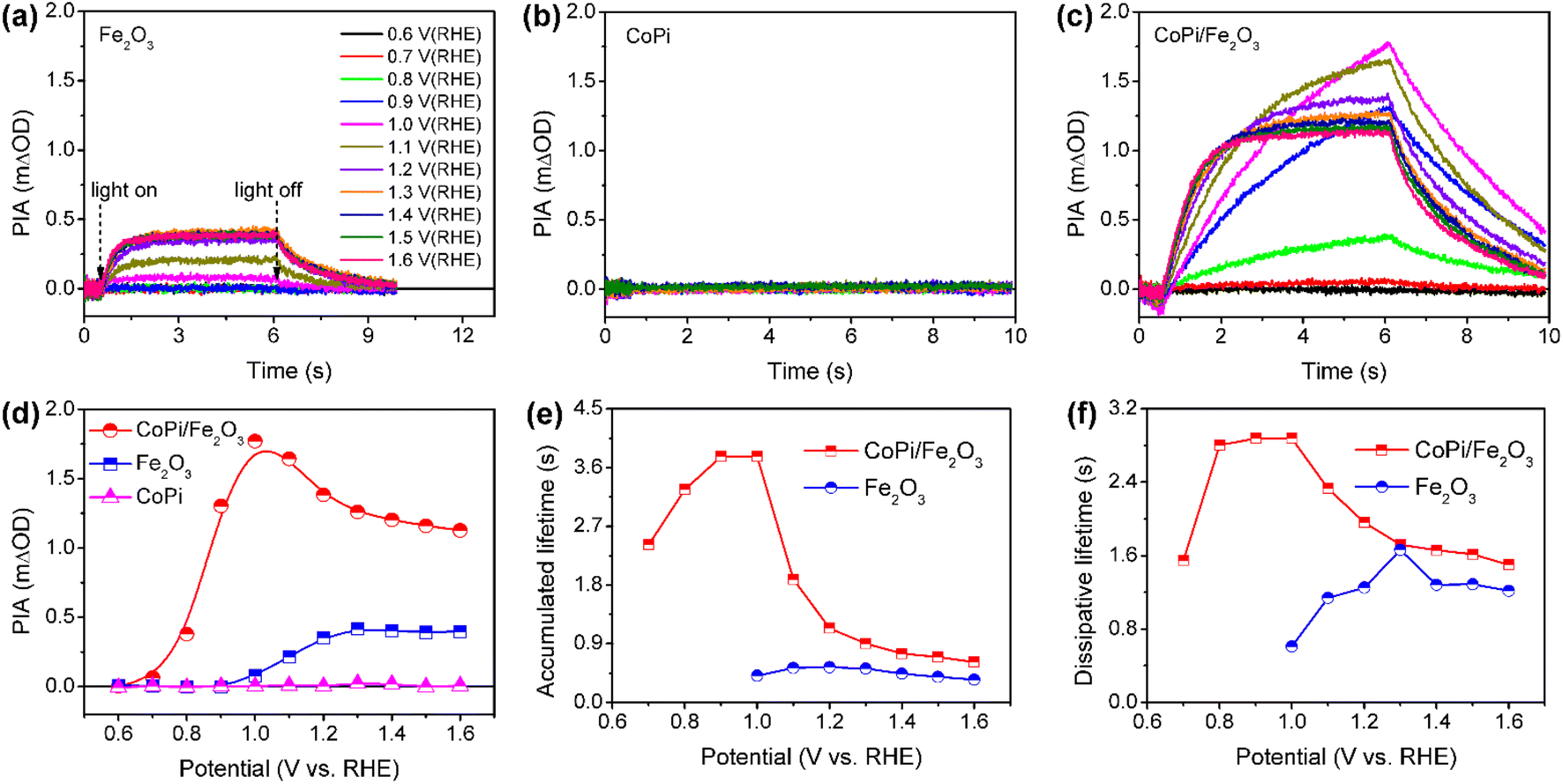 | ||
| Fig. 3 PIA responses at various potentials for bare Fe2O3 (a), CoPi (b) and CoPi/Fe2O3 (c). (d) PIA amplitudes following increasing potential extracted from Fig. 2a–2c. Average lifetimes (τave) of (e) accumulated and (f) dissipative processes for bare Fe2O3 and CoPi/Fe2O3 photoanodes in (a and c). Pump wavelength is 456 nm (5 mW cm−2); probe wavelength is 694.3 nm; colors of curves at different potentials in (a−c) are identical. | ||
Effect of stored holes in CoPi on charge transfer
After knowing the potentiodynamic hole storage mechanisms of loaded CoPi, the dissipation paths of holes in CoPi are further studied to uncover the function of CoPi in PEC water oxidation. By comparing the potential-dependent PIA responses of bare Fe2O3 and CoPi/Fe2O3 in Fig. 3a and c, the PIA spectra of the CoPi/Fe2O3 photoanode display slower accumulation and dissipation processes. In order to specifically compare the kinetic change, the lifetimes of PIA accumulation and dissipation processes for bare Fe2O3 and CoPi/Fe2O3 photoanodes are fitted using a bi-exponential function in Fig. 3e and f (fitted results are shown in Table S1†). At 0.7–0.9 VRHE, drastic recombination leads to no accumulation processes in bare Fe2O3. After loading CoPi on the Fe2O3 surface, the sequential hole transfer through surface states to oxidize Co2+/3+ results in the long accumulation lifetimes of 2.4∼3.8 s. With potential above 1.0 VRHE, the accumulation processes of the bare Fe2O3 photoanode show an average lifetime of ∼0.45 s, which is faster than the shortest accumulation lifetime (∼0.62 s) in the CoPi/Fe2O3 photoanode. Due to concentrated electrolyte (1 M NaOH) and weak illumination (5 mW cm−2) in PIA measurements, the slow accumulation lifetimes in the CoPi/Fe2O3 photoanode shouldn't be due to the CoPi deposition caused by the local pH change under light irradiation.32 Moreover, with the increase of applied potential, the first increasing then decreasing accumulation lifetimes in Fig. 3e also exclude diffusion-controlled CoPi deposition. Thus, this slow accumulation process is attributed to slow hole hopping from the CoPi/Fe2O3 interface to the CoPi bulk after cobalt oxidation at the millisecond timescale. The prolonged accumulation lifetimes indicate that loaded CoPi induces new charge separation paths near the Fe2O3 surface for promoting charge separation via cobalt oxidation by photoinduced holes. The smaller accumulation lifetimes with potential above 1.2 VRHE than that with potential below 1.0 VRHE suggest that the direct hole transfer to oxidize Co3+/4+ is faster than the sequential hole transfer to oxidize Co2+/3+.For dissipation processes, except the short lifetime (∼0.6 s) near the onset potential, bare Fe2O3 displays an almost constant dissipation lifetime about 1.2 s, in line with the understanding of the water oxidation reaction by free holes in the VB.3 Namely, the increased potential for bare Fe2O3 is used for suppressing recombination and increasing density of free holes in the VB, rather than changing the enthalpy or activation energy. In contrast, the dissipation lifetimes of CoPi/Fe2O3 exhibit first an