
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-controllable heterostructured crystals of metal–organic frameworks via reversible photocycloaddition†
Xin-Da
Huang‡
a,
Ben-Kun
Hong‡
 b,
Ge-Hua
Wen
a,
Shu-Hua
Li
*b and
Li-Min
Zheng
*a
b,
Ge-Hua
Wen
a,
Shu-Hua
Li
*b and
Li-Min
Zheng
*a
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Collaborative Innovation Centre of Advanced Microstructures, Nanjing University, Nanjing 210023, China. E-mail: lmzheng@nju.edu.cn
bInstitute of Theoretical and Computational Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: shuhua@nju.edu.cn
First published on 20th January 2023
Abstract
Metal–organic framework (MOF)-based heterostructures are attractive because they can provide versatile platforms for various applications but are limited by complex liquid epitaxial growth methods. Here, we employ photolithography to fabricate and control MOF-based heterostructured crystals via [4 + 4] photocycloaddition. A layered dysprosium–dianthracene framework, [Dy(NO3)3(depma2)1.5]·(depma2)0.5 (2) [depma2 = pre-photodimerized 9-diethylphosphonomethylanthracene (depma)] underwent a single-crystal-to-single-crystal transition at 140 °C to form [Dy(NO3)3(depma)(depma2)]·(depma2)0.5 (3). The dissociated anthracene moieties are face-to-face π–π interacted allowing a reversible photocycloaddition between 2 and 3. This structural transformation causes a luminescence switch between blue and yellow-green and thus can be used to fabricate erasable 2 + 3 heterostructured crystals for rewritable photonic barcodes. The internal strain at the heterostructure interface leads to photobending and straightening of the crystal, a photomechanical response that is fast, reversible and durable, even operating at 140 °C, making it promising for photoactuation. This work may inspire the development of intelligent MOF-based heterostructures for photonic applications.
Introduction
Heterostructures have attracted considerable attention in recent years because they can create novel and multifunctional platforms to achieve desired functions and applications. Metal–organic frameworks (MOFs) are a promising class of materials that have tunable structures and can be designed to introduce specific physicochemical properties.1 MOF-based heterostructured crystals are typically prepared by a bottom-up epitaxial growth method, which is the crystallization of one structure on the surface of another structure.2 This method has some limitations, such as the need to select highly lattice-matched MOFs, the difficulty in controlling heterostructure patterns, the inability to erase and reuse them, and the complicated preparation process. To overcome these limitations, an effective approach is to prepare MOF-based heterostructured crystals precisely by external stimulation, but to our knowledge, related studies have scarcely been reported.3We envision that MOFs incorporating photosensitive and photochromic components are excellent choices for this purpose, as light offers unique advantages such as easy access, and rapid and precise operability.4,5 To this end, anthracene and its derivatives are appealing ligands because they can experience reversible [4 + 4] photocycloaddition reactions upon UV light irradiation, accompanied by significant photochromic behaviour.6 However, a prerequisite for such a reversible photocycloaddition reaction to occur is a face-to-face π–π stacking of anthracene units with a spacing of less than 4.2 Å, which is hard to satisfy in the anthracene-based MOF system because the self-assembly of the MOF is usually dominated by coordination bonds rather than π–π interactions.7 An alternative approach is to use pre-synthesized dianthracene ligands as linkers to construct the metal–dianthracene frameworks.8 After UV light irradiation or thermal annealing, the dianthracene ligands dissociate to form a pair of anthracene units, which can be further used for the photocycloaddition reaction. Unfortunately, previous studies have demonstrated that de-dimerization of dianthracene ligand often leads to the slip of the two anthracene groups and the destruction of single crystallinity,8a–e making it difficult to attain a reversible photocycloaddition reaction for the fabrication of erasable heterostructures.
We envisage that reversible photocycloaddition reactions can be achieved in the dianthracene-based MOFs when the framework reaches a delicate balance of rigidity and flexibility. Herein, we report a dysprosium–dianthracene framework, [Dy(NO3)3(depma2)1.5]·(depma2)0.5·CH3OH (1), where depma2 is pre-photodimerized 9-diethylphosphonomethylanthracene (depma). Compound 1 has a two-dimensional (2D) honeycomb-like layer structure in which the DyIII ions are connected by flexible depma2 linkers. These layers are stacked to form a framework containing hexagonal channels where the lattice depma2 and methanol molecules reside. The encapsulated guest depma2 molecule enhances the backbone rigidity of 1. Interestingly, 1 underwent two consecutive thermally induced single-crystal-to-single-crystal (SC–SC) phase transitions, first forming [Dy(NO3)3(depma2)1.5]·(depma2)0.5 (2) below 80 °C and then [Dy(NO3)3(depma)(depma2)]·(depma2)0.5 (3) at 140 °C. Further increase in temperature led to complete dissociation of the dianthracene ligands and collapse of the framework structure, forming the mononuclear compound Dy(NO)3(depma)3 (4) at 160 °C.9 Impressively, the partially dissociated phase 3 has a one-dimensional (1D) zigzag chain structure, where the dissociated anthracene moieties are face-to-face π–π interacted. As a result, a reversible photocycloaddition reaction was realized between 2 and 3 in an SC–SC manner, accompanied by a photoluminescence (PL) change between blue and yellow-green. Based on this reversible photocycloaddition reaction, we developed a strategy via the photolithography technique for the precise and controllable fabrication of MOF-based heterostructures, which is further used for photonic applications such as photonic barcoding and photomechanical response. To our knowledge, this is the first example of MOF-based heterostructures that can be used for rewritable photonic barcoding. It is also the first example of MOFs showing photomechanical response that can repeatedly operate above 100 °C.
Results and discussion
Crystal structure of 1
The stoichiometric reaction of Dy(NO3)3·6H2O and depma2 in CH3OH/CH2Cl2 at room temperature resulted in compound 1. Single crystal X-ray structure analysis revealed that compound 1 crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Table S1†). The asymmetric unit consists of one DyIII, three NO3−, four half depma2 and one methanol molecule (Fig. S1a†). Each DyIII is coordinated to nine oxygen atoms, three from the three independent depma2 and six from three chelated nitrate anions. The Dy–O bond lengths fall in the range 2.295(4)–2.487(3) Å, and the O–Dy–O bond angles lie between 51.6(2)and 153.2(1) ° (Table 1 and S2†). There are four crystallographically distinct depma2; each one is located at an inversion center. Three of them (depma2-A, -B, -C) serve as two-connected linkers to bridge metal centers into a honeycomb-like layer in the ac plane (Fig. 1a). The fourth (depma2-D) is encapsulated as a guest molecule in the channel generated by the layer stacking. The Dy–Dy distances over the depma2 ligands are 14.88, 15.21 and 14.24 Å for depma2-A, -B, and -C, respectively. The central C–C bond lengths are 1.638(6) Å (C2–C9A) for depma2-A, 1.634(5) Å (C21–C28B) for depma2-B, 1.640(6) Å (C40–C47C) for depma2-C, and 1.638(7) Å (C59–C66D) for depma2-D (Table 1). The layer has a (3,2)-connecting topology and can be viewed as composed of edge-sharing hexamer rings of [Dy6(depma2)6] (Fig. 1a and S1b†). The size of the hexamer ring is ca. 11.4 Å × 15.4 Å, which is sufficient to accommodate the guest molecules of one depma2 and two methanols. These guest molecules are stabilized in the lattice by C–H⋯O and O–H⋯O hydrogen bonds and C–H⋯π interactions with the coordinated depma2-C ligands (Fig. 1a and S1c†).
