
Merging photochemistry with electrochemistry in organic synthesis†
Yi
Yu‡
,
Peng
Guo‡
,
Jun-Song
Zhong
,
Yaofeng
Yuan
and
Ke-Yin
Ye
 *
*
Key Laboratory for Molecule Synthesis and Function Discovery (Fujian Province University), College of Chemistry, Fuzhou University, Fuzhou 350116, P.R. China. E-mail: kyye@fzu.edu.cn
First published on 1st November 2019
Abstract
Photochemistry and electrochemistry are two powerful tools in organic synthesis as reflected by the recent resurgence of both research areas. Despite the impressive advances achieved so far, both synthetic technologies suffer from innate disadvantages. The constructive merging of photochemistry and electrochemistry, therefore, offers the potential to overcome the distinct flaws of one method through the advantages of the other, so that novel reaction pathways that are unachievable with individual methods can be envisioned. This Highlight article describes recent breakthroughs using this promising concept of merging photochemistry with electrochemistry in organic synthesis.
The recent resurgence of photoredox catalysis1 and synthetic electrochemistry2 has changed the landscape of modern organic synthesis tremendously, by bringing forth a multitude of innovative chemical transformations previously thought to be impossible. Both research areas share the common feature of using electrons as reagents to generate open-shell radical intermediates, and thus many transformations have translated from photoredox catalysis to electrochemistry and vice versa. Photoredox catalysis can take advantage of its redox-neutral nature to facilitate net-redox-neutral transformations. However, such a characteristic inevitably requires a stoichiometric amount of oxidant/reductant with each turnover of the photocatalyst in the net-oxidative/reductive transformation. This flaw is not seen in green electrochemical synthesis because oxidation and reduction occur simultaneously on the anode and cathode, respectively. On the other hand, radicals that are electrochemically generated in direct electrolysis may easily undergo undesirable over-reduction/oxidation, radical homocoupling and electrode passivation due to inefficient mass transfer at the interface of the electrode surface and bulk solution. Redox mediators, that shift the electron transfer step from a heterogeneous process at an electrode to a homogeneous process, circumvent these drawbacks of direct electrochemical synthesis.3