increased then decreased trend, resulting in the longest lifetime about 2.9 s at 1.0 VRHE. Due to the weak contribution to water oxidation of CoPi (Fig. 2c and d), the slow PIA decay should be attributed to the recombination process between stored holes in CoPi and photoinduced electrons on the Fe2O3 surface. The dissipation lifetimes of 1.6 ∼ 2.9 s (0.7–1.0 VRHE) directly demonstrate the effective suppression of charge recombination by Co3+ states. When external potential increases to 1.6 VRHE, the dissipative average lifetime (∼1.6 s) is still longer than the dissipation process of bare Fe2O3 (∼1.2 s), indicating that the oxidized Co4+ states also suppress the charge recombination effectively. The suppressed charge recombination is also supported by the TAS and TPC results (Fig. S7 and S11†).
Then, the charge recombination between oxidized CoPi and Fe2O3 was investigated at high potential, to confirm the roles of oxidized Co4+ in PEC processes. In principle, if the Co4+ concentration is high enough in CoPi for the CoPi/Fe2O3 photoanode at the ground state, photogenerated hole transfer from Fe2O3 to CoPi under illumination will be blocked. In consequence, more recombination events of photoinduced electrons and holes in CoPi will be observed, which will lead to decreased PIA amplitude. In order to confirm this hypothesis, CoPi was loaded on the porous Fe2O3. Since the conductive substrate (FTO) is partially bare, there exist triphasic interfaces of FTO/CoPi, FTO/Fe2O3 and CoPi/Fe2O3, as shown in Fig. S13a.† When potential is applied to CoPi/porous-Fe2O3, CoPi can be directly oxidized to high-valent cobalt species (Co4+) resulting in typical redox waves (Fig. S13b†). Then the charge transfer processes of CoPi/porous-Fe2O3 are studied using PIA. CoPi/porous-Fe2O3 shows similar potential-dependent PIA spectra to that of the above dense CoPi/Fe2O3 under low light intensity irradiation of 5 mW cm−2 (Fig. S14†). As expected, the PIA spectra in Fig. 4a display a negative amplitude at high potential (1.5 VRHE) and under illumination (30 mW cm−2). Additionally, the chopped photocurrent at 1.5 VRHE in Fig. 4b shows a disappearing transient spike, which can be understood by electron shunt transfer. Photoinduced electrons don't move from the porous Fe2O3 surface to the bulk then to FTO, but from the porous Fe2O3 surface to CoPi then to FTO resulting in a positive current. The oxidized CoPi can effectively capture photoinduced electrons, resulting in a flow of electrons at the moment of suddenly turning the light on/off. Because the flow of electrons induced by CoPi is opposite to the flow of back electrons, the disappearing transient spikes are observed. Thus, both PIA and photocurrent results of porous Fe2O3 with loaded CoPi directly demonstrate the possibility of charge recombination between Co4+ species and photoinduced electrons of Fe2O3. Therefore, at high potential, loaded CoPi on dense/porous-Fe2O3 serves as a mediator to fast capture holes in the VB of Fe2O3 followed by slow consumption by photogenerated electrons of the Fe2O3 surface, which can slow down the recombination process and produce more long-lived holes for water oxidation.