(Table S1†). The asymmetric unit consists of one DyIII, three NO3−, four half depma2 and one methanol molecule (Fig. S1a†). Each DyIII is coordinated to nine oxygen atoms, three from the three independent depma2 and six from three chelated nitrate anions. The Dy–O bond lengths fall in the range 2.295(4)–2.487(3) Å, and the O–Dy–O bond angles lie between 51.6(2)and 153.2(1) ° (Table 1 and S2†). There are four crystallographically distinct depma2; each one is located at an inversion center. Three of them (depma2-A, -B, -C) serve as two-connected linkers to bridge metal centers into a honeycomb-like layer in the ac plane (Fig. 1a). The fourth (depma2-D) is encapsulated as a guest molecule in the channel generated by the layer stacking. The Dy–Dy distances over the depma2 ligands are 14.88, 15.21 and 14.24 Å for depma2-A, -B, and -C, respectively. The central C–C bond lengths are 1.638(6) Å (C2–C9A) for depma2-A, 1.634(5) Å (C21–C28B) for depma2-B, 1.640(6) Å (C40–C47C) for depma2-C, and 1.638(7) Å (C59–C66D) for depma2-D (Table 1). The layer has a (3,2)-connecting topology and can be viewed as composed of edge-sharing hexamer rings of [Dy6(depma2)6] (Fig. 1a and S1b†). The size of the hexamer ring is ca. 11.4 Å × 15.4 Å, which is sufficient to accommodate the guest molecules of one depma2 and two methanols. These guest molecules are stabilized in the lattice by C–H⋯O and O–H⋯O hydrogen bonds and C–H⋯π interactions with the coordinated depma2-C ligands (Fig. 1a and S1c†).
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| a Ref. 9. b The Dy–Dy distance over the dianthracene bridge. | ||||
| Space group |
P |
P |
P |
P |
| a (Å) | 12.058(1) | 12.017(2) | 12.132(1) | 11.547(1) |
| b (Å) | 13.858(1) | 13.485(2) | 13.334(1) | 13.173(1) |
| c (Å) | 23.233(1) | 23.445(3) | 23.308(2) | 21.025(1) |
| V (Å3) | 3835.5(2) | 3762.6(10) | 3745.4(5) | 2891.2(2) |
| Dy–O (Å) | 2.295(4)–2.487(3) | 2.288(5)–2.481(5) | 2.290(4)–2.487(4) | 2.314(2)–2.510(2) |
| O–Dy–O (°) | 51.6(2)–153.2(1) | 51.7(2)–153.5(2) | 51.0(2)–153.1(1) | 51.5(1)–153.9(1) |
| C2–C9A (Å) (depma2-A) | 1.638(6) | 1.634(7) | 1.632(6) | 3.730 |
| C21–C28B (Å) (depma2-B) | 1.634(5) | 1.627(8) | 1.631(6) | 5.782 |
| C40–C47C (Å) (depma2-C) | 1.640(6) | 1.622(9) | 3.60 | 5.049 |
| C59–C66D (Å) (depma2-D) | 1.638(7) | 1.657(14) | 1.632(6) | — |
| Intralayer Dy–Dy (Å)b | 14.88, 15.21, 14.24 | 14.71, 15.21, 14.11 | 14.63, 15.24, 13.83 | 15.97, 19.41, 13.69 |
| Shortest interlayer Dy–Dy (Å) | 12.06 | 12.02 | 12.13 | 9.356 |
| Interlayer distance (Å) | 8.40 | 8.34 | 8.37 | 8.35 |
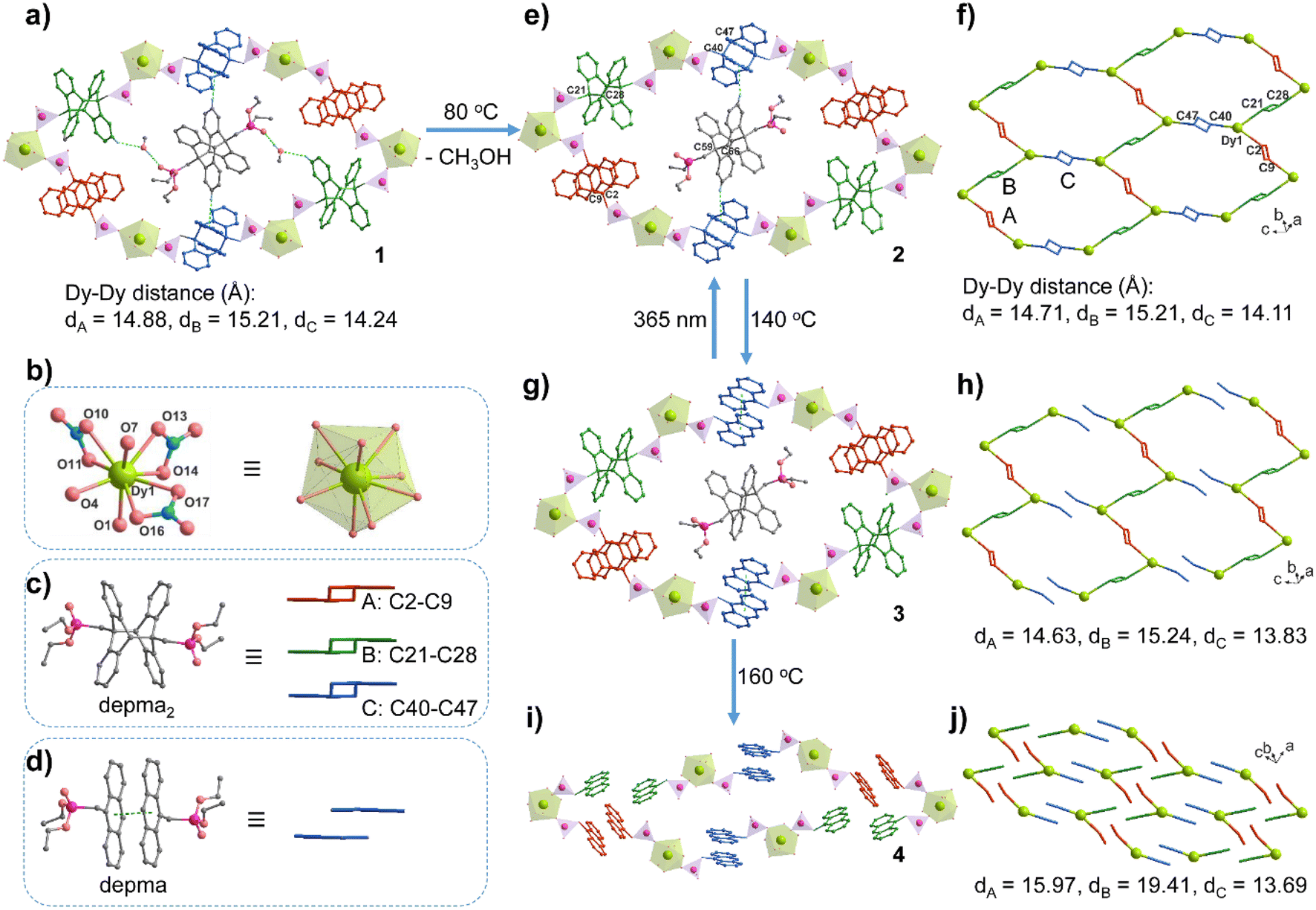 | ||
| Fig. 1 The structural transformation from 1 to 2 to 3 to 4. (a) The [Dy6(depma2)6] hexamer ring with encapsulated depma2 and methanol guests in the layered structure of 1. The green dotted lines represent the hydrogen bonds between the guest and framework depma2 ligands. (b) The coordination environment of the Dy1 atom in structure 1. (c) The three types of depma2 linkers in the framework of 1 with the simplified forms in different colors (red for depma2-A, green for depma2-B, and blue for depma2-C). (d) The dissociated depma2-C ligand in structure 2 and its simplified form. (e and f) The hexamer ring and layer topology in structure 2. (g and h) The hexamer ring and supramolecular layer topology in structure 3, where zigzag chains contain alternating depma2-A and -B linkers, which are further stacked by π–π interactions between depma ligands dissociated from depma2-C. (i and j) The mononuclear structure of 4 stacking in the lattice. Adapted from ref. 9. | ||
SC–SC structural transformation
Thermal gravimetric (TG) analysis showed that the as-prepared compound 1 lost one molecule of methanol below 100 °C (obs. 1.97%, calcd. 1.89%), followed by a plateau between 100 and 200 °C (Fig. S2a†). The differential scanning calorimetry (DSC) curve showed an endothermic peak near 80 °C, and two exothermic peaks near 130 °C and 166 °C (Fig. S2b†). The endothermic peak is due to the removal of methanol molecules (enthalpy ΔH = 51.7 kJ mol−1). In contrast, the two exothermic peaks are attributed to the de-dimerization of depma2 ligands (enthalpy ΔH = −25.0 and −72.1 kJ mol−1). The dissociation of the depma2 ligand is supported by the infrared (IR) spectra of the samples treated at 140 °C and 160 °C (Fig. S3†), which show a peak at 1247 cm−1 characteristic of the C–H in-plane bending vibration of dissociated anthracene units.6 Apparently, the thermally triggered de-dimerization of depma2 proceeds in two steps rather than simultaneously. Noting that there are four independent depma2 in the structure, the question arises as to which depma2 in the structure dissociates first.To examine the structural changes during the thermal treatment, we kept the crystals of 1 in air at room temperature for two days (or heating at 80 °C for 5 min) to obtain the desolvation phase [Dy(NO3)3(depma2)1.5]·(depma2)0.5 (2). When the temperature was increased to 140 °C and 160 °C, respectively, and held for 5 min, we obtained the new phases [Dy(NO3)3(depma)(depma2)]·(depma2)0.5 (3) and Dy(NO)3(depma)3 (4) (major phase, Fig. S4†), respectively. Impressively, the structural transformation from 1 to 2 to 3 was carried out in an SC–SC manner, which allowed the structural analyses of 2 and 3 by the single-crystal diffraction method.