Though photoredox catalysis and electrochemistry share the common feature of radical intermediate generation and thus are often mentioned together and compared alongside one another, little attention has been paid to the concept of merging photochemistry with electrochemistry in organic synthesis.4 The adoption of photochemistry and electrochemistry in organic synthesis has been known since the 1980s (vide infra) but the development in this field has been slow. The recent renaissance in photocatalysis and electrocatalysis has led synthetic chemists to reconsider this approach in organic synthesis. The constructive merging of photochemistry with electrochemistry can create a scenario in which each techniques’ flaws can be perfectly compensated for by their respective advantages, thus making possible novel reaction pathways which are unachievable with individual methods (Scheme 1).
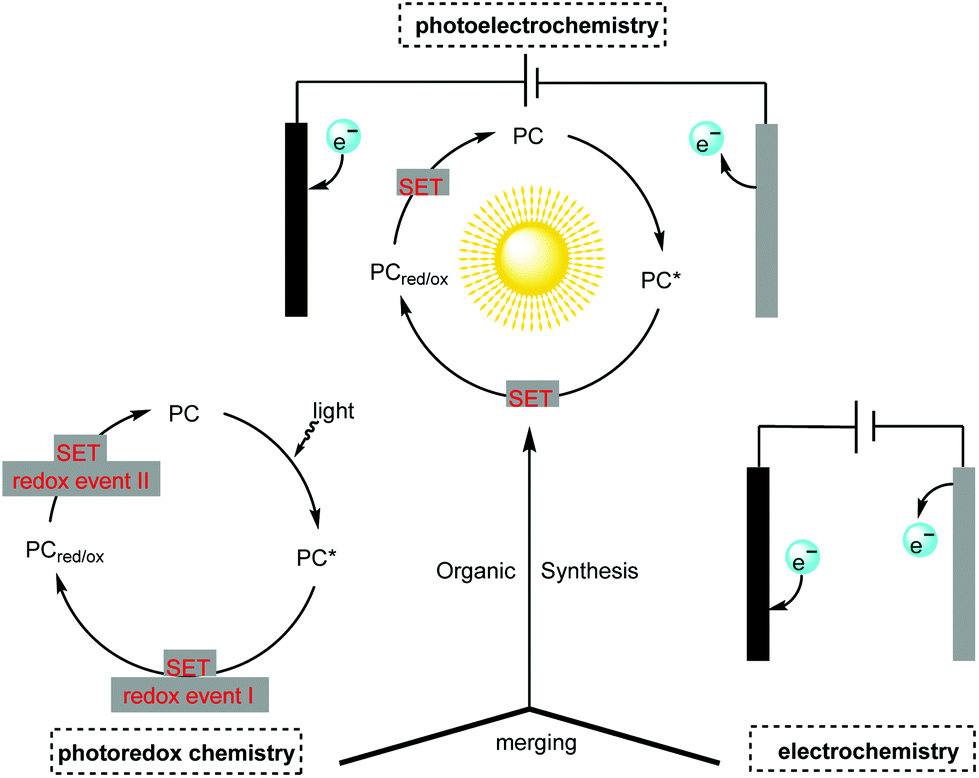 | ||
| Scheme 1 Concept of merging photochemistry with electrochemistry in organic synthesis, PC = photocatalyst, SET = single-electron transfer. | ||
The adoption of this emerging but highly promising strategy using photoelectrochemical cells (PECs) and photoelectrochemical catalyst-mediated electrolysis in organic synthesis is still in its infancy. However, recent breakthroughs suggest that it is a very promising technique well suited to the continuous pursuit of greener and more sustainable organic synthesis. Herein, we highlight recent advances in organic synthesis featuring the creative adoption of this emerging concept. We also cover the fundamental aspects, reaction scopes and mechanisms of the integration of photochemistry with electrochemistry in organic synthesis.
One salient feature of electrochemistry, as discussed previously, is the replacement of the chemical oxidant/reductant in the net-oxidative/reductive photochemistry process with a traceless electron “reagent”. Minisci alkylation5 of heteroarenes with photoredox catalysis generally requires a stoichiometric amount of oxidizing reagent such as a peroxydisulfate salt (S2O82−) or hypervalent iodine. Replacing those oxidants with reagent-free electrochemistry as demonstrated by Xu and co-workers6 provided a multitude of functionalized heteroarenes using primary, secondary and tertiary alkyltrifluoroborates (or 1,4-dihydropyridine) with excellent regio- and chemoselectivity (Scheme 2). Various heteroarenes (substituted quinolines, isoquinoline, acridine, benzothioazole, imidazo[1,2-b]pyridazine and purine) as well as some bioactive compounds such as voriconazole, camptothecin, fasudil and quinine were all well tolerated.