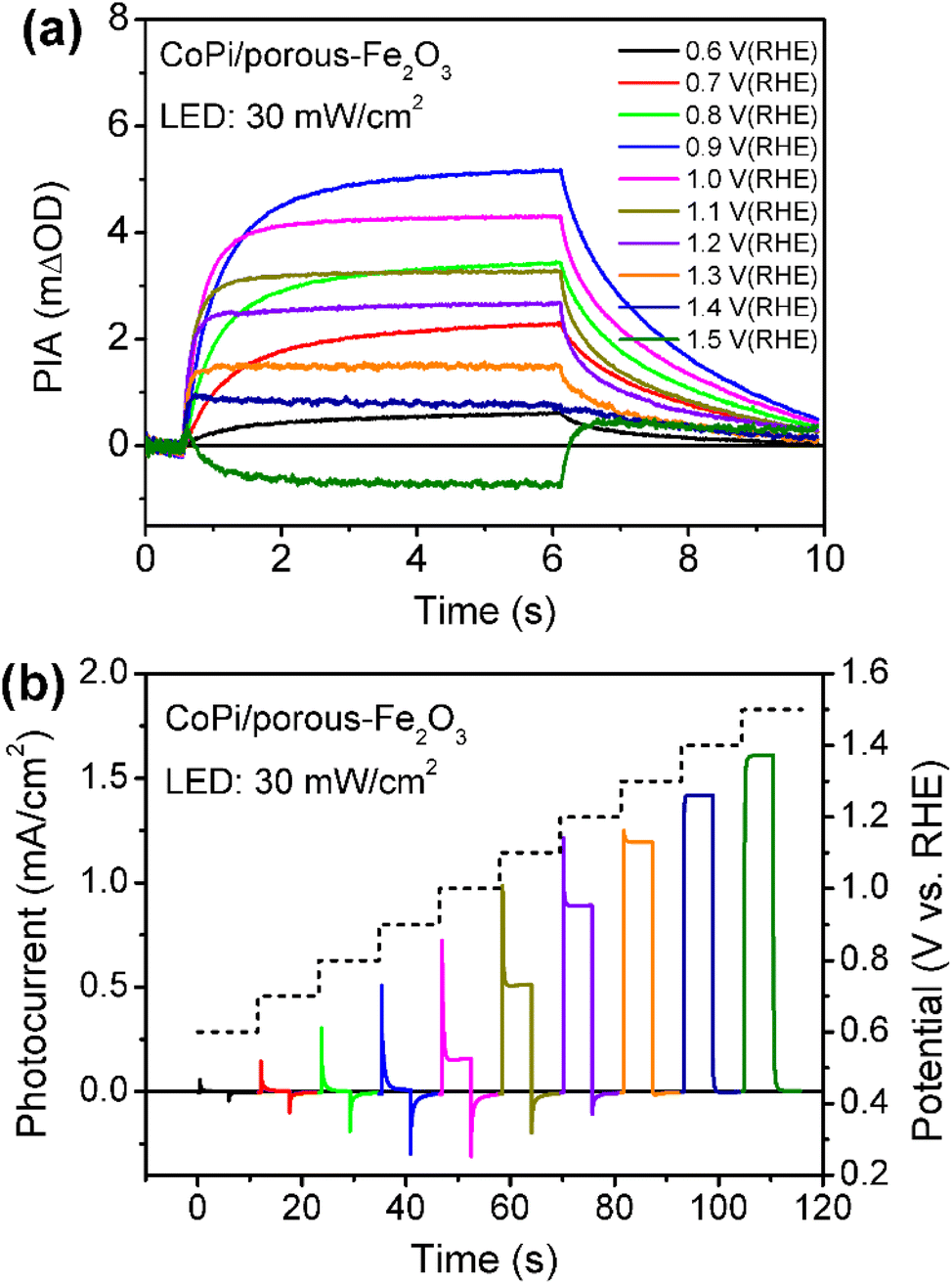 | ||
| Fig. 4 Potential-dependent PIA spectra (a) and chopped photocurrents (b) for CoPi/porous-Fe2O3. Pump wavelength is 456 nm (30 mW cm−2); probe wavelength is 694.3 nm; colors of curves at different potentials in (a and b) are identical. | ||
To further understand the roles of electrocatalyst-mediated charge transfer processes in the PEC water oxidation reaction, we correlate reaction rate and hole density for Fe2O3 without/without CoPi loading at increased potential. The light-intensity-dependent PIA spectra were recorded for bare Fe2O3 and CoPi/Fe2O3 photoanodes. In Fig. 5a and b, PIA amplitudes for the two photoanodes increase with light intensity increasing at a fixed potential, suggesting more holes on the Fe2O3 surface and promotional oxidation for CoPi. According to previous reports,4,12 the photoinduced hole can directly be regarded as a reactant, and its reaction parameters can be quantified by rate analysis (r = J = k × [h+]β, where r is the reaction rate and equivalent to current density (J), k is the rate constant, [h]+ is the hole density and β is the reaction order). Due to the linear relation of PIA intensity and hole concentration in Fig. S15,† the reaction orders at changed potential from 0.6 VRHE to 1.6 VRHE are calculated and shown in Fig. 5c and d. Although PIA amplitudes of Fe2O3 under fixed illumination display saturation above 1.2 VRHE due to surface states, there is an effective rate analysis at the fixed potential and changed illumination in Fig. 5a. This result demonstrates that there is a linear scaling relationship of photoinduced hole density distribution between the VB and surface states in the present study. With potential increase, the reaction order of Fe2O3 displays first an increase to a maximum value (from zero to fourth) and then a decrease to a constant (from fourth to third) value in Fig. 5e, implying that surface states participate in or modulate the surface reaction. After loading CoPi on the Fe2O3 surface, in line with the scaling relationship of hole density with PIA (Fig. S15d†), a similar linear shape for the reaction order–potential relation is obtained. Particularly, the maximum (fourth) and constant (third) reaction orders for CoPi/Fe2O3 arise at lower potential than bare Fe2O3 (Fig. 