Like 1, compound 2 adopts the triclinic space group P (Table S1†). It shows a similar layer structure to 1 except for the absence of lattice methanol molecules. The release of the methanol guest leads to cell volume contraction [3762.6(10) Å3vs. 3835.9(2) Å3 for 1]. However, the differences in bond lengths and angles of the dysprosium ion are small. For 2, the Dy–O bond lengths and O–Dy–O angles are 2.288(5)–2.481(5) Å [vs. 2.295(4)–2.487(3) Å for 1] and 51.7(2)–153.5(1)° [vs. 51.6(2)–153.2(1)° for 1], respectively (Table 1 and S2). The Dy–Dy distances over the depma2 (-A, -B, -C) bridges are 14.71, 15.21, and 14.11 Å, among which that over the depma2-C is the shortest. The C–H⋯π interactions are still present between the guest depma2-D molecules and coordinated depma2-C ligands (Fig. 1e and S5†). Notably, the DSC curve of 2 showed two exothermic peaks attributed to the de-dimerization of depma2 ligands (enthalpy ΔH = −24.4 and −73.8 kJ mol−1) (Fig. S2b†), which is consistent with that of 1.
Remarkable structural change was observed for compound 3. This compound remains in the same space group P. In contrast to 1 and 2, one kind of dianthracene linkers (depma2-C) de-dimerizes in 3, forming face-to-face stacked anthracene moieties (Fig. 1g and S6†). The dissociation of depma2-C also leads to a slight contraction of the unit cell in 3 [3745.4(5) Å3vs. 3762.6(10) Å3 for 2], and a small change of the Dy–O bond lengths [2.290(4)–2.487(4) Å] and O–Dy–O angles [51.0(2)–153.1(1) °] (Table 1 and S3†). But the central C40–C47C distance of depma2-C is strikingly elongated from 1.640(6) to 3.60 Å. The Dy–Dy distance over depma2-C is also shortened from 14.11 Å in 2 to 13.83 Å in 3. No C–H⋯π interactions between the guest depma2-D and the de-dimerized depma2-C can be found. Clearly, upon heating to 140 °C the layer structure of 2 was dissociated into a zigzag chain structure of 3 in which the Dy atoms are alternatively bridged by depma2-A and depma2-B ligands (Fig. 1h). The reason why it is depma2-C and not the other three kinds of ligands that first undergoes thermally induced de-dimerization is rationalized by theoretical calculations as shown below. Noting that the dissociated anthracene groups in 3 are face-to-face interacted with a separation of 3.60 Å, it is possible to reverse the process via a photocycloaddition reaction to form 2. Indeed, irradiation of crystals of 3 with 365 nm LED UV light (ca. 100 mW cm−2) at room temperature for 0.5 h gave rise to the colorless crystals of 2, confirmed by the single crystal structural analysis (Tables S1 and S5†). We conjecture that the realization of the reversible SC–SC photocycloaddition reaction between 2 and 3 may be related to the presence of the guest depma2-D, which pillars the adjacent depma2-C ligands within the [Dy6(depma2)6] hexamer ring and interacts with the depma2-C ligands via weak C–H⋯π contacts, whereby a delicate balance of rigidity and flexibility may have been achieved. To our knowledge, the reversible SC–SC photocycloaddition reaction in anthracene-based coordination polymers has not been documented in the literature.10
Compound 4 was obtained as a yellow powder, the structure of which was previously reported.9 It has a mononuclear structure wherein each dysprosium ion is linked by three NO3− and three depma (Fig. 1i and j). As for its three asymmetric anthracene units, only one of them maintains face-to-face π–π interactions with the anthracene ring in the adjacent monomer, thus ruling out the possibility of a reverse structural transformation from 4 to 3.
Finally, one might ask whether UV light shorter than 300 nm can trigger the structural transformation from 2 to 3. We prepared 2UV by exposing compound 2 to 280 nm LED UV light (ca. 100 mW cm−2) for 5 min. The UV-vis spectrum of 2UV is similar to that of 3 (Fig. S7†). The IR spectrum of 2UV shows weak vibrational bands at 1247 and 903 cm−1, ascribed to the in-plane and out-of-plane bending vibrations of the C–H bonds of the anthracene unit (Fig. S8†). However, the PXRD pattern of 2UV is identical to the original 2 (Fig. S9†), indicating that the light-induced dissociation of dianthracene in 2 is far from complete and may occur only on the surface. Extending the light exposure time to 1 h did not bring significant changes.