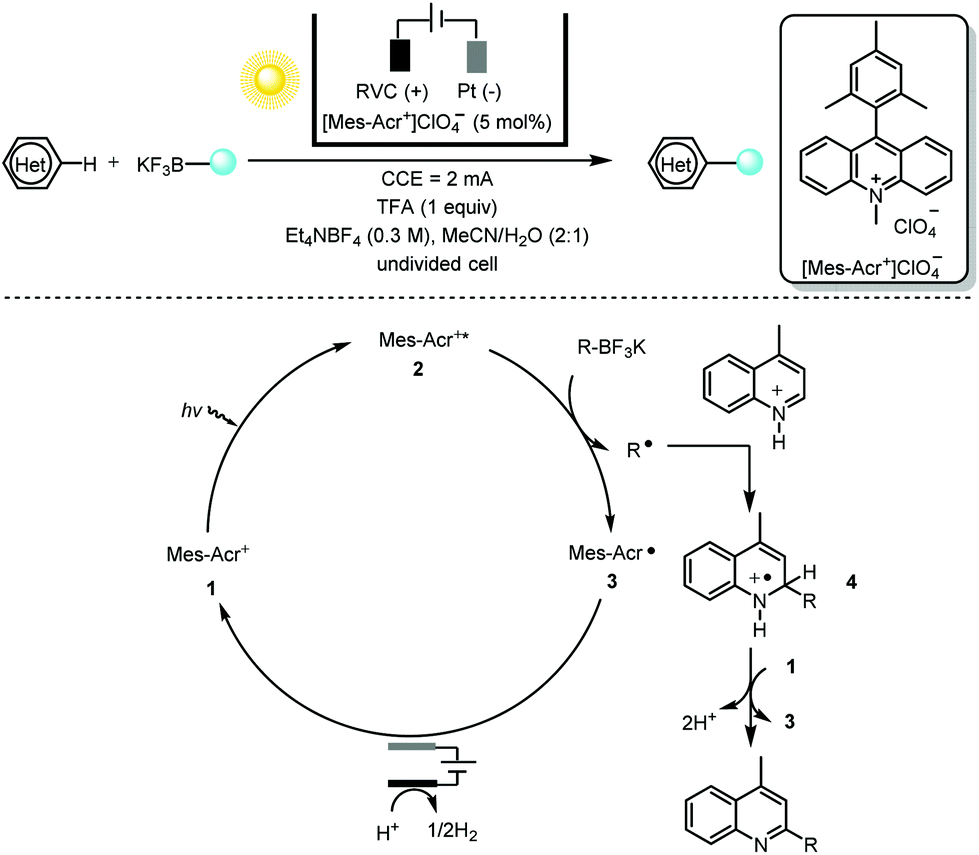 | ||
| Scheme 2 Photoelectrochemical C–H alkylation of heteroarenes with organotrifluoroborates, 34–99% yields. RVC = reticulated vitreous carbon; CCE = constant-current electrolysis; TFA = trifluoroacetic acid. | ||
With respect to the mechanism, the photoelectrochemical redox catalyst [Mes-Acr+]ClO4−1 is initially irradiated (with blue LEDs) to its highly oxidizing excited state 2 (Ered = 2.06 V vs. SCE in MeCN). This undergoes a single-electron transfer (SET) with the organotrifluoroborate leading to the acridinyl radical 3 and an alkyl radical. Regeneration of the organic dye [Mes-Acr+]ClO4−1 takes place at the anode via another SET with the stable radical 3. The alkyl radical, on the other hand, proceeds via a typical Minisci-type radical addition to the protonated heteroarene. The short lifetime of radical cation 4 suggests that it is oxidized by the ground state of [Mes-Acr+]ClO4− (Eredp/2 = −0.57 V vs. SCE in MeCN) rather than at the anode. This creative merger of photochemistry and electrochemistry using the photoelectrochemical redox catalyst [Mes-Acr+]ClO4− not only obviates the need for a chemical oxidant but also overcomes the drawback of over-oxidation of electrochemically generated electron-rich alkyl radicals.
Alcohol oxidation using riboflavin photocatalysts has been extensively investigated in past decades.7 However, this catalytic method is limited to benzylic alcohols, as aliphatic alcohols are generally not applicable. To address this challenge, Lin and co-workers carried out an extensive mechanistic study of flavin-catalyzed alcohol oxidation using a thiourea co-catalyst (Scheme 3).8 They found that the major decomposition pathway of thiourea during the canonical flavin/thiourea co-catalysis under an O2 atmosphere involved the formation of thiourea dioxide. With this key mechanistic understanding in mind, they creatively used electricity to replace O2 in the photocatalytic oxidation of alcohols to circumvent the problem of thiourea degradation. The photochemical oxidation of alcohols by flavin alone relies heavily on the oxidation potential of the flavin excited state, and thus only activated alcohols could be used. In contrast, the thiourea-mediated photoelectrochemical flavin catalysis takes advantage of the HAT (hydrogen atom transfer) of thiourea-based radicals generated by means of flavin oxidation, and so previously untouched aliphatic alcohols could be oxidized using this novel photoelectrochemical protocol.