5e), illustrating similar functions of surface states and coated CoPi electrocatalyst. Furthermore, Fig. 5f demonstrates that the relationship of hole density (electro-induced absorption (EIA) intensity) and current in the CoPi electrocatalyst obeys an Eying-like equation (log(J) ∝ [h+]) reported by Hong Nhan Nong,33 rather than rate analysis that shows a high-order reaction (β ≈ 9) for electrocatalytic current from microampere to milliampere. These results demonstrate that surface states and the loaded electrocatalyst can directly receive and store a part of holes from the VB of hematite, but don't affect the reaction kinetics of reaction sites.
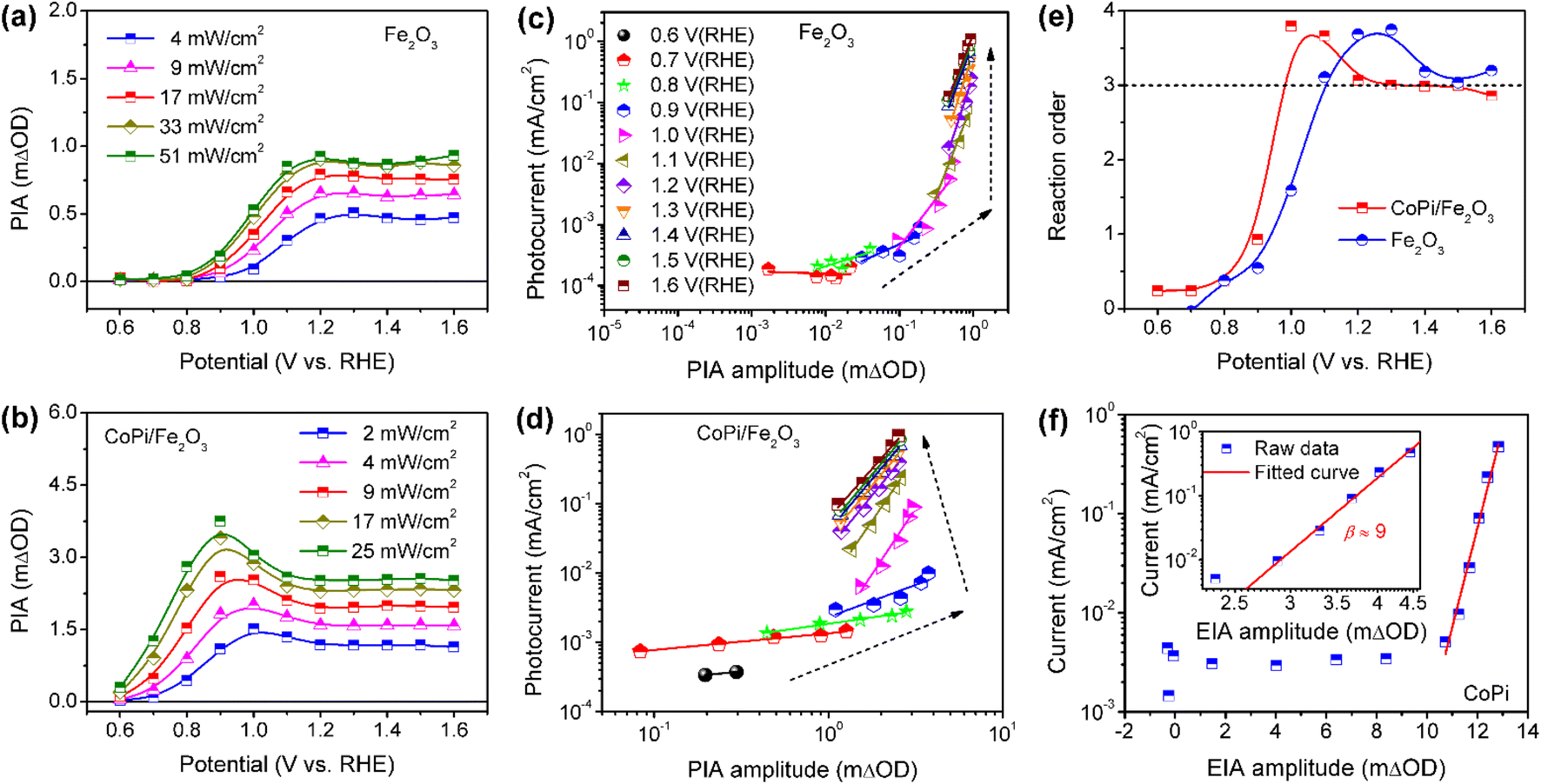 | ||
| Fig. 5 Light-intensity-dependent PIA spectra of (a) bare Fe2O3 and (b) CoPi/Fe2O3 at increased potential. Pump wavelength is 456 nm; probe wavelength is 694.3 nm. Rate law analysis for (c) Fe2O3 and (d) CoPi/Fe2O3. The colors of lines at various potentials in (c and d) are identical. (e) Reaction order of the two photoanodes as a function of potential. (f) Log(current) versus hole density from EIA spectra for the CoPi electrocatalyst. The inset shows the rate law analysis with corrected hole density for the CoPi electrocatalyst. | ||
Discussion
The charge transfer processes for three kinds of hole storage sites at the interface of Fe2O3, CoPi and water under illumination and at applied potential are summarized in Scheme 1. As discussed above, there are surface states for bare Fe2O3 with a potential distribution in the 0.8–1.4 VRHE range. These surface states are occupied by intrinsic electrons, which can only be removed by illumination (trapping holes). Due to limited oxidation activity, the surface states play a role in aggravating recombination of photoinduced charges resulting in large onset potential of bare Fe2O3. Moreover, the holes of surface states at low potential (≤0.8 VRHE) can only oxidize Co2+ to Co3+ rather than Co3+ to Co4+ and water oxidation, which is the main charge transfer path in the CoPi/Fe2O3 photoanode (the left panel of Scheme 1). This process is a sequential hole transfer from the VB to surface states (ps ∼ ns)34 then to CoPi (∼0.6 s), followed by recombination with photoinduced electrons (≥1.3 s) resulting in dissipation of hole energy. Slower recombination in CoPi/Fe2O3 is the origin of the negative shift of onset potential.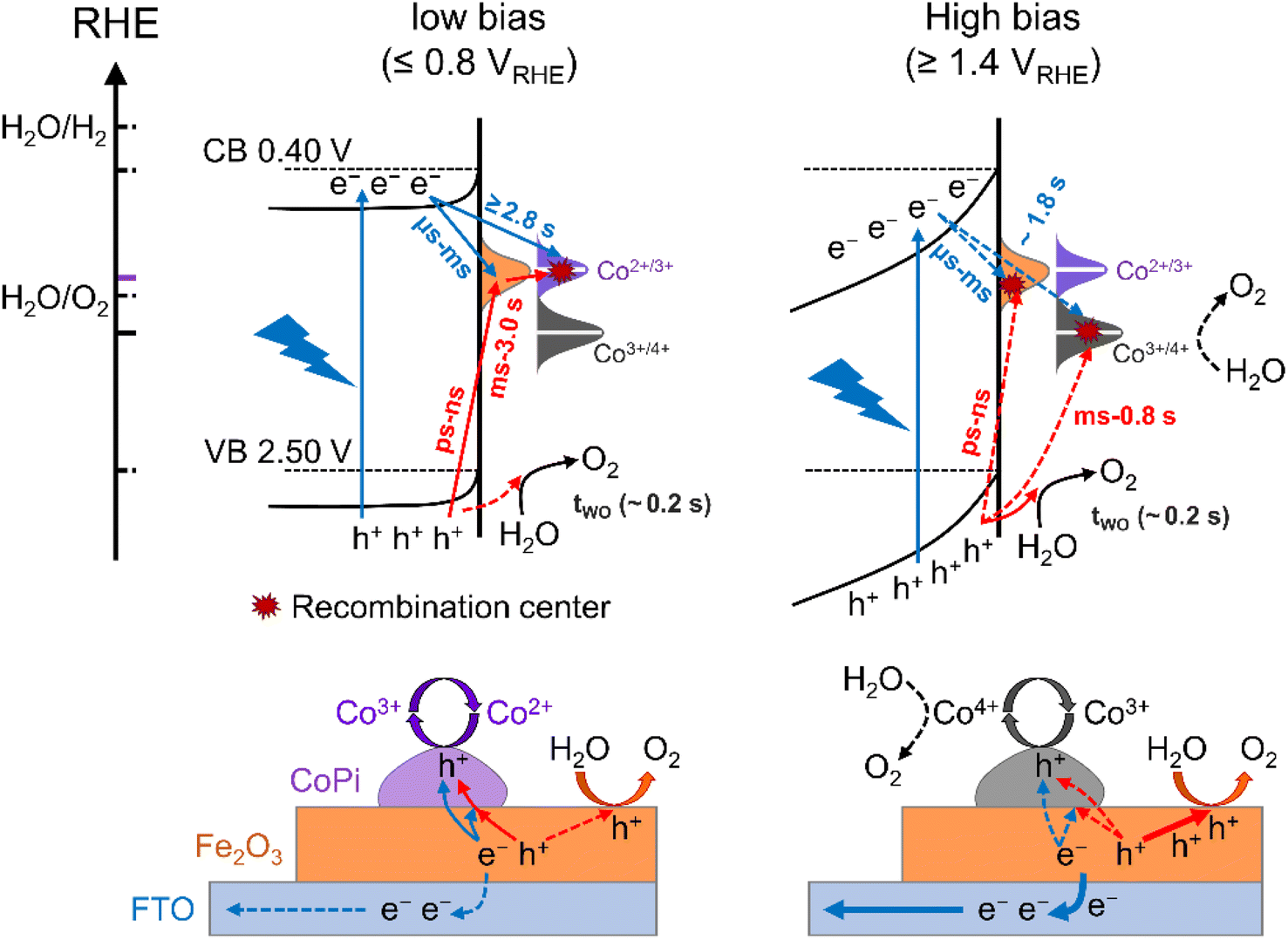 | ||