Theoretical calculations
The selective dissociation of depma2-C in compound 2 after thermal treatment is crucial for our material because it makes it possible to achieve a reversible structural transformation between 2 and 3 in an SC–SC manner. In contrast, other reported coordination polymers with dianthracene units suffered from a fatal collapse of the framework upon thermally induced de-dimerization. Therefore, uncovering the cause of selective dissociation is essential for further exploration of light-responsive coordination polymers with dianthracene units. To this end, periodic DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP) code11 with the projector augmented wave (PAW) method12 at the gamma point. The PBE method13 was adopted with D3 (BJ) dispersion correction.14 The kinetic energy cutoff was chosen to be 450 eV and the Hubbard Uf = 5.0 eV was employed for Dy.15 The non-spin-polarized calculations have been tested to be reasonable in this work.Crystals 2 and 3 (dissociated depma2-C) were fully optimized with their X-ray structures (obtained at 140 °C) as initial structures. Based on the optimized structure of 2, depma2-C was scanned along the double reaction coordinates C40–C47′ and C40′-C47 (distance from 1.637 Å to 3.552 Å) and the energy profile is shown in Fig. 2. This process is highly exothermic by 37.3 kJ mol−1, with a reasonable electronic energy barrier of 50.6 kJ mol−1. Similarly, the optimized structures of the dissociated depma2-A, -B and -D, as well as the energy profile of the dissociation of depma2-A, were estimated and the results are listed in Tables S6–S8.† For the dissociation of depma2-A, the electronic energy barrier is estimated to be 67.6 kJ mol−1, and the overall reaction is exothermic by 8.4 kJ mol−1. The dissociation processes of depma2-B (ΔE = −1.9 kJ mol−1) and depma2-D (ΔE = 34.8 kJ mol−1) are less energetically favorable or unfavorable. Overall, the pathway with the lowest electronic energy barrier is the dissociation of depma2-C, which follows the experimental result.
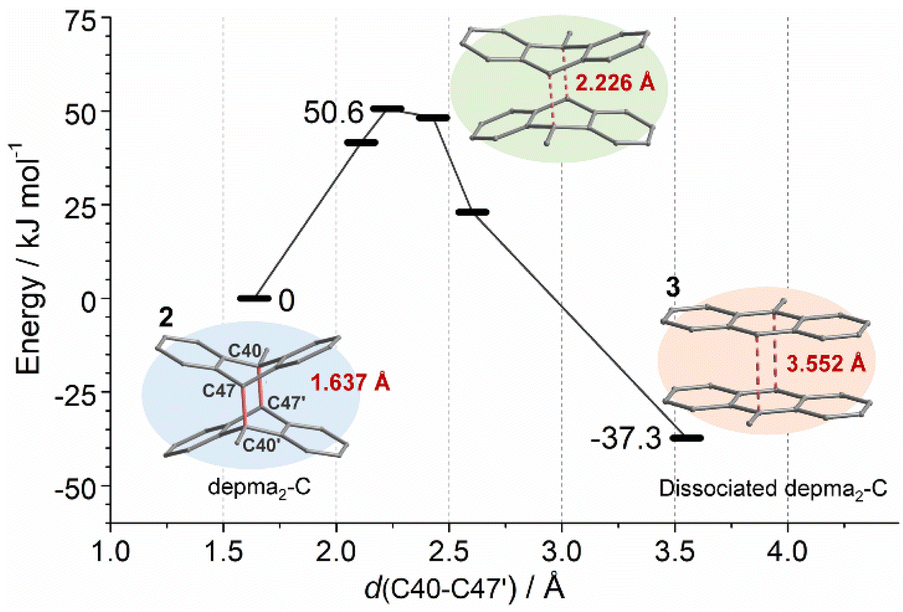 | ||
| Fig. 2 DFT-computed energy profile for the dissociation of depma2-C. | ||
The selectivity is dominated by the steric hindrance. By analyzing the optimized structure of 3, we found that the distance between the anthracene planes d(C40–C47′) is 3.552 Å, which is consistent with the distance (3.600 Å) between two anthracene planes in the experimental crystal of 3 and the calculated equilibrium distance of two anthracene molecules in the gas phase (3.551 Å) at the same theoretical level. For the dissociated depma2-A, -B, and -D, the distances between the two anthracene planes in the optimized crystal structures are 3.388 Å, 3.217 Å and 3.399 Å, respectively, indicating that there exists steric hindrance between two anthracene planes. This is because the dissociations of the dianthracene planes of depma2-A, -B or -D (Fig. S10–S12†) are hindered by the rigid framework while the same process in the case of depma2-C is not hindered by the neighboring framework. Hence, our calculations can account for the fact that only the dissociation of depma2-C was observed at 140 °C.
The dissociation temperature of depma2-C is lower than that of other coordination polymers linked by dianthracene units,8a–c but slightly higher than that of dianthracene compounds given by the solid-state photodimerization reaction of metal–anthracene complexes.6,7a The latter may stem from the fact that in situ topochemical photocycloaddition of two coplanar anthracene units may release a looser lattice environment than depma2-C. The unique structural transformation and the resulting change in luminescence properties allow the material to have a variety of applications, such as molecular switches, barcodes, and photomechanical actuators based on optically programmable heterostructures.
Switching of photoluminescence
The reversible structural transformation associated with the central C–C cleavage/formation of the dianthracene ligand would lead to a significant change in its optical properties. Compounds 1 and 2 are colorless while 3 is yellow. The fluorescence images under 365 nm UV light clearly reveal that compounds 1 and 2 emit blue light while compound 3 displays yellow-green emission (Fig. 3a). The results indicate that the emission color can be reversibly switched between 2 and 3via thermal treatment at 140 °C and 365 nm light illumination.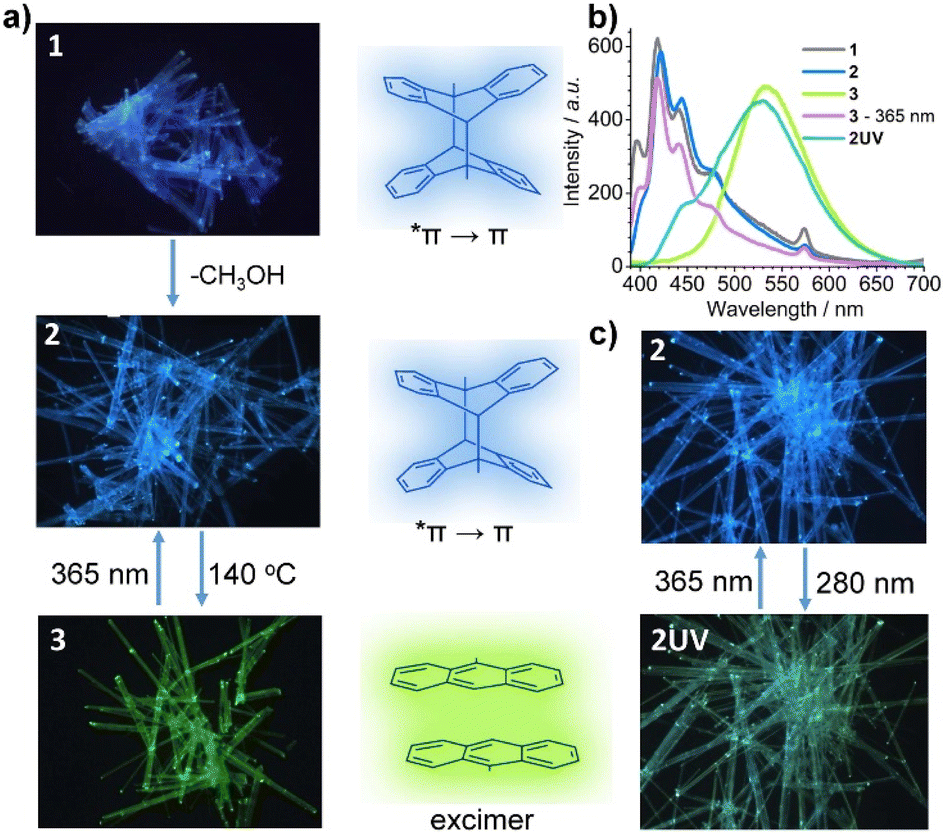 | ||
| Fig. 3 (a) The photoluminescence switching from 1 to 2 to 3. (b) The emission spectra excited at 365 nm for compounds 1, 2, 2UV, 3 and its irradiated sample with 365 nm UV light. (c) The reversible photoluminescence switching from 2 to 2UV upon UV light irradiation. | ||
To investigate the optical properties of 1–3 in-depth, we measured their UV-vis and PL spectra. As shown in Fig. S7,† the UV-vis diffuse reflectance spectrum for compound 1 exhibits several absorption peaks in the range of 300–420 nm because of the Frank–Condon vibration. The strong absorption band at ca. 286 nm is attributed to the π → *π transition of the ligand depma2. The small peaks at 757 nm are assigned to the f–f transition of DyIII ion from 6H15/2 to 6F3/2 states. Fig. 3b and S13† give the PL spectra of 1 which show five peaks at 398, 420, 442, 475 and 574 nm. The first four originate from the vibrational manifold of the π ← *π transition of dianthracene, and the lifetime of the peak at 420 nm is τ = 5.3 ns. The peak at 573 nm is attributed to the f–f transition of the DyIII ion from 4F9/2 to 6H13/2, which is verified by its long lifetime of 72.7 μs (Table S9 and Fig. S14†). Apparently, the excited dimeric ligand depma2 can sensitize the dysprosium emission, but the sensitization efficiency is low, so that blue light can be observed with the naked eye.