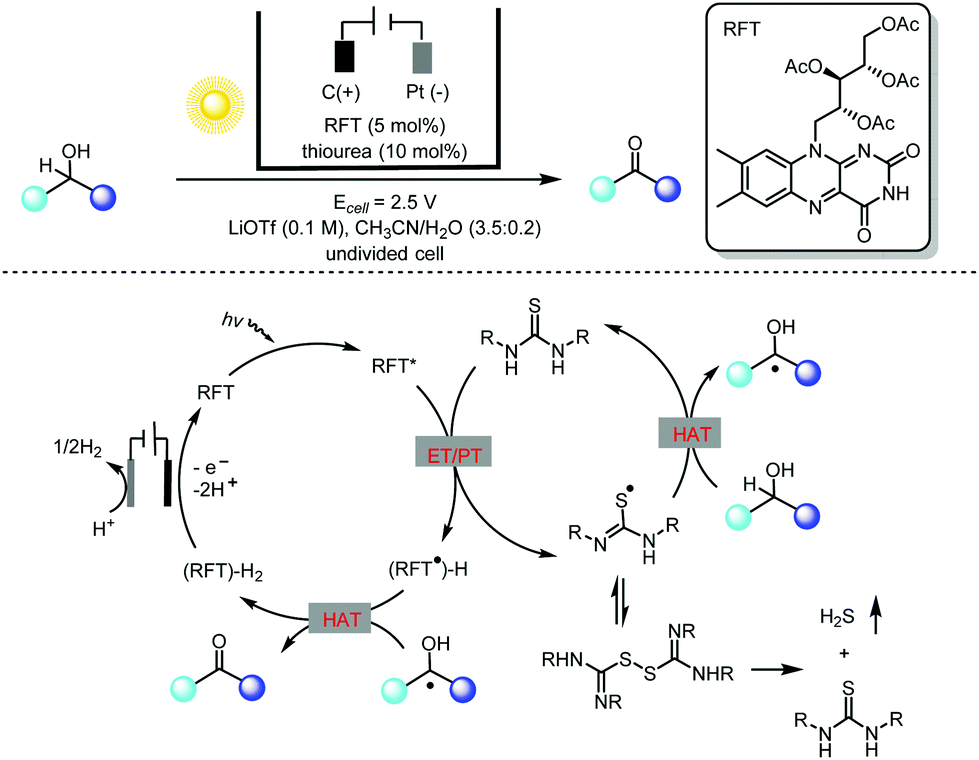 | ||
| Scheme 3 Photoelectrocatalytic oxidation of unactivated alcohols via flavin/thiourea co-catalysis, 53–96% yields. RFT = riboflavin tetraacetate; HAT = hydrogen atom transfer; ET = electron transfer; PT = proton transfer. | ||
The energy-transfer relay of electrochemistry and photochemistry is capable of generating much more potent catalysts than can be obtained using either photochemistry or electrochemistry alone. The direct C–H amination reaction constitutes a straightforward route to important organic compounds bearing an aryl C–N moiety; these are prevalent in pharmaceuticals, agrochemicals and natural products. Typical synthetic methods for these targets require the substrate to bear a directing group9 and/or the reactions to be conducted at elevated temperatures.10 Lambert and co-workers11 showed that the combination of photochemistry with electrochemistry could be applied to direct C–N coupling reactions between arenes and various azoles with moderate to good para-selectivity (Scheme 4). The bench-stable, colorless trisaminocyclopropenium (TAC) ion catalyst 5 was electrochemically oxidized to the corresponding stable radical di-cations 6, which could undergo photoexcitation using visible light to produce the deep red, highly oxidizing excited species 7 (3.33 V vs. SCE) capable of oxidizing the inert benzene (2.48 V vs. SCE) and halogenated benzenes under very mild reaction conditions. The authors also showed that this electrophotocatalytic system could efficiently facilitate SNAr reactions of unactivated aryl fluorides at ambient temperatures in the absence of base.12 The generation of a potent photoelectrochemical catalyst via the integration of photochemistry and electrochemistry is reminiscent of the N,N,N′,N′-tetraphenyl-p-phenylenediamine (TPPD) cation-radical-catalyzed alcohol oxidation described by Reverdy13 in 1982 and the methylene blue (MB)-mediated sulfide oxidation described by Chiba14 in 1998.
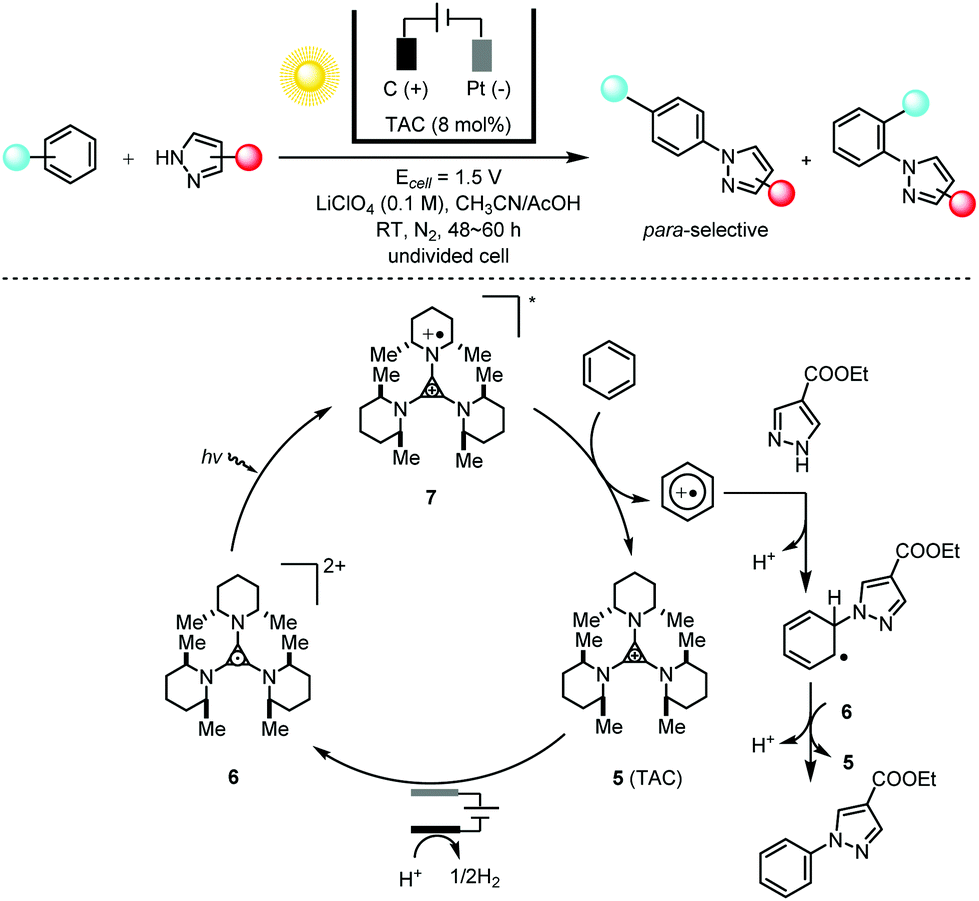 | ||
Scheme 4 Direct C–H amination via electrophotocatalysis using a TAC radical di-cation catalyst, 30–83% yields, para/ortho selectivity of up to 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. TAC = trisaminocyclopropenium. :1. TAC = trisaminocyclopropenium. | ||
In addition to the homogeneous redox-mediated photoelectrochemical synthesis, heterogeneous photoelectrodes can be employed. The transformation of solar energy to chemical fuels using PECs is a widely studied topic due to the high oxidizing/reducing capacity of the photoanode/photocathode generated upon illumination.15 Typically, a photoanode is used to oxidize water to oxygen while a photocathode is used for the reduction of water to hydrogen. The advanced concept of using PECs in organic synthesis has recently been adopted by organic chemists in the oxidation of benzyl alcohols, tetralin,16 furans,17 cyclohexane18 and biomass-derived 5-hydroxylmethylfurfural19 due to its high oxidizing power in the presence of a photoanode (such as BiVO4 and WO3).
Hu and co-workers found that the abundant and robust haematite photoanode was applicable in the highly ortho-selective, non-directed arene C–H amination reaction with a wide range of azoles in hexafluoroisopropanol (Scheme 5).20 Key to the observed high ortho-selectivity seemed to be the formation of intermediate 8 which possessed a hydrogen bond between hexafluoroisopropanol and pyrazole. This method was also successfully applied in the late-stage functionalization of several pharmaceutical compounds. In a manner that was mechanistically distinct from that of canonical photoredox catalysis, the light absorption was achieved by a heterogeneous haematite semiconductor and thus the photogenerated oxidizing hole was localized in the valence band (VB) of the haematite photoanode. Regeneration of the catalyst was then realized via the migration of photoexcited electrons to a counter electrode (Pt in this case) to form hydrogen. In addition, the charge transfer in this photoelectrocatalysis from the edge of the VB to the substrate for oxidation differed from that in electrochemistry in which the electrons are transferred from the substrate to the Fermi level of the electrode.