| Scheme 1 Kinetic schematic of photoinduced charge transfer at the interface of Fe2O3 and CoPi under illumination and at applied potential from energy (top panel) and space (bottom panel) scenarios. The potential distribution of surface states (orange), Co2+/3+ (purple) and Co3+/4+ (grey) are displayed as Gaussian curves. Colored (solid) lines “—” are representative of redox potentials of cobalt species, hydrogen generation and oxygen generation. Red and blue arrows refer to transfer processes of photogenerated electrons and photoinduced holes, respectively (the dashed and solid lines indicate the transferred amount of photoinduced charges). | ||
At high potential (≥1.4 VRHE), the space charge region and band-bending of Fe2O3 will be broadened and increased, respectively (the right panel of Scheme 1). Under quasi-steady-state illumination and at high potential, photoinduced holes can completely fill the surface states and oxidize Co2+ to Co3+via the sequential hole transfer process. Although surface states on the bare Fe2O3 surface can be completely occupied by trapping holes, the photocurrent is still lower than the theoretical value due to inevitable recombination loss by back electrons (Fig. S11†). Because of the delayed recombination process at high potential, more high-energy and long-lived holes in the VB will be generated to directly contribute to water oxidation (OER: ∼0.2 s), as well as Co3+ oxidation (Co3+/4+: ∼0.8 s). Nevertheless, although CoPi acts as the hole storage site, CoPi still doesn't contribute to PEC water oxidation due to low Co4+ density. However, the stored holes in CoPi are not static spectators but dynamic charge mediators, which can again capture photoinduced electrons for a long time. Owing to spatial charge separation of photogenerated electrons (Fe2O3 surface) and trapped holes (CoPi electrocatalyst), CoPi/Fe2O3 displays longer recombination lifetimes resulting in enhanced PEC performances.