The desolvation phase 2 shows almost the same absorption and emission spectra as 1. For compound 3, the absorption intensity at ca. 286 nm decreases and a strong and broad absorption band appears between 300 and 480 nm which is red-shifted compared to those of 1 and 2. In addition, compound 3 shows a completely different emission profile (Fig. 3b and S13c†), where a broad peak emerges at 535 nm, in line with the observed yellow-green emission. The broad peak is ascribed to the excimer emission arising from the two face-to-face π–π interacted anthracene moieties with a lifetime of 42.3 ns (Fig. S14†). Interestingly, although only one of the four dianthracene ligands was dissociated in 3, no emission peak corresponding to dianthracene was visualized. Continuous irradiation of 3 with 365 nm UV light caused the excimer emission at 535 nm to fade out, and the vibronic peaks of dianthracene at 398, 420, 442, and 475 nm to reappear. This is consistent with the photodimerization of the anthracene units in 3 leading to the regeneration of compound 2.
We also measured the emission spectrum of 2UV. As shown in Fig. 3b and S15,† it has two main peaks at 446 and 526 nm and two shoulders at 485 and 574 nm. The peak at 446 nm is attributed to the π ← *π transition of the undissociated dianthracence units. The peak at 526 nm originates from the excimer emission of the face-to-face π–π interacted anthracene moieties. The two very weak shoulder peaks at 485 and 574 nm originate from the DyIII center. The simultaneous observation of the three types of emission in 2UV further demonstrates that only a small amount of the dianthracene ligands in 2 dissociates under 280 nm light. As a result of the combination of the three types of emission, compound 2UV emits green light (Fig. 3c), unlike 3 which emits yellow-green light. Interestingly, 2UV can be converted back to 2 after exposure to 365 nm UV light and the emission spectrum reverts to that of 2. Thus, reversible luminescence switching between 2 and 2UV can be achieved by alternating exposure to 280 nm and 365 nm UV light.
Photogenerated heterostructured crystals
The reversible SC–SC structural transformation between 2 and 3 and the resulting luminescence switch suggest that this MOF material may be used to construct erasable heterostructures with potential applications in photonic barcoding.We first selected a rod-shaped crystal (A: 466 × 27 × 20 μm3) of 1 and heated it to 140 °C to obtain a crystal of 3. Then, the crystal was covered with a photomask with parallel slits and exposed to 365 nm UV light for 2 min (Fig. 4a and S16†). The resulting crystal maintained a smooth surface and transparency. The photograph taken in the reflectance mode showed continuous segments that are alternately yellowish and colorless (Fig. 4b, inset). The faint yellow color comes from compound 3, whereas the colorless species is the photogenerated compound 2. The fluorescence image of the crystal clearly showed alternating yellow-green (bright) and blue (dark) domains corresponding to compounds 3 and 2, respectively. The result indicates that the multi-segment heterostructured crystal of 3 + 2 with bright and dark stripes can be successfully fabricated by photolithography. More interestingly, single yellow-green emission can recover after thermal annealing of heterostructured crystal (Fig. S16†), meaning that the heterostructure can be generated and erased through alternative UV light irradiation and thermal annealing of the crystal of 3.
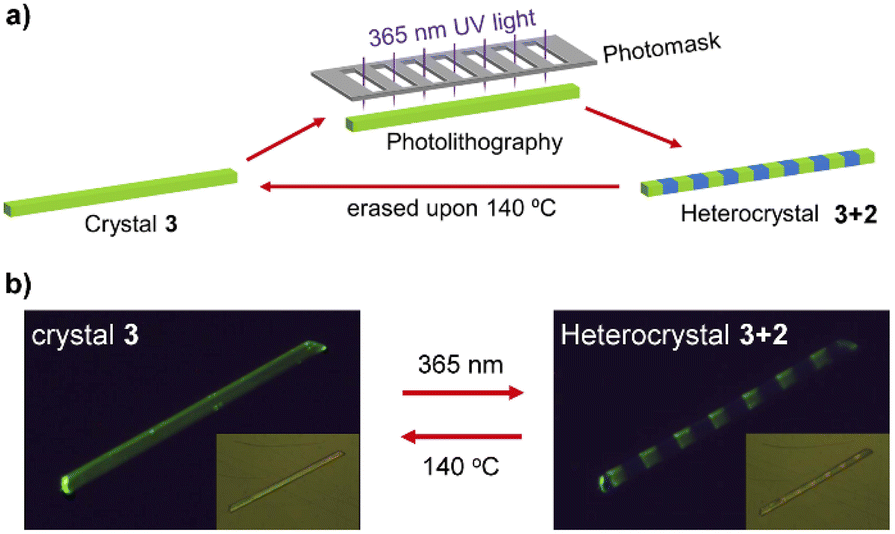 | ||
| Fig. 4 (a) Scheme to show the photolithography process to fabricate and erase the heterostructured crystal 3 + 2. (b) The fluorescence graphs showing the switching between the crystal 3 (left) and heterostructured crystal consisting of sequent 2 and 3 blocks (right). The inset displays their optical pictures. | ||
We then investigated the possibility of producing heterostructured crystals by irradiating the photomask crystal 2 (B: 701 × 32 × 16 μm3) with 280 nm light. As shown in Fig. S17 and S18,† the fluorescence image of the resulting crystal shows a clear sequence of green and blue regions, where the green color corresponds to the partially de-dimerized product 2UV. Interestingly, this new heterostructured (2 + 2UV) crystal can be erased and regenerated by alternately exposing it to 365 nm and 280 nm UV light.
Noting that the known MOF-based heterostructures were normally fabricated in solution via the epitaxial growth method, the photolithography method developed in this work possesses several significant advantages.16 First, the fabrication process is much faster and more convenient. The heterostructure can be generated in situ simply by UV light irradiation for several minutes. Second, the pattern of the heterostructure can be easily controlled by specific design of the photomask. Finally, the heterostructure is erasable via thermal annealing or UV light irradiation. These advantages make the material an outstanding candidate for the fabrication of rewritable barcodes with complicated stripes on-demand via photolithography technology.
