
Merging photochemistry with electrochemistry in organic synthesis†
Ke-Yin
Ye  *a
*a
*a
Abstract
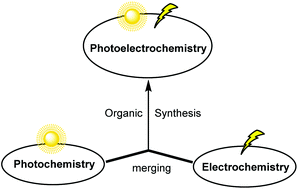
Photochemistry and electrochemistry are two powerful tools in organic synthesis as reflected by the recent resurgence of both research areas. Despite the impressive advances achieved so far, both synthetic technologies suffer from innate disadvantages. The constructive merging of photochemistry and electrochemistry, therefore, offers the potential to overcome the distinct flaws of one method through the advantages of the other, so that novel reaction pathways that are unachievable with individual methods can be envisioned. This Highlight article describes recent breakthroughs using this promising concept of merging photochemistry with electrochemistry in organic synthesis.
 Please wait while we load your content...
Please wait while we load your content...