As shown by the spectroscopic results, the delayed recombination between photogenerated electrons of Fe2O3 and trapped holes of CoPi electrocatalyst due to spatial charge separation will improve PEC performances of CoPi/Fe2O3, which can be applied to all potentials with photo-response. There are connections and differences between this new viewpoint and previous reports. Although surface state passivation to decrease recombination is common understanding,26,35,36 the EIS results in Fig. 1d reveal that loaded CoPi leads to more surface states, demonstrating negligible contribution of surface state passivation. Using TAS and PIA, Durrant found consistent spectral shapes and long-lived holes after loading CoPi on the Fe2O3 surface, which are attributed to enhanced electron depletion by broadening the space charge region.14,22,24,25 Besides reproducing the reported results, our potential- and light-intensity-dependent spectra further reveal that loaded CoPi on the Fe2O3 surface displays potentiodynamic hole storage mechanisms and acts as a hole storage site of delayed recombination. On the other hand, although Boettcher claimed the direct catalysis evidence of CoPi by tracking surface potential of loaded CoPi in PEC water oxidation,18 the microscopic measurement of AFM might not reflect the overall character of loaded CoPi electrocatalyst. Nevertheless, because of the ensemble-averaged measurements in spectroscopy, present experiments can't completely exclude that a loaded electrocatalyst (like CoPi) on the Fe2O3 surface has no contribution to water oxidation. Instead, we are convinced that a large proportion of the electrocatalyst only acts as a mediator that stores photoinduced holes and recombines with photogenerated electrons. Considering the key role of photogenerated electrons in PEC performance of Fe2O3, several strategies of influencing photogenerated electrons on/near the surface were employed, which display considerable PIA difference (Fig. S16†), such as decreasing density of electron trap states by surface covalent doping with fluorine (Fig. S16a†),37 reducing reverse recombination of electrons via surface insulator modification (SnO2) (Fig. S16b†)38 and enhancing separation efficiency of photoinduced charges by constructing a built-in electric field using a homojunction (Fig. S16c†).15 These results demonstrate that controlling recombination of photogenerated electrons plays a remarkable role in charge dynamics, highlighting the importance of regulating charge transfer processes at the semiconductor–electrolyte interface.
Conclusions
We studied the charge transfer mechanisms among surface states, reaction sites and the coated electrocatalyst in Fe2O3 without/with CoPi by PIA spectroscopy. Surface states in Fe2O3 are not removed by loading CoPi, but serve as a mediator. At low potential, photoinduced holes of the VB are preferentially trapped into surface states and trapped holes further move to CoPi, presenting a sequential hole transfer. Photoinduced holes at high potential are prone to direct water oxidation by holes of the VB on the Fe2O3 surface, resulting in constant and decreasing PIA signals for Fe2O3 and CoPi/Fe2O3 photoanodes, respectively. Namely, there appears a dynamic transition in collecting holes for CoPi from surface states to the VB with increasing potential. More importantly, loaded CoPi did not serve as a catalyst to change third-order water oxidation reaction kinetics on the Fe2O3 surface, but promotes spatial separation of photoinduced charges resulting in long-lived holes for facilitating PEC water oxidation performance. The dynamic charge collecting mechanism not only deepens the understanding of the roles of the loaded electrocatalyst in PEC, but also helps the designing of the microstructure of electrocatalyst loaded photoanodes.Data availability
All experimental and characterization data are available in the ESI.†Author contributions
D. L., X. W. and C. L. conceived the research. D. L. carried out the experiments, analyzed data and wrote the manuscript. R. W. and H. Y. analyzed data. H. Z. provided the homojunction sample. X. W. and C. L. discussed the data and revised the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Key R&D Program of China (2021YFA1500600), the National Natural Science Foundation of China (21872143), the Fundamental Research Center of Artificial Photosynthesis (FReCAP), and the DICP Foundation of Innovative Research (DICP I202122).Notes and references
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 1–13 CrossRef.
- C. Ding, J. Shi, Z. Wang and C. Li, ACS Catal., 2016, 7, 675–688 CrossRef.
- S. Corby, R. R. Rao, L. Steier and J. R. Durrant, Nat. Rev. Mater., 2021, 6, 1136–1155 CrossRef CAS.
- C. A. Mesa, L. Francàs, K. R. Yang, P. Garrido−Barros, E. Pastor, Y. Ma, A. Kafizas, T. E. Rosser, M. T. Mayer, E. Reisner, M. Grätzel, V. S. Batista and J. R. Durrant, Nat. Chem., 2020, 12, 82–89 CrossRef CAS PubMed.
- C. Li, Z. Luo, T. Wang and J. Gong, Adv. Mater., 2018, 30, 1707502 CrossRef PubMed.
- D. R. Gamelin, Nat. Chem., 2012, 4, 965–967 CrossRef CAS PubMed.
- D. A. Grave, N. Yatom, D. S. Ellis, M. C. Toroker and A. Rothschild, Adv. Mater., 2018, 30, 1706577 CrossRef PubMed.
- D. Zhou and K. Fan, Chin. J. Catal., 2021, 42, 904–919 CrossRef CAS.
- O. Zandi and T. W. Hamann, Nat. Chem., 2016, 8, 778–783 CrossRef CAS PubMed.
- Y. Zhang, H. Zhang, A. Liu, C. Chen, W. Song and J. Zhao, J. Am. Chem. Soc., 2018, 140, 3264–3269 CrossRef CAS PubMed.
- B. Klahr and T. Hamann, J. Phys. Chem. C, 2014, 118, 10393–10399 CrossRef CAS.
- F. Le Formal, E. Pastor, S. D. Tilley, C. A. Mesa, S. R. Pendlebury, M. Gratzel and J. R. Durrant, J. Am. Chem. Soc., 2015, 137, 6629–6637 CrossRef CAS PubMed.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, T. Hamann and J. Bisquert, J. Am. Chem. Soc., 2012, 134, 4294–4302 CrossRef CAS PubMed.
- M. Barroso, C. A. Mesa, S. R. Pendlebury, A. J. Cowan, T. Hisatomi, K. Sivula, M. Gratzel, D. R. Klug and J. R. Durrant, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15640–15645 CrossRef CAS PubMed.