Reversible photomechanical response
The reversible SC–SC structural transformation between 2 and 3 also allows for their application as photomechanical actuators. In order to observe the photomechanical effect more clearly, we chose elongated crystals of 3 for our study. We fixed one end of a needle-like crystal of 3 (C: 477 × 10 × 7 μm3) on a glass plate and monitored its photo-induced deformation through a microscope under light (bright field) or darkness (dark field) (Fig. 5a). After irradiation with 365 nm UV light, crystal C bent towards the incident light, reaching a maximum displacement of 70.6 μm at its other end at 14 s, with a deflection angle of 9.8° (Fig. 5a, S19 and Video S1†). It then bent away from the incident light and recovered its straight shape, but with a significant deviation from the original position, with a slight contraction of 6.2 μm (ca. 1.3%) along the long axis of the crystal.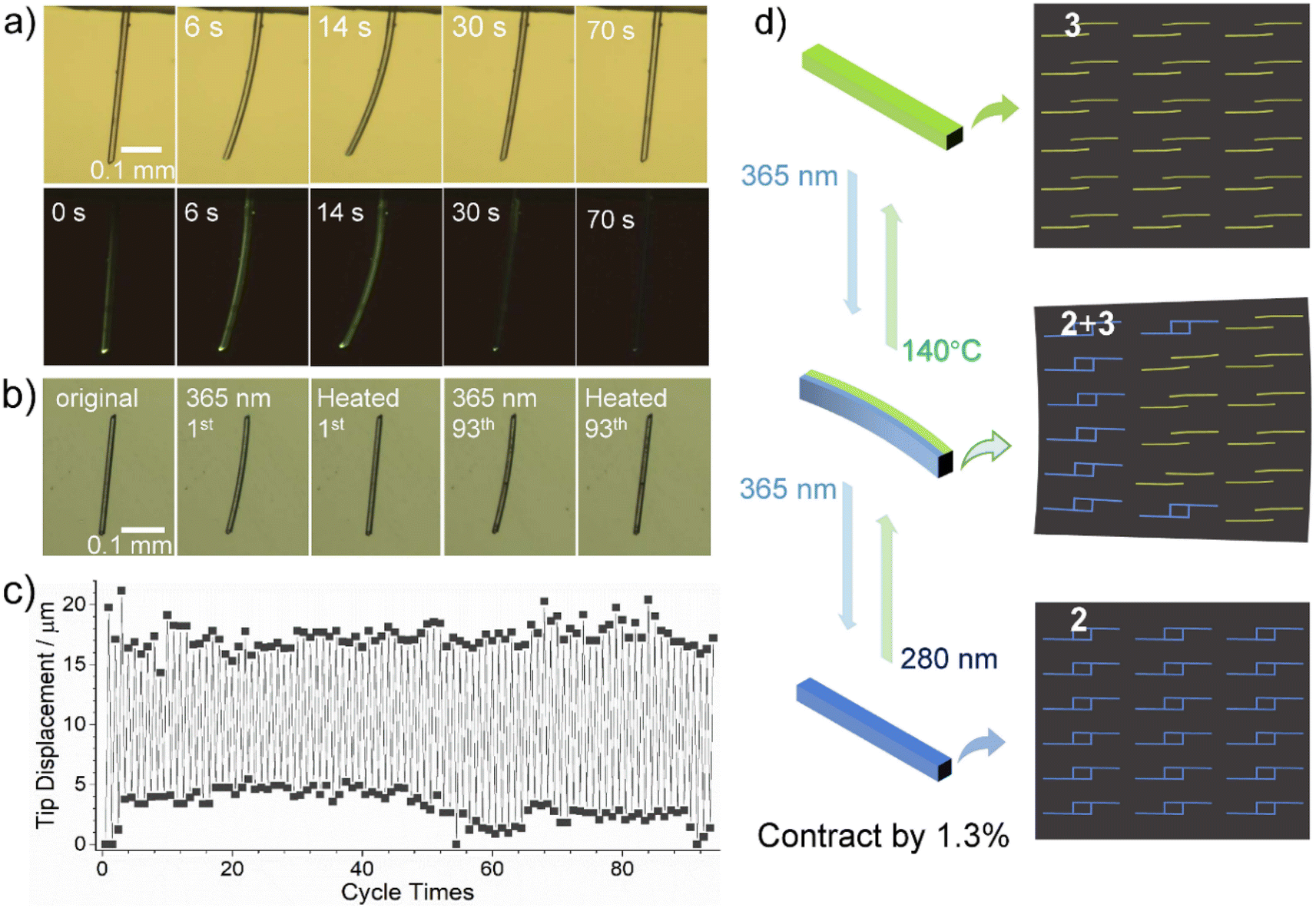 | ||
| Fig. 5 (a) The observed photobending deformation of crystal C upon irradiation with 365 nm UV light two times, respectively taken under lamp light (top) and in the dark (down). (b) The photograph and (c) tip displacement of the repeated photobending deformation for crystal E (310 × 7 × 6 μm3) of compound 3 through alternate irradiation with 365 nm UV light for 4 s and thermal annealing at 140 °C for 2 min. (d) Photomechanical bending and contraction of the rod-like crystal resulted from gradual changes from pure 3 to the heterostructure of 2 and 3, to pure 2. | ||
Photo-induced bending of crystals usually arises from internal strain at the interface of the reactant–product heterostructure after an incomplete photochemical reaction.17 When the rod-like crystal was irradiated with vertical UV light, the crystal bent toward or away from incident light depending on lattice contraction or elongation of the product phase along the long axis of the crystal. The crystal face indexing shows that the long axis of our needle-like crystal extends along the direction of the a-axis, which is also the layer stacking direction, while the b-axis and c-axis are almost perpendicular to the long-axis direction of the crystal (Fig. S20†). According to the above-mentioned crystal structure description, the photodimerization reaction of compound 3 to form 2 is accompanied by unit cell expansion (by 0.4%) and shortening of the a-axis (by 0.9%) (Table 1). Thus, the incomplete photodimerization reaction of crystal 3 can produce 2 + 3 heterostructures and accumulate internal strains, which drive the bending deformation of the crystal (Fig. 5d). When the photodimerization reaction is completed to produce 2, this internal strain is released, leading to the re-straightening of the crystal.18 The resulting straightened crystal of 2 shrinks by 1.3% along the long axis (a-axis) compared to the original crystal of 3.
Clearly, the complete transformation from 3 to 2 causes the shortening of the needle-like crystal and significant deviation from the initial position. Thermal annealing at 140 °C can restore the crystal of 3 to its initial state. Due to the reversibility of the photocycloaddition reaction, the thermally recovered crystal of 3 can be bent and straightened again under 365 nm light (Fig. 5a and Video S2†), accompanied by a change in luminescence from bright yellow-green to blue.
To further investigate the photo-driven bending effect of crystal 3 at elevated temperature, we put another needle-like crystal D (426 × 7 × 6 μm3) onto a hot stage with the temperature kept at 140 °C and irradiated with a 365 nm UV light from the upper left (Fig. S21a and Video S3†). It was found that crystal D underwent rapid photo-induced bending and strengthening at 140 °C, with the degree of bending reaching a maximum at 3 s of exposure and returning to a straight shape at 12 s of exposure. This response is much faster than that at room temperature (Fig. S21b and Video S4†). It is of interest that although the temperature of 140 °C leads to dissociation of the dianthracence unit, the photodimerization reaction still occurs at 140 °C and its reaction rate is faster than that at room temperature. After turning off the UV light after the exposure of 3 s, the bent crystal straightened within 3 min at 140 °C (Fig. S22a and Video S5†). These results indicate that crystals of 3 can achieve a rapid and reversible photomechanical motion through alternating 365 nm light illumination and thermal annealing, both at 140 °C. The reversible photomechanical behavior can also be realized at other temperatures except for different annealing times, i.e., 12 min at 120 °C, 5 min at 130 °C, and 2 min at 150 °C (Fig. S22b†).