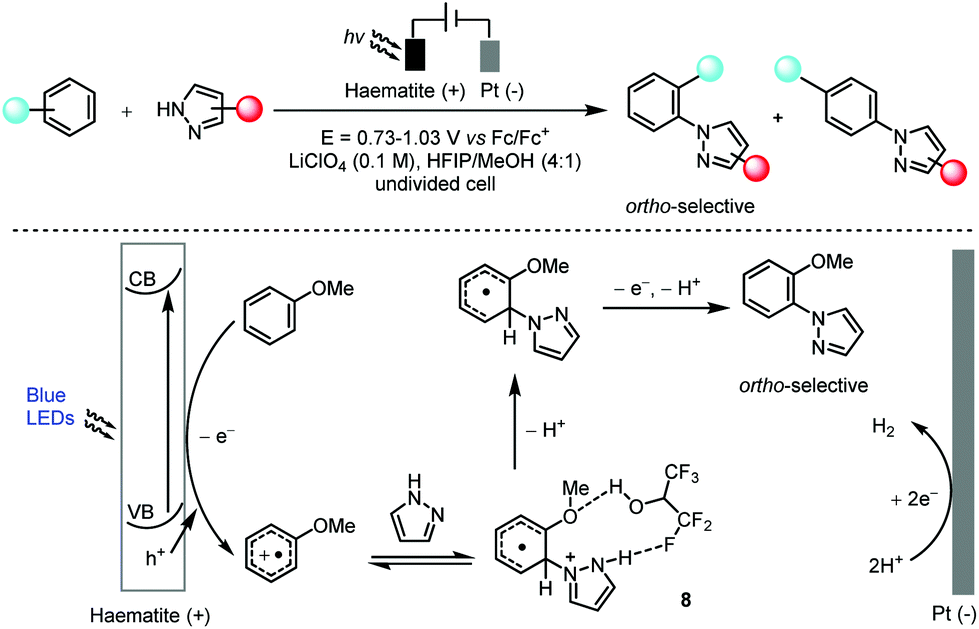 | ||
| Scheme 5 Photoelectrocatalytic arene C–H amination using a haematite photoanode, 10–89% yields, ortho/para selectivity of up to 14:1. Fc = ferrocene; HFIP = hexafluoroisopropanol; CB = conduction band; VB = valence band. | ||
To probe the potential for energy saving in electrochemical synthesis using PECs, Wu and co-workers conducted a direct comparison of a PEC (using BiVO4 as the photoanode) and an electrochemical approach in the P–H/C–H cross-coupling reaction. They found that nearly 90% external bias input is saved when the PEC drives the desired C–P bond formation in comparison to similar yields achieved with an electrochemical cell. Specifically, to produce a current of 5.0 mA required an external applied voltage of 0.1 V (vs. Ag/AgCl) for the BiVO4 PEC system while 1.5 V (vs. Ag/AgCl) was needed for a glassy carbon electrochemical system.21
Photochemistry and electrochemistry can also be effectively coupled in a custom-made manner. For instance, Stahl and co-workers22 adopted a combined electrochemical/photochemical method for dehydrogenative C(sp3)–H/N–H coupling with sp2 and sp3 N–H bonds in a Hofmann–Löffler–Freytag-type reaction23 (Scheme 6). The rationale for choosing iodine as the electrochemical mediator stems from the fact that the I−/I3−/I2 redox couple is about 0.4 V lower than that for bromide and 1–1.5 V lower than those for the electrochemical PCET (proton-coupled electron transfer) and ET-PT-ET (electron transfer-proton transfer-electron transfer) reactions. On the other hand, the light-induced homolysis of the N–I bond of intermediate 9 mitigates the tendency of the N-iodo intermediates to undergo β-elimination to form HI and imine under thermal conditions. Therefore, this photoelectrochemical Hofmann–Löffler–Freytag reaction offers significantly improved functionality compatibility (especially for electron-rich aromatics) in the direct C–H amination reaction. This constructive merging of electrochemistry (for a desired redox chemical transformation) and photochemistry (for the effective cleavage of certain chemical bonds) is again reminiscent of the early contributions from Scheffold and co-workers in the nucleophilic alkylation and acylation of electron-deficient olefins using vitamin B12 as the reductive photoelectrocatalyst in the 1980s.24
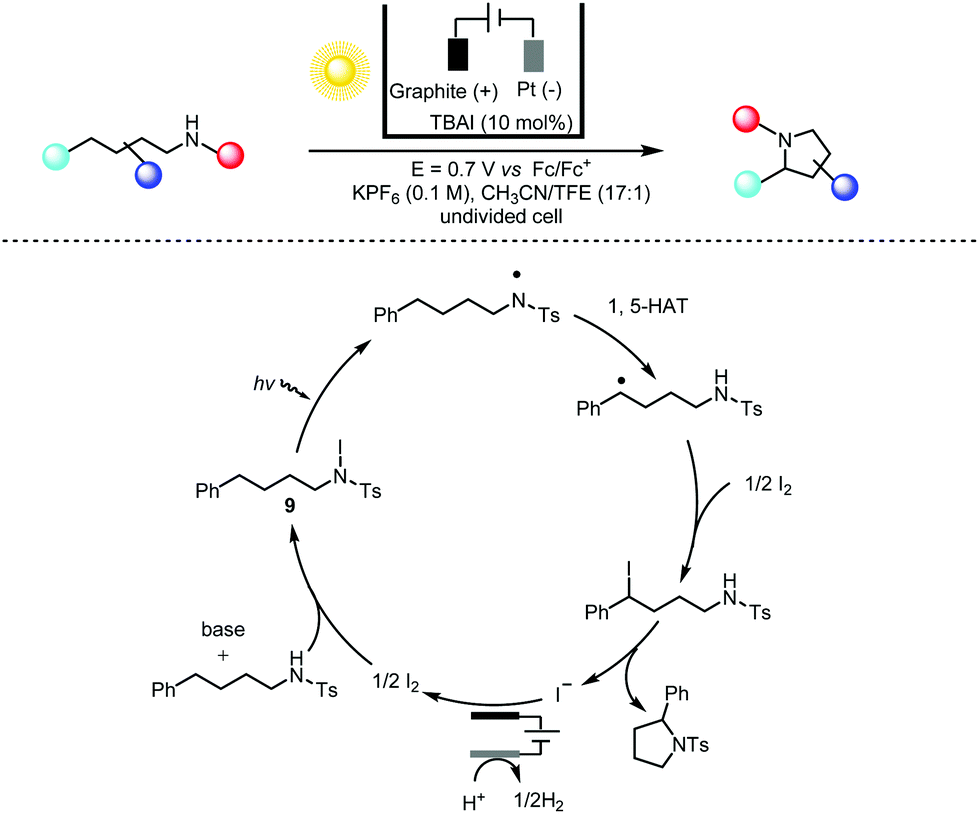 | ||
| Scheme 6 Photoelectrochemical amination of C(sp3)–H bonds, 44–86% yields. TBAI = tetrabutylammonium iodide; HAT = hydrogen atom transfer; TFE = trifluoroethanol. | ||