- H. Zhang, D. Li, W. J. Byun, X. Wang, T. J. Shin, H. Y. Jeong, H. Han, C. Li and J. S. Lee, Nat. Commun., 2020, 11, 4622 CrossRef PubMed.
- J. Qiu, H. Hajibabaei, M. R. Nellist, F. A. L. Laskowski, S. Z. Oener, T. W. Hamann and S. W. Boettcher, ACS Energy Lett., 2018, 3, 961–969 CrossRef CAS.
- P. Shadabipour and T. W. Hamann, Chem. Commun., 2020, 56, 2570–2573 RSC.
- M. R. Nellist, F. A. L. Laskowski, J. Qiu, H. Hajibabaei, K. Sivula, T. W. Hamann and S. W. Boettcher, Nat. Energy, 2017, 3, 46–52 CrossRef.
- M. R. Nellist, J. Qiu, F. A. L. Laskowski, F. M. Toma and S. W. Boettcher, ACS Energy Lett., 2018, 3, 2286–2291 CrossRef CAS.
- G. M. Carroll, D. K. Zhong and D. R. Gamelin, Energy Environ. Sci., 2015, 8, 577–584 RSC.
- J. Xiao, L. Fan, Z. Huang, J. Zhong, F. Zhao, K. Xu, S.-F. Zhou and G. Zhan, Chin. J. Catal., 2020, 41, 1761–1771 CrossRef CAS.
- Y. Ma, A. Kafizas, S. R. Pendlebury, F. Le Formal and J. R. Durrant, Adv. Funct. Mater., 2016, 26, 4951–4960 CrossRef CAS.
- J. Li, W. Wan, C. A. Triana, Z. Novotny, J. Osterwalder, R. Erni and G. R. Patzke, J. Am. Chem. Soc., 2019, 141, 12839–12848 CrossRef CAS PubMed.
- M. Barroso, A. J. Cowan, S. R. Pendlebury, M. Gratzel, D. R. Klug and J. R. Durrant, J. Am. Chem. Soc., 2011, 133, 14868–14871 CrossRef CAS PubMed.
- Y. Ma, F. Le Formal, A. Kafizas, S. R. Pendlebury and J. R. Durrant, J. Mater. Chem. A, 2015, 3, 20649–20657 RSC.
- C. Zachaus, F. F. Abdi, L. M. Peter and R. van de Krol, Chem. Sci., 2017, 8, 3712–3719 RSC.
- Z. Wang, G. Liu, C. Ding, Z. Chen, F. Zhang, J. Shi and C. Li, J. Phys. Chem. C, 2015, 119, 19607–19612 CrossRef CAS.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, J. Am. Chem. Soc., 2012, 134, 16693–16700 CrossRef CAS PubMed.
- C. A. Mesa, L. Steier, B. Moss, L. Francas, J. E. Thorne, M. Gratzel and J. R. Durrant, J. Phys. Chem. Lett., 2020, 11, 7285–7290 CrossRef CAS PubMed.
- J. Yuan, Y. Yuan, J. Zhang, H. Xu, Z. Mao and Y. Ma, ChemSusChem, 2022, 15, e202102313 CAS.
- S. R. Pendlebury, A. J. Cowan, M. Barroso, K. Sivula, J. Ye, M. Grätzel, D. R. Klug, J. Tang and J. R. Durrant, Energy Environ. Sci., 2012, 5, 6304–6312 RSC.
- Y. Surendranath, D. A. Lutterman, Y. Liu and D. G. Nocera, J. Am. Chem. Soc., 2012, 134(14), 6326–6336 CrossRef CAS PubMed.
- H. N. Nong, L. J. Falling, A. Bergmann, M. Klingenhof, H. P. Tran, C. Spöri, R. Mom, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Pérez-Ramírez, B. R. Cuenya, R. Schlögl, P. Strasser, D. Teschner and T. E. Jones, Nature, 2020, 587, 408–413 CrossRef CAS PubMed.
- M. Barroso, S. R. Pendlebury, A. J. Cowan and J. R. Durrant, Chem. Sci., 2013, 4, 2724 RSC.
- J. E. Thorne, J.-W. Jang, E. Y. Liu and D. Wang, Chem. Sci., 2016, 7, 3347–3354 RSC.
- J. Lee, D. Seo, S. Won and T. D. Chung, Sustainable Energy Fuels, 2021, 5, 501–508 RSC.
- S. Chen, J. Bai, X. Nurimaimaiti, J. Wang, Y. Zhang, T. Zhou, J. Li and B. Zhou, Nano Energy, 2020, 78, 105396 CrossRef CAS.
- T. Hisatomi, H. Dotan, M. Stefik, K. Sivula, A. Rothschild, M. Gratzel and N. Mathews, Adv. Mater., 2012, 24, 2699–2702 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental section and characterization methods. See DOI: https://doi.org/10.1039/d2sc05802b |
| This journal is © The Royal Society of Chemistry 2023 |