To verify the excellent fatigue resistance of the process, we selected another crystal of 3 (E: 310 × 7 × 6 μm3) and studied its photomechanical properties by 365 nm light illumination at 20% power for 4 s and thermal annealing at 140 °C for 2 min alternatively. The results demonstrate that this reversible process can be repeated for at least 93 cycles without significant tip displacement change (Fig. 5b and c). It is worth mentioning that coordination polymers showing reversible photomechanical motion have not been reported before,19 though a few discrete metal–organic complexes were found to show photomechanical effects, such as bending, popping, jumping, and peeling (Table S10†).20 Impressively, the reversible photomechanical motion can be processed at high temperatures above 100 °C; this phenomenon has never been documented for metal–organic systems.
The photomechanical properties can also be observed using alternative 280 nm and 365 nm light sources due to the slight dissociation of depma2 upon exposing the crystal of 2 to 280 nm UV light. As shown in Fig. S23,† when the crystal of 2 (F: 368 × 7 × 5 μm3) was exposed to 280 nm UV light (20% powder) from the left side, it bent away from the light source and reached a maximum within 8 s with a tip displacement of d2 = 42 μm, corresponding to the formation of a 2+3 heterostructure. Then, we switched the light source to 365 nm UV light, the bent crystal F became almost straight again within 10 s, but with a small deviation from the original one (d1 = 6 μm). Thus, the reversible light-induced bending and straightening can be achieved by alternating irradiation with 280 nm and 365 nm UV light (Video S5†). We repeated this process at least 16 times and found that the tip displacement change (Δd) decreased linearly after each cycle (t), obeying the equation Δd = −0.851 t + 37.8 μm (Fig. S24†). This means that the reversibility will disappear around t = 44 cycles. The faster fatigue of this photobending driven only by UV light could come from unavoidable damage to the crystal. Nevertheless, our deformable crystal stands out among MOFs for its multi-stimulus response, bidirectional motion and fluorescence changes.
Conclusions
We report a new two-dimensional dysprosium-organic framework [Dy(NO3)3(depma2)1.5]·(depma2)0.5·CH3OH (1), where the DyIII ions are linked by three types of dianthracene ligands (depma2-A, -B, -C). One of them (depma2-C) in the desolvation phase [Dy(NO3)3(depma2)1.5]·(depma2)0.5 (2) can be selectively dissociated by thermal annealing at 140 °C, which is rationalized by the DFT calculation, forming [Dy(NO3)3(depma)(depma2)]·(depma2)0.5 (3). Compound 3 has a one-dimensional zigzag chain structure in which the dissociated anthracene moieties are face-to-face π–π interacted. Reversible SC–SC structural transformation between 2 and 3 was visualized by alternative thermal annealing and 365 nm UV light irradiation, accompanied by the luminescence change between blue and yellow-green. In addition, incomplete structural transformation between 2 and 3 was achieved using light irradiation only with alternative 280 and 365 nm light sources, accompanied by the luminescence change between blue and green. By taking advantages of this fascinating photo-responsive property, the material has been used to fabricate 2 + 3 heterostructured crystals using a photolithography technique, which can serve as an erasable photonic barcode. Moreover, photomechanical response was observed for 3 and 2 due to the internal strain at the interface of the reactant–product heterostructure, making it possible for the fabrication of photoactuators. This work provides a feasible strategy for the fabrication of MOF-based heterostructured crystals via the reversible [4 + 4] photocycloaddition reaction of anthracene and may shed light on the development of intelligent MOF-based heterostructures for photonic applications.Data availability
The experimental and computational data have been provided in the ESI.†Author contributions
X.-D. H. and B.-K. H. contributed equally to this work. X.-D. H. designed the study, conducted most of the experiments and wrote the draft of the manuscript. B.-K. H. and S.-H. L. performed the calculation and wrote the related discussion. G.-H. W. assisted in performing the single-crystal X-ray diffraction data collection. L.-M. Z. conceived the project and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (21731003, 22273037) and the National Key R&D Program of China (2018YFA0306004).Notes and references
- (a) M. Pan, W.-M. Liao, S.-Y. Yin, S.-S. Sun and C.-Y. Su, Chem. Rev., 2018, 118, 889–918 CrossRef PubMed; (b) J. M. Cain, W. He, I. Maurin, M. W. Meisel and D. R. Talham, J. Appl. Phys., 2021, 129, 160903 CrossRef CAS; (c) M. L. Foo, R. Matsuda and S. Kitagawa, Chem. Mater., 2014, 26, 310–322 CrossRef CAS.
- (a) R. Haldar and C. Wöll, Nano Res., 2021, 14, 355–368 CrossRef CAS; (b) X.-T. Liu, K. Wang, Z. Chang, Y.-H. Zhang, J. Xu, Y.-S. Zhao and X.-H. Bu, Angew. Chem., Int. Ed., 2019, 58, 13890–13896 CrossRef CAS PubMed; (c) Y. Yao, Z. Gao, Y. Lv, X. Lin, Y. Liu, Y. Du, F. Hu and Y. S. Zhao, Angew. Chem., Int. Ed., 2019, 58, 13803–13807 CrossRef CAS PubMed; (d) G. Lee, S. Lee, S. Oh, D. Kim and M. Oh, J. Am. Chem. Soc., 2020, 142, 3042–3049 CrossRef CAS PubMed; (e) C. R. R. Adolf, S. Ferlay, N. Kyritsakas and M. W. Hosseini, J. Am. Chem. Soc., 2015, 137, 15390–15393 CrossRef CAS PubMed; (f) K. C. Jayachandrababu, D. S. Sholl and S. Nair, J. Am. Chem. Soc., 2017, 139, 5906–5915 CrossRef CAS PubMed; (g) J. A. Boissonnault, A. G. Wong-Foy and A. Matzger, J. Am. Chem. Soc., 2017, 139, 14841–14844 CrossRef CAS PubMed.
- Y.-J. Ma, X. Fang, G. Xiao and D. Yan, Angew. Chem., Int. Ed., 2022, 61, e20211410 Search PubMed.
- (a) D. Venkatakrishnarao, M. A. Mohiddon, N. Chandrasekhar and R. Chandrasekar, Adv. Opt. Mater., 2015, 3, 1035–1040 CrossRef CAS; (b) H. Xia, J. Cheng, L. Zhu, K. Xie, Q. Zhang, D. Zhang and G. Zou, ACS Appl. Mater. Interfaces, 2019, 11, 15969–15976 CrossRef CAS PubMed.
- (a) A. M. Rice, C. R. Martin, V. A. Galitskiy, A. A. Berseneva, G. A. Leith and N. B. Shustova, Chem. Rev., 2020, 120, 8790–8813 CrossRef CAS PubMed; (b) W. Danowski, T. v. Leeuwen, W. R. Browne and B. L. Feringa, Nanoscale Adv., 2021, 3, 24–40 RSC.