In summary, recent advances in organic synthesis that merge photochemistry with electrochemistry using PECs and redox-mediated electrolysis have been accomplished. This emerging yet promising concept integrates the merits of both photochemistry and electrochemistry while overcoming their flaws: (1) no stoichiometric amount of oxidant/reductant is needed. (2) The photoelectrochemistry is capable of generating much more potent catalysts than can be obtained using either photochemistry or electrochemistry alone. (3) It is possible to incorporate modified photoelectrodes in organic synthesis (with PECs). (4) The approach offers great opportunities to creatively integrate photochemistry with electrochemistry to meet the specific requirements of certain reactions. Following these exciting breakthroughs, future studies should focus on the adoption of this new concept in forming chemical bonds that are otherwise difficult or impossible to produce using conventional synthetic methods. The mild reaction conditions, rich sources of photoelectrochemical redox mediators and photoelectrodes, and convenient energy-input tuning via the light source and applied potential make the merging of photochemistry and electrochemistry a highly promising strategy for meeting the green and sustainable requirements of modern organic synthesis. We envisage that more applications and developments of this emerging concept in organic synthesis will arise in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21901041 and 21772023) is gratefully acknowledged. We thank Prof. Yi Li (Fuzhou University) for helpful discussions. We highly appreciate the insightful opinions and suggestions from all the reviewers that have helped us to improve this manuscript.Notes and references
- (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (c) S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059–6071 RSC; (d) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS; (e) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS; (f) C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS.
- (a) K. D. Moeller, Tetrahedron, 2000, 56, 9527–9554 CrossRef CAS; (b) J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605–621 RSC; (c) J.-i. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef CAS; (d) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (e) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706–6765 CrossRef CAS; (f) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS; (g) S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CrossRef CAS; (h) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC; (i) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS; (j) Q.-L. Yang, P. Fang and T.-S. Mei, Chin. J. Chem., 2018, 36, 338–352 CrossRef CAS.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- (a) R. H. Verschueren and W. M. De Borggraeve, Molecules, 2019, 24, 2122 CrossRef; (b) L. Capaldo, L. L. Quadri and D. Ravelli, Angew. Chem., Int. Ed., 2019, 58 DOI:10.1002/anie.201910348.
- (a) F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575–3579 CrossRef CAS; (b) M. A. J. Duncton, Med. Chem. Commun., 2011, 2, 1135–1161 RSC.
- H. Yan, Z.-W. Hou and H.-C. Xu, Angew. Chem., Int. Ed., 2019, 58, 4592–4595 CrossRef CAS.
- (a) R. Lechner, S. Kümmel and B. König, Photochem. Photobiol. Sci., 2010, 9, 1367–1377 RSC; (b) B. Mühldorf and R. Wolf, Chem. Commun., 2015, 51, 8425–8428 RSC; (c) B. Mühldorf and R. Wolf, Angew. Chem., Int. Ed., 2016, 55, 427–430 CrossRef; (d) G. Tang, Z. Gong, W. Han and X. Sun, Tetrahedron Lett., 2018, 59, 658–662 CrossRef CAS.
- W. Zhang, K. Carpenter and S. Lin, Angew. Chem., Int. Ed., 2019, 58 DOI:10.1002/anie.201910300.
- (a) L. D. Tran, J. Roane and O. Daugulis, Angew. Chem., Int. Ed., 2013, 52, 6043–6046 CrossRef CAS; (b) T. Matsubara, S. Asako, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2014, 136, 646–649 CrossRef CAS.
- (a) H. J. Kim, J. Kim, S. H. Cho and S. Chang, J. Am. Chem. Soc., 2011, 133, 16382–16385 CrossRef CAS; (b) A. A. Kantak, S. Potavathri, R. A. Barham, K. M. Romano and B. DeBoef, J. Am. Chem. Soc., 2011, 133, 19960–19965 CrossRef CAS.
- H. Huang, Z. M. Strater, M. Rauch, J. Shee, T. J. Sisto, C. Nuckolls and T. H. Lambert, Angew. Chem., Int. Ed., 2019, 58, 13318–13322 CrossRef CAS.
- H. Huang and T. H. Lambert, Angew. Chem., Int. Ed., 2019, 58 DOI:10.1002/anie.201909983.
- (a) J.-C. Moutet and G. Reverdy, J. Chem. Soc., Chem. Commun., 1982, 654–655 RSC; (b) J.-C. Moutet and G. Reverdy, Tetrahedron Lett., 1979, 20, 2389–2392 CrossRef.
- K. Chiba, Y. Yamaguchi and M. Tada, Tetrahedron Lett., 1998, 39, 9035–9038 CrossRef CAS.
- (a) M. Grätzel, Nature, 2001, 414, 338–344 CrossRef; (b) Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS; (c) A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- (a) T. Li, T. Kasahara, J. He, K. E. Dettelbach, G. M. Sammis and C. P. Berlinguette, Nat. Commun., 2017, 8, 390 CrossRef; (b) H. Tateno, Y. Miseki and K. Sayama, ChemElectroChem, 2017, 4, 3283–3287 CrossRef CAS.
- H. Tateno, Y. Miseki and K. Sayama, Chem. Commun., 2017, 53, 4378–4381 RSC.
- H. Tateno, S. Iguchi, Y. Miseki and K. Sayama, Angew. Chem., Int. Ed., 2018, 57, 11238–11241 CrossRef CAS PubMed.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328 CrossRef CAS PubMed.
- L. Zhang, L. Liardet, J. Luo, D. Ren, M. Grätzel and X. Hu, Nat. Catal., 2019, 2, 366–373 CrossRef CAS.
- J.-H. Wang, X.-B. Li, J. Li, T. Lei, H.-L. Wu, X.-L. Nan, C.-H. Tung and L.-Z. Wu, Chem. Commun., 2019, 55, 10376–10379 RSC.
- F. Wang and S. S. Stahl, Angew. Chem., Int. Ed., 2019, 58, 6385–6390 CrossRef CAS PubMed.
- (a) M. E. Wolff, Chem. Rev., 1963, 63, 55–64 CrossRef CAS; (b) X. Hu, G. Zhang, F. Bu, L. Nie and A. Lei, ACS Catal., 2018, 8, 9370–9375 CrossRef CAS; (c) S. Herold, D. Bafaluy and K. Muñiz, Green Chem., 2018, 20, 3191–3196 RSC.
- (a) L. Auer, C. Weymuth and R. Scheffold, Helv. Chim. Acta, 1993, 76, 810–818 CrossRef CAS; (b) R. Orliński and T. Stankiewicz, Tetrahedron Lett., 1988, 29, 1601–1602 CrossRef; (c) R. Scheffold and R. Orlinski, J. Am. Chem. Soc., 1983, 105, 7200–7202 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Li-Xin Dai on the occasion of his 95th birthday. |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2020 |