- (a) X.-D. Huang, Y. Xu, K. Fan, S.-S. Bao, M. Kurmoo and L.-M. Zheng, Angew. Chem., Int. Ed., 2018, 57, 8577–8581 CrossRef CAS PubMed; (b) X.-D. Huang, J.-G. Jia, M. Kurmoo, S.-S. Bao and L.-M. Zheng, Dalton Trans., 2019, 48, 13769–13779 RSC; (c) X.-D. Huang, G.-H. Wen, S.-S. Bao, J.-G. Jia and L.-M. Zheng, Chem. Sci., 2021, 12, 929–937 RSC.
- (a) X.-D. Huang, X.-F. Ma, T. Shang, Y.-Q. Zhang and L.-M. Zheng, Inorg. Chem., 2022, 61, DOI:10.1021/acs.inorgchem.2c01210; (b) X.-F. Ma, Y. Guo, X.-D. Huang, G.-H. Wen, S.-S. Bao, Y.-Q. Zhang and L.-M. Zheng, Dalton Trans., 2022, 51, 12026–12030 RSC.
- (a) J.-C. Liu, X.-D. Huang, Q. Zou, S.-S. Bao, X.-Z. Wang, J.-Y. Ma and L.-M. Zheng, J. Mater. Chem. C, 2020, 8, 7369–7377 RSC; (b) Q. Zou, T. Shang, X.-D. Huang, Q.-Q. Guo, J.-G. Jia, S.-S. Bao, Y.-Q. Zhang and L.-M. Zheng, J. Mater. Chem. C, 2021, 9, 10749–10758 RSC; (c) Q. Zou, J.-C. Liu, X.-D. Huang, S.-S. Bao and L.-M. Zheng, Chin. Chem. Lett., 2021, 32, 1519–1522 CrossRef CAS; (d) Q. Zou, S.-S. Bao, X.-D. Huang, G.-H. Wen, J.-G. Jia, L.-Q. Wu and L.-M. Zheng, Chem.–Asian J., 2021, 16, 1456–1465 CrossRef CAS PubMed; (e) G. Collet, T. Lathion, C. Besnard, C. Piguet and S. Petoud, J. Am. Chem. Soc., 2018, 140, 10820–10828 CrossRef CAS PubMed; (f) Q. Zou, G.-L. Wang, Y.-Q. Chen, X.-D. Huang, G.-H. Wen, M.-F. Qin, S.-S. Bao, Y.-Q. Zhang and L.-M. Zheng, Chem.–Eur. J., 2023, e202203454 Search PubMed.
- X.-D. Huang, M. Kurmoo, S.-S. Bao, K. Fan, Y. Xu, Z.-B. Hu and L.-M. Zheng, Chem. Commun., 2018, 54, 3278–3281 RSC.
- (a) X.-Y. Chen, H. Chen, L. Đorđevic, Q.-H. Guo, H. Wu, Y. Wang, L. Zhang, Y. Jiao, K. Cai, H. Chen, C. L. Stern, S. I. Stupp, R. Q. Snurr, D. Shen and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 9129–9139 CrossRef CAS PubMed; (b) M. Tu, H. Reinsch, S. Rodríguez-Hermida, R. Verbeke, T. Stassin, W. Egger, M. Dickmann, B. Dieu, J. Hofkens, I. F. J. Vankelecom and N. Stock, Angew. Chem., Int. Ed., 2019, 58, 2423–2427 CrossRef CAS PubMed.
- (a) G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed; (b) G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS; (c) G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11185 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed; (b) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- (a) S. Grimme and J. Antony, et. al. , J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. Topsakal and R. M. Wentzcovitch, Comput. Mater. Sci., 2014, 95, 263–270 CrossRef CAS.
- (a) Y. Zhang, L. Zhang, R. Deng, J. Tian, Y. Zong, D. Jin and X. Liu, J. Am. Chem. Soc., 2014, 136, 4893–4896 CrossRef CAS PubMed; (b) Z. Gao, B. Xu, T. Zhang, Z. Liu, W. Zhang, X. Sun, Y. Liu, X. Wang, Z. Wang, Y. Yan, F. Hu, X. Meng and Y.-S. Zhao, Angew. Chem., Int. Ed., 2020, 59, 19060–19064 CrossRef CAS PubMed; (c) M. Pan, Y.-X. Zhu, K. Wu, L. Chen, Y.-J. Hou, S.-Y. Yin, H.-P. Wang, Y.-N. Fan and C.-Y. Su, Angew. Chem., Int. Ed., 2017, 56, 14582–14586 CrossRef CAS PubMed; (d) G. Zhan and H.-C. Zeng, Nat. Commun., 2018, 9, 3778 CrossRef PubMed.
- (a) P. Naumov, S. Chizhik, M. K. Panda, N. K. Nath and E. Boldyreva, Chem. Rev., 2015, 115, 12440–12490 CrossRef CAS PubMed; (b) T. Kim, L. Zhu, R. O. Al-Kaysi and C. J. Bardeen, ChemPhysChem, 2014, 15, 400–414 CrossRef CAS PubMed; (c) B. Zhou and D. Yan, Appl. Phys. Rev., 2021, 8, 041310 CrossRef CAS; (d) F. Tong, W. Xu, M. Al-Haidar, D. Kitagawa, R. O. Al-Kaysi and C. J. Bardeen, Angew. Chem., Int. Ed., 2018, 57, 7080–7084 CrossRef CAS PubMed; (e) Q. Yu, M. Li, J. Gao, P. Xu, Q. Chen, D. Xing, J. Yan, M. J. Zaworotko, J. Xu, Y. Chen, P. Cheng and Z. Zhang, Angew. Chem., Int. Ed., 2019, 58, 18634–18640 CrossRef CAS PubMed.
- (a) T. Kim, L. L. Zhu, J. Mueller and C. J. Bardeen, J. Am. Chem. Soc., 2014, 136, 6617–6625 CrossRef CAS PubMed; (b) A. Fujimoto, N. Fujinaga, R. Nishimura, E. Hatano, L. Kono, A. Nagai, A. Sekine, Y. Hattori, Y. Kojima, N. Yasuda, M. Morimoto, S. Yokojima, S. Nakamura, B. L. Feringa and K. Uchida, Chem. Sci., 2020, 11, 12307–12315 RSC.
- (a) Y.-X. Shi, W.-H. Zhang, B. F. Abrahams, P. Braunstein and J.-P. Lang, Angew. Chem., Int. Ed., 2019, 58, 9453–9458 CrossRef CAS PubMed; (b) B. B. Rath and J. J. Vittal, J. Am. Chem. Soc., 2020, 142, 20117–20123 CrossRef CAS PubMed.
- (a) J.-s. Geng, L. Mei, Y.-y. Liang, L.-y. Yuan, J.-p. Yu, K.-q. Hu, L.-h. Yuan, W. Feng, Z.-f. Cha and W.-q. Shi, Nat. Commun., 2022, 13, 2030 CrossRef CAS PubMed; (b) A. K. Bartholomew, I. B. Stone, M. L. Steigerwald, T. H. Lambert and X. Roy, J. Am. Chem. Soc., 2022, 144, 16773–16777 CrossRef CAS PubMed; (c) B. B. Rath and J. J. Vitta, CrystEngComm, 2021, 23, 5738–5752 RSC; (d) S. Khan, Akhtaruzzaman, R. Medishetty, A. Ekka and M. H. Mir, Chem.–Asian J., 2021, 16, 2806–2816 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, crystallographic details, and full characterization (SC-XRD, PXRD, IR, UV-vis and photoluminescence spectra, thermal analysis, photographs) of all described compounds. CCDC 2214247–2214250. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06732c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |