
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBio-inspired interfacial chemistry for the fabrication of a robust and functional graphene oxide composite film†
Yoo-Bin Kwon a,
Seongwon Chob,
Dal-Hee Min*a and
Young-Kwan Kim*b
a,
Seongwon Chob,
Dal-Hee Min*a and
Young-Kwan Kim*b
aDepartment of Chemistry, Seoul National University, Seoul 08826, Republic of Korea. E-mail: dalheemin@snu.ac.kr
bDepartment of Chemistry, Dongguk University, 30 Pildong-ro, Jung-gu, Seoul, 04620, Republic of Korea. E-mail: kimyk@dongguk.edu
First published on 5th March 2024
Abstract
A strong and functional artificial nacre film is developed by using polyethyleneimine-functionalized GO (PEI-GO) and pyrogallol (PG) inspired by insect exoskeleton sclerotization. PEI-GO is macroscopically assembled into the laminated films and then reacted with PG under the optimized condition for their efficient cross-linking through Schiff-base reactions. The internal structure and physicochemical properties of PG-treated PEI-GO (PG@PEI-GO) films are systematically explored with various analytical tools. The optimized PG@PEI-GO films exhibit excellent tensile strength, modulus, and toughness of 216.0 ± 12.9 MPa, 17.0 ± 1.1 GPa, and 2192 ± 538.5 kJ m−3 which are 2.7, 2.8, and 2.3-fold higher than those of GO films, respectively. Furthermore, silver nanoparticles (AgNPs) are densely immobilized on the PG@PEI-GO films harnessing their abundant amine groups, and the AgNPs immobilized PG@PEI-GO films exhibit a high catalytic activity in the conversion of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) with maintaining structural integrity. Based on the results, we demonstrate that the rational design of interfaces, inspired by natural materials, is an efficient approach to achieving strong and functional GO laminated composite films.
1 Introduction
Biomimetic nanohybrid structures have gained significant interest due to their hierarchical structures, resulting in outstanding mechanical properties. Thus, numerous natural materials with excellent mechanical strength have been extensively studied to develop high-performance composite materials.1–10 A material that shows promise for various purposes is nacre, which has a brick-and-mortar structure. The high mechanical properties of nacre come from its unique laminated structure consisting of inorganic platelets (CaCO3) as a hard component and biopolymers as a soft component.11,12 Graphene oxide (GO) is a promising material for mimicking natural nacre. It possesses unique physicochemical properties such as strong mechanical strength, flexibility, extremely high aspect ratio, aqueous processability and chemically-tailorable surface.13–16The presence of oxygen-containing functional groups on the surface of GO is particularly important in developing a high-performance composite material, especially when it comes to nacre-mimetic structures. This is because the mechanical properties of such structures are mostly influenced by chemical interactions at the interfaces between their hard and soft components.17–19 Therefore, there have been numerous attempts to create strongly interactive interfaces by cross-linking the oxygen-containing functional groups of GO sheets through various chemical interactions. For example, An et al. have utilized borate ions as an intercalator to produce a robust and stiff nacre-like GO film. They achieved this by forming borate orthoester covalent bonds between interlayers of the laminated GO films. These bonds are commonly found in higher-order plants.20 Cui et al. synthesized polydopamine-functionalized GO sheets and fabricated strong, tough nacre-like GO films with cross-linked structures based on mussel-inspired universal adhesiveness.21 Chen et al. demonstrated that phytic acid can cross-link nacre-like GO films, resulting in comprehensively reinforced mechanical properties through chelating interactions commonly found in biomaterials.22
Recently, phenol-amine chemistry found in insect exoskeletons has been widely explored to develop a strong and universal adhesive layer.23,24 This phenol-amine chemistry is inspired by the sclerotization process of insects to form their hard cuticle layer. The crucial chemistry involved in the sclerotization process is cross-linking reactions of amine-rich polymers with quinone, which is an oxidative form of phenol.25,26 Since this cross-linking process not only quickly occurs under mild conditions but also results in highly cross-linked structure, we aimed to incarnate this simple process in the rational interface design and fabrication of GO composite films with strongly enhanced mechanical properties.23 However, to the best of our knowledge, this interesting bio-inspired phenol-amine chemistry has been not fully harnessed to fabricate a high-performance artificial nacre despite its strong potential. Therefore, the hybridization of interfacial chemistry in insect sclerotization and hierarchical structure in a nacre can be a novel and efficient bio-inspired strategy to fabricate a robust composite material based on the evolutionary strategies of living organisms. For the realization of this bio-inspired fabrication strategy, amine-rich GO derivatives and polyphenol analogues are utilized as a chemically-reactive brick-and-mortar, respectively.
Herein, polyethyleneimine functionalized GO (PEI-GO) was synthesized by the ring-opening reaction between amine groups of PEI and epoxy groups of GO, and the synthesized PEI-GO sheets were vacuum-filtered to construct a free-standing film mimicking a nacre-like laminated structure. The PG treated PEI-GO (PG@PEI-GO) films were prepared through two different methods for mimicking sclerotization process. First, the PEI-GO suspension, which is being filtered, was mixed with PG when it turned to gel-like state, and the resulting films were named PG@PEI-GO (wet) films. Second, the dried PEI-GO films were incubated in an aqueous solution of PG, and the resulting films were named PG@PEI-GO (dry) films (Fig. 1a). The prepared PG@PEI-GO films were thoroughly analysed using various analytical tools to explore their internal structures and physicochemical properties. The results clearly revealed that PEI-GO sheets were efficiently cross-linked with PG through Schiff-base reactions, and the degree of cross-linking was higher in the case of PG@PEI-GO (wet) films than PG@PEI-GO (dry) films. Consequently, the PG@PEI-GO (wet) films presented the significantly enhanced overall mechanical properties including tensile strength, modulus, and toughness compared to GO, PEI-GO, PG treated GO (PG@GO), and PG@PEI-GO (dry) films. Furthermore, silver NPs (AgNPs) were densely immobilized on the surface of PG@PEI-GO (wet) films, and the AgNPs immobilized PG@PEI-GO films showed a high catalytic activity and structure stability in the conversion of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP). The results clearly demonstrate that by mimicking natural materials, the rationally designed interfacial chemistry of GO films can not only reinforce their overall mechanical properties but also endow a novel functionality such as catalytic activity through the incorporation of catalytic nanomaterials. The catalytic nanocomposite films can be utilized as a versatile platform for the synthesis of valuable compounds and decomposition of environmental pollutants owing to their catalytic activity, strong mechanical properties, and recyclability. In addition, the PG@PEI-GO (wet) films can be diversely hybridized with other functional nanomaterials by simple dip- or spray-coating through electrostatic interaction and hydrogen bonding based on their surface properties. Furthermore, the high robustness of PG@PEI-GO films can provide recyclability of AgNP/PG@PEI-GO films for application as a bulk heterogeneous catalytic material without loss of their structural integrity and catalytic activity. This robustness is also prerequisite for various industries including aerospace, automobile, flexible electronics, and packaging/protective materials.27
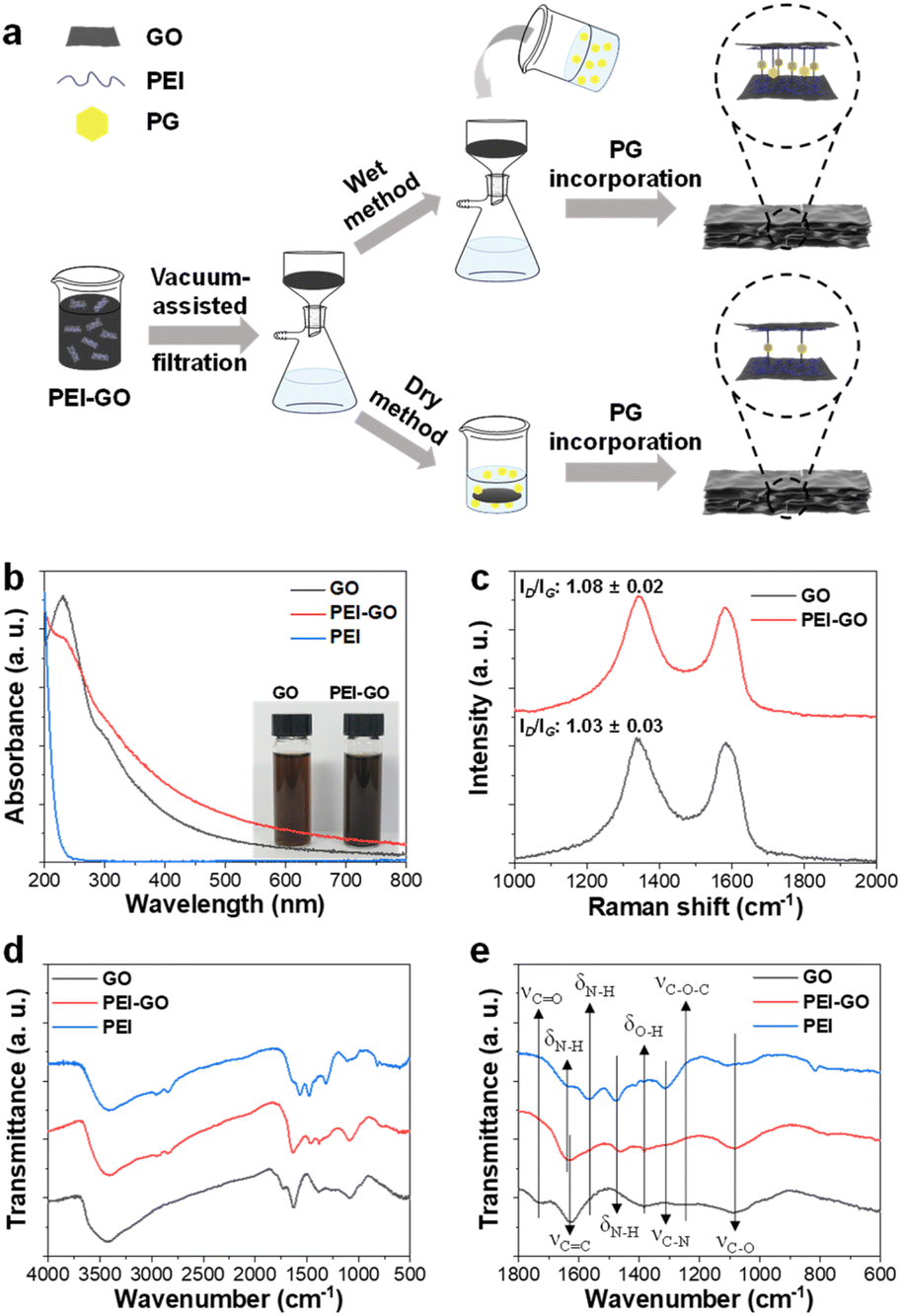 | ||
| Fig. 1 (a) Schematic diagram for the fabrication of PG@PEI-GO (wet) and PG@PEI-GO (dry) films. (b) UV-vis spectra of GO, PEI, and PEI-GO with inset of the optical images of GO and PEI-GO suspensions. (c) Raman spectra of GO and PEI-GO, (d and e) FT-IR spectra of GO, PEI-GO, and PEI with different spectral regions. | ||
2 Experimental section
2.1 Materials
Graphite was purchased from Kropfmühl AG (Herzenberg, Germany). Potassium permanganate, sodium nitrate, sulfuric acid, hydrogen peroxide (30%), pyrogallol, and hydrochloric acid (35–37%) were purchased from Daejung Chemicals (Siheung, Korea). Polyethylene imine (MW: 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 50000) was purchased from Sigma-Aldrich (St. Louis, MO, USA).
50000) was purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.2 Preparation of GO
1.5 g of graphite and 0.5 g of NaNO3 were added to 23 mL of 98% H2SO4 in an ice bath with magnetic stirring. 3 g of KMnO4 was slowly added into the reaction mixture to keep the temperature below 20 °C. After thoroughly mixed, the mixture was transferred to a 35 °C oil bath and stirred for 1 h, resulting in the formation of a dense paste. Then, 40 mL of water was carefully added into the mixture and stirred for 30 min (this step requires caution due to the rapid temperature increase). Finally, 100 mL of water was added, followed by the addition of 3 mL of 30% H2O2 (the color shift from dark brown to yellow). For purification, the mixture was washed with a 3.4% HCl solution to eliminate residual salts and washed with water until the supernatant is neutralized.282.3 Synthesis of PEI-GO
GO was prepared in 500 mL of water at 0.1 mg mL−1, and as-prepared GO suspension was slowly added into 1000 mL of aqueous solution of PEI at 0.25 mg mL−1 with vigorous stirring. Then, the reaction mixture was stirred for 12 h at room temperature. After the reaction, the reaction mixture was centrifuged 5 times at 15770 rcf for washing and re-suspended in water for further use.
2.4 Fabrication of GO and PEI-GO films
For the fabrication of GO and PEI-GO films, GO and PEI-GO suspensions were prepared in 10 mL of water at 5 mg mL−1, and the resulting suspensions were vacuum-assisted filtered with a cellulose ester (CE) membrane of 0.45 μm pore size, respectively. After the completion of the filtration, the GO and PEI-GO films were taken off from a CE membrane and dried in an oven at 60 °C for 12 h.2.5 Fabrication of PG@PEI-GO (wet) films
5 mL of 0.4 M aqueous solution of PG was added into the PEI-GO suspension, which is being vacuum-filtered, when the PEI-GO suspension turned to gel-state. After the completion of the filtration, the cross-linked PEI-GO films were taken off from a CE membrane and dried in an oven at 60 °C for 12 h.2.6 Fabrication of PG@PEI-GO (dry) films
After fabrication of PEI-GO films, as prepared and dried PEI-GO films were immersed in a 0.4 M aqueous solution of PG for 1 h and dried in an oven at 60 °C for 12 h.2.7 Synthesis of AgNPs
0.5 mL of 59 mM AgNO3 solution and 1 mL of 34 mM trisodium citrate solution were added to 98 mL of water and stirred for several min. Subsequently, 0.5 mL of a 20 mM NaBH4 solution, aged for 2 h, was added into the mixture under continuous stirring. The reaction mixture was further stirred for 1 h and then left to age for 24 h at room temperature before use.292.8 Introduction of AgNPs onto GO and PG@PEI-GO (wet) films
GO and PG@PEI-GO (wet) films were incubated in aqueous suspensions of AgNPs for 1 h, washed thoroughly with water, and dried in an oven at 60 °C for 12 h.2.9 Catalytic reduction of 4-NP to 4-AP
The AgNPs immobilized PG@PEI-GO (wet) films (AgNP/PG@PEI-GO (wet)) film was added to 10 mL of a freshly prepared 0.05 mM aqueous solution of 4-NP, adjusted to pH 6.0. Subsequently, 1 mL of a freshly prepared 1 M aqueous solution of NaBH4 was rapidly added to the mixture at room temperature. The catalytic reaction was then monitored using UV-vis spectroscopy based on the absorption peak at around 400 nm.302.10 Characterization
UV-vis spectra were measured using a Varian Cary 50 Conc (Varian, Inc. USA). Fourier transform infrared (FT-IR) analysis was carried out using an FTIR-7600 (Lambda scientific systems, Inc. USA). Raman analysis was carried out using HEDA (WEVE, Korea) with a diode laser (532 nm) focused through an integral microscope (BX53M, Olympus) equipped with a 20× objective lens (MPLFLN20×, Olympus). X-ray photoelectron spectroscopy (XPS) spectra were obtained using a Thermo Scientific K-alpha (Thermo VG, USA) with monochromated Al Kα (1486.6 eV). X-ray diffraction (XRD) patterns were acquired using an X-ray diffractometer (D/MAX 2500, Rigaku) with Cu Kα radiation (λ = 0.15418 nm) at 40 kV and 50 mA. Scanning electron microscopy (SEM) images were acquired using a NOVA Nano scanning electron microscope 450 (FEI company, Netherlands). Tensile tests for evaluating the mechanical properties of GO, PEI-GO, PG@PEI-GO (wet), PG@PEI-GO (dry), and PG@GO (wet) films were conducted using a universal testing machine (UTM) (ST-1000, SALT Co., Ltd) equipped with a 200 N load cell with a 15 mm gauge length and a 0.7 mm min−1 loading rate, respectively.2.11 Data analysis
A completely randomized design was adopted in this study. For statistical analysis, all experiments were independently performed at least in triplicate for each sample, and the results were indicated as average values with their standard deviation. The significance of the experimental results was validated using one-way analysis of variance (ANOVA) test, and significantly different means were evaluated using a Tukey honest significant difference (HSD) test at the p < 0.05 level.3 Results and discussion
The synthesized GO has an average lateral size of 8.4 μm and was functionalized with PEI through a ring opening reaction between epoxy groups of GO and primary amine groups of PEI (Fig. S1†).31 The as-synthesized PEI-GO was thoroughly characterized using various spectroscopic tools to confirm the successful preparation. The UV-vis spectrum of GO exhibited a typical peak at 231 nm from the π–π* transition of conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds with a shoulder at around 294 nm from the n–π* transition of CO bonds, respectively. After the reaction with PEI, a new absorption peak at around 200 nm, which was observed from PEI solution, was observed from UV-vis spectrum of PEI-GO with a partial decrease of n–π* transition shoulder and overall increase of absorption in visible region (Fig. 1b).32 Those changes indicated that PEI-GO was successfully synthesized with a partial restoration of the sp2 carbon networked structure of GO during the PEI functionalization.31,33 Consequently, the resulting PEI-GO suspension exhibited a high colloidal dispersibility and dark color compared to GO suspension (Fig. 1b). It has been previously reported that GO can be reduced by the reaction with amine-rich polymers in aqueous media.31
C bonds with a shoulder at around 294 nm from the n–π* transition of CO bonds, respectively. After the reaction with PEI, a new absorption peak at around 200 nm, which was observed from PEI solution, was observed from UV-vis spectrum of PEI-GO with a partial decrease of n–π* transition shoulder and overall increase of absorption in visible region (Fig. 1b).32 Those changes indicated that PEI-GO was successfully synthesized with a partial restoration of the sp2 carbon networked structure of GO during the PEI functionalization.31,33 Consequently, the resulting PEI-GO suspension exhibited a high colloidal dispersibility and dark color compared to GO suspension (Fig. 1b). It has been previously reported that GO can be reduced by the reaction with amine-rich polymers in aqueous media.31
In their Raman spectra, both GO and PEI-GO showed characteristic D- and G-bands at 1336 and 1583 cm−1, corresponding to disordered and ordered sp2 carbon networked structures. The relative intensity ratio of the D- and G-bands (ID/IG) served as a typical indicator for the degree of defect in graphene derivatives.28 The ID/IG value of GO increased from 1.03 to 1.08 after functionalization with PEI, suggesting the formation of defects in the sp2 carbon domains during PEI functionalization (Fig. 1c).33 In their FT-IR spectra, GO showed its representative peaks at 3428 cm−1 from O–H stretching, 1727 cm−1 from CO stretching, 1625 cm−1 from aromatic CC stretching, 1384 cm−1 from O–H bending, 1255 cm−1 from C–O–C stretching, and 1087 cm−1 from C–O stretching.34 PEI exhibited its characteristic peaks at 3409 cm−1 from N–H stretching, 2952 cm−1 from –CH2– symmetric stretching, 2846 cm−1 from –CH2– asymmetric stretching, 1635, 1565, and 1479 cm−1 from N–H bending, and 1317 cm−1 from C–N stretching.35 As expected, PEI-GO showed the overlapped peaks which originated from both GO and PEI at 3415 cm−1 from O–H stretching, 1631 cm−1 from CC stretching, and 1083 cm−1 from C–O stretching of GO and at 3409 cm−1 from N–H stretching and 1465 cm−1 from N–H bending of PEI, respectively (Fig. 1d and e).36,37 All the spectroscopic analyses collectively confirmed that the PEI was successfully functionalized on the surface of GO sheets.
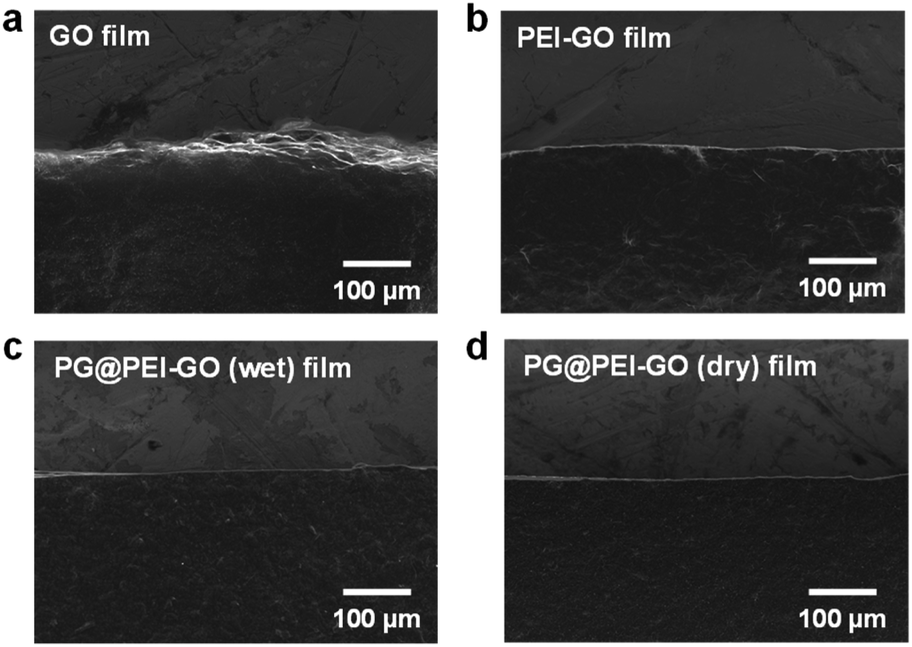
The GO and PEI-GO dispersions were prepared at a concentration of 5 mg mL−1 and filtered under a reduced pressure to fabricate the nacre-like laminated films. The resulting PEI-GO films were treated with PG through two different methods to find the optimal cross-linking process for mimicking insect sclerotization. In the first method, the PG@PEI-GO (wet) films were prepared through addition of PG solution to the PEI-GO suspension, which is being filtered, when the suspension turned gel-like state before completing the filtration process. In the second method, the PG@PEI-GO (dry) films were prepared through immersing as-prepared dry PEI-GO films into PG solution for 1 h and dried in an oven. The cross-sectional SEM images of GO, PEI-GO, PG@PEI-GO (wet), and PG@PEI-GO (dry) films showed that they were successfully assembled into nacre-like laminated films (Fig. 2a–d). XRD analysis was next conducted to further investigate their laminated structures. GO films exhibited the typical (001) diffraction peak at 2θ = 10.36° corresponding to d-spacing of 8.53 Å, and the peak was shifted in the XRD pattern of PEI-GO films to 2θ = 9.48° corresponding to d-spacing of 9.32 Å (Fig. 2e). This peak shift was due to the functionalized PEI on the surface of GO sheets, which enlarged the d-spacing between GO sheets.32,38 Additionally, a new peak appeared at 2θ = 17.52° corresponding to d-spacing of 5.05 Å, which originated from the reduced interlayer distance of GO sheets due to the partial deoxygenation during PEI functionalization (Fig. 2e). The PG@PEI-GO (dry) films showed a nearly identical pattern to that of PEI-GO films with the only slight shift of the (001) diffraction peak to 2θ = 9.14° corresponding to 9.67 Å due to partial PG intercalation. However, the (001) diffraction peak of PG@PEI-GO (wet) films showed a greater down-shift compared to PG@PEI-GO (dry) films, which lead to broadening its shape compared to PEI-GO and PG@PEI-GO (dry) films. Additionally, the new diffraction peak at 2θ = 17.52° derived from partial deoxygenation was shifted to 2θ = 23.86° corresponding to 3.73 Å derived from the reduced interlayer distance by further deoxygenation of GO sheets owing to the intrinsic reducing power of PG (Fig. 2e).39 All those results harmoniously indicated that PG@PEI-GO (wet) films have a higher degree of PG intercalation into the interlayer of laminated PEI-GO sheets than PG@PEI-GO (dry) films, which confirmed that the PG@PEI-GO (wet) films have the optimized interfacial structure to efficiently mimic insect sclerotization.
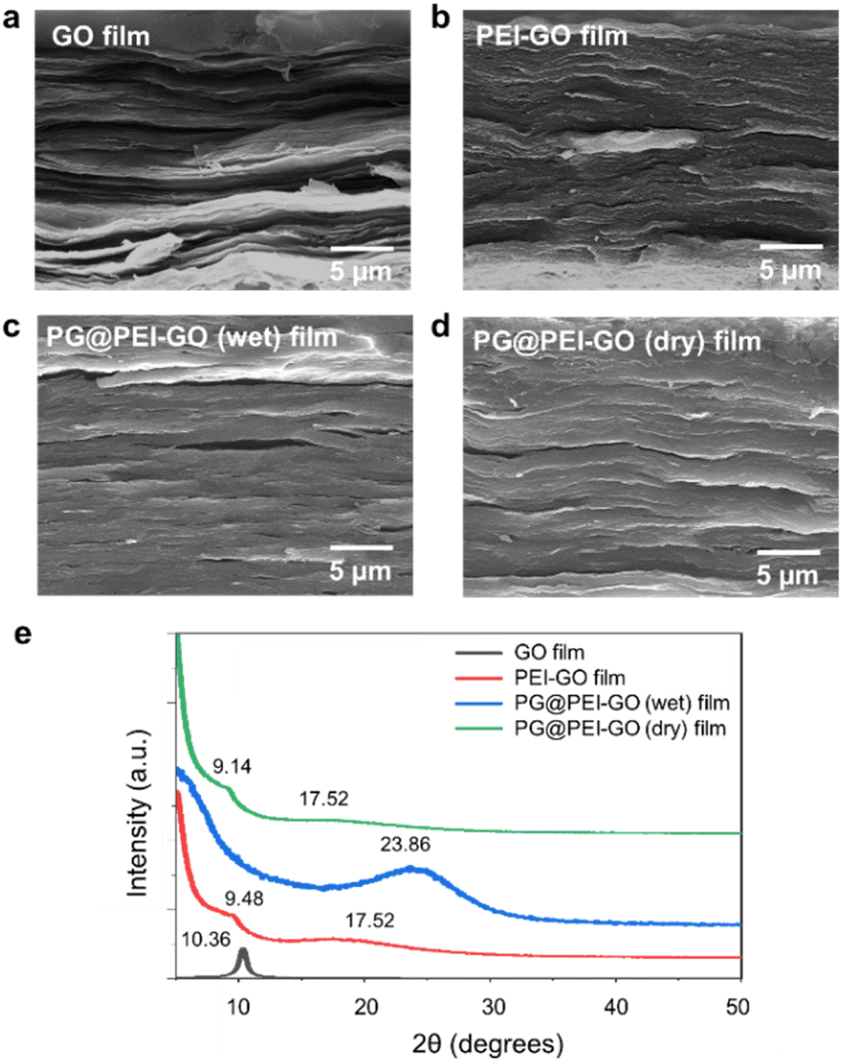 | ||
| Fig. 2 Cross-sectional SEM images of (a) GO, (b) PEI-GO, (c) PG@PEI-GO (wet), and (d) PG@PEI-GO (dry) films, and their (e) XRD patterns. | ||
The chemical structures of GO, PEI-GO, PG@PEI-GO (wet), and PG@PEI-GO (dry) films were analysed using XPS. In C 1s XPS spectrum of GO films, typical peaks were observed at 283.7, 285.8, 287.2, and 288.2 eV originated from C–C and CC bonds (40%), C–O bonds (45.2%), CO bonds (10%), and O–CO bonds (4.8%), respectively (Fig. 3).34 After PEI functionalization, the composition of C–C and CC bonds increased to 51.6%, while oxygen containing bonds such as C–O, CO, and O–CO decreased to 15.8%, 5.0%, and 2.6%, respectively. Importantly, a new peak was observed at 285.1 eV corresponding to C–N bonds of PEI (Fig. 3).40 This result also indicated PEI was successfully functionalized onto the surface of GO sheets with partial reduction of GO during PEI functionalization. In the C 1s XPS spectrum of PG@PEI-GO (wet) films, the composition of C–O bonds notably increased to 23.6%, and a new peak was observed at 284.9 eV corresponding to CN bonds (2.5%) (Fig. 3). These changes originated from the formation of C–O bonds through intercalation of PG and the Schiff-base reactions between PEI and PG at the interlayer of PEI-GO films.25 However, PG@PEI-GO (dry) films showed only slight increase in the composition of C–O and CN bonds to 18.8% and 0.7%, respectively (Fig. 3). Additionally, N 1s XPS spectrum of GO films showed no signal, while that of PEI-GO films showed a single peak at 397.8 eV corresponding to N–C bonds. In stark contrast, PG@PEI-GO (wet) films exhibited two peaks at 397.8 and 400.2 eV corresponding to N–C bonds (70%) and NC bonds (30%). Although the PG@PEI-GO (dry) films also showed peaks corresponding to the N–C and NC bonds, the composition of N–C bond (95%) was much higher than that of NC bonds (5%) (Fig. S2†).41 The comprehensive XPS analysis clearly revealed that PG intercalation and subsequent Schiff-base reaction were much more efficient in PG@PEI-GO (wet) films than PG@PEI-GO (dry) films, which is well matched with the results of XRD analysis.
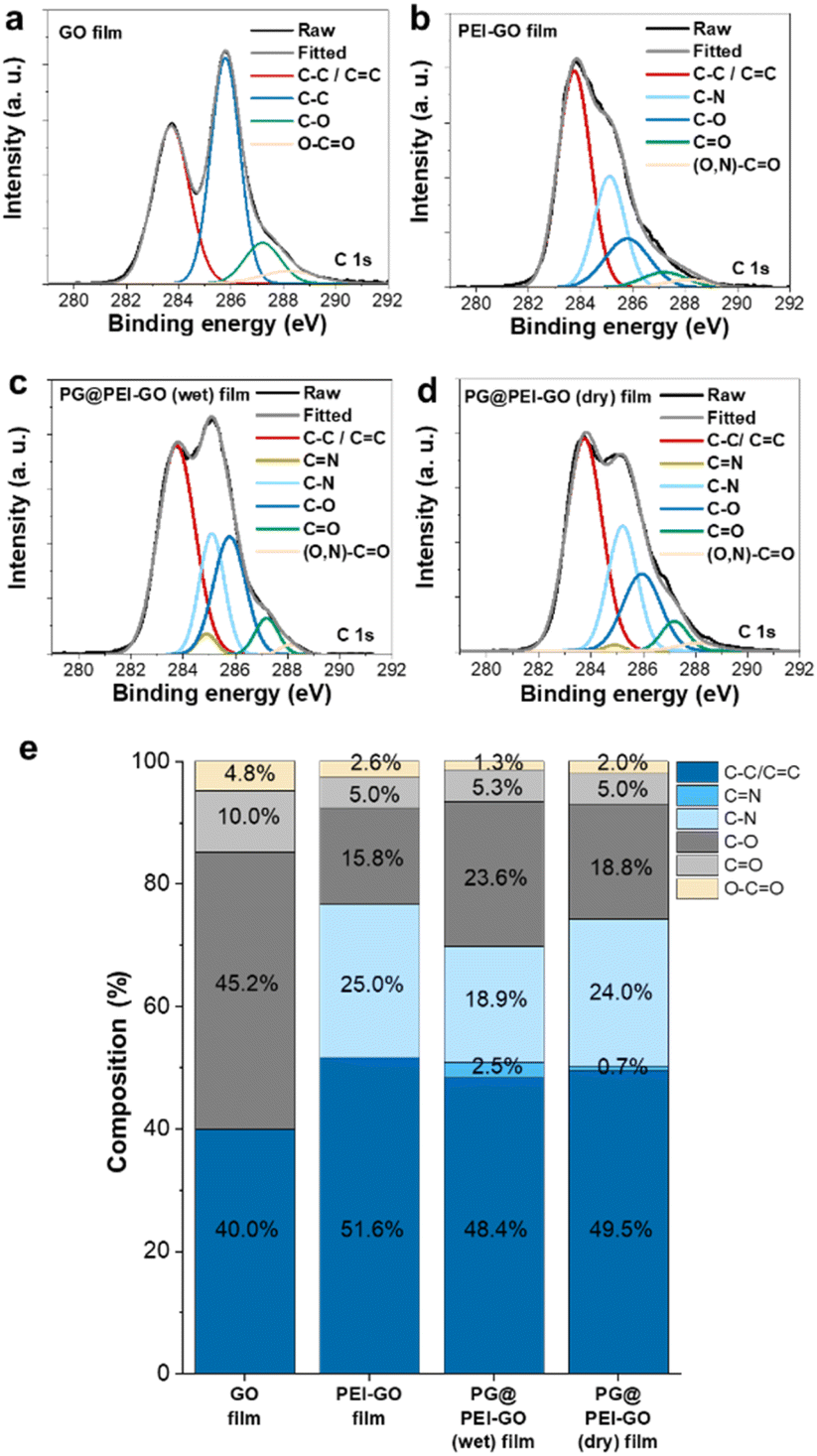 | ||
| Fig. 3 C 1s XPS spectra of (a) GO, (b) PEI-GO, (c) PG@PEI-GO (wet), and (d) PG@PEI-GO (dry) films and (e) composition of C–C/CC, CN, C–N, C–O, CO, and O–CO bonds in those films. | ||
The mechanical properties of GO, PEI-GO, PG@PEI-GO (wet), and PG@PEI-GO (dry) films were thoroughly investigated with tensile experiments to explore reinforcement effect of the bio-inspired interfacial structure. The tensile strength, modulus, and toughness of GO films were 79.5 ± 5.5 MPa, 6.1 ± 0.4 GPa, and 937.2 ± 76.8 kJ m−3, and after PEI functionalization, those values increased to 149.0 ± 16.6 MPa, 11.4 ± 0.4 GPa, and 1035.0 ± 203.1 kJ m−3, respectively (Fig. 4). These changes of mechanical properties implied that the interfacial interaction of GO sheets was strengthened by the surface adsorbed PEI through the enhanced π–π interaction and intramolecular hydrogen bonding between abundant amine groups.42 After intercalation of PG into PEI-GO films, the tensile strength, modulus, and toughness of PG@PEI-GO (wet) films were measured to be 216.0 ± 12.9 MPa, 17.0 ± 1.1 GPa, and 2192.9 ± 538.5 kJ m−3, which were 1.5, 1.5 and 2.1-fold higher than those of PEI-GO films, respectively (Fig. 4). However, as expected, the mechanical properties of PG@PEI-GO (dry) films were only 160.3 ± 18.5 MPa, 15.4 ± 1.1 GPa, and 1106.0 ± 610.0 kJ m−3 which were comparable to those of PEI-GO films (Fig. 4). This higher enhancement of PG@PEI-GO (wet) films than PG@PEI-GO (dry) films can be attributed to the more efficient intercalation of PG into the laminated PEI-GO films which lead to the formation of cross-linked structures, as confirmed by the XRD and XPS analysis.
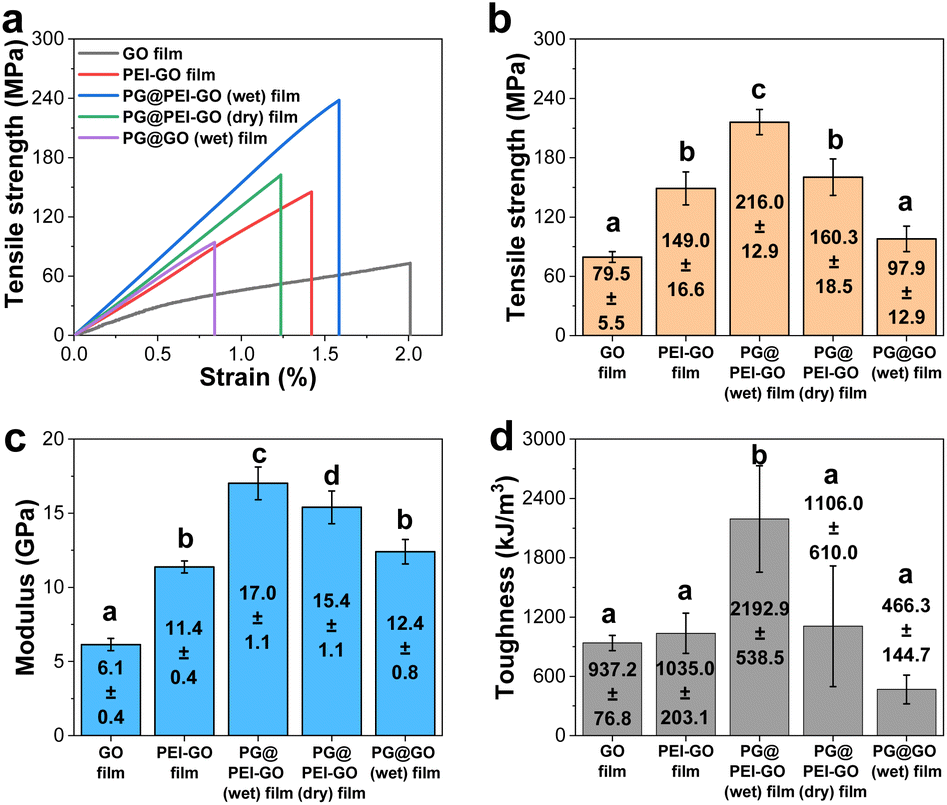 | ||
| Fig. 4 (a) Stress–strain curves, (b) tensile strength, (c) modulus, and (d) toughness of GO, PEI-GO, PG@PEI-GO (wet), PG@PEI-GO (dry), and PG@GO (wet) films. Error bars represent the standard deviation (at least n = 3); different letters represent significantly different means (p < 0.05). | ||
Then, the fracture analysis was carried out to further explore their interfacial structures. In general, the laminated GO sheets are pulled-out during tensile experiments when the applied shear force exceeded their friction force. Therefore, the degree of cross-linking in the GO films is inversely proportional to the extent of pull-out when their fracture occurs. As a result, GO films showed the typical pull-out behaviour, whereas this pull-out behaviour was not observed from the fractured PEI-GO, PG@PEI-GO (wet), and PG@PEI-GO (dry) films (Fig. 5). These results showed that the PEI functionalization and subsequent PG treatment resulted in the reinforcement of interfacial shear stress between the laminated GO sheets by the multiple interactions through π–π interaction, hydrogen bonding, and covalent bonds. As a control, the GO films were treated through the equal process to PG@PEI-GO (wet) films, and the resulting PG@GO (wet) films exhibited the tensile strength, modulus, and toughness of 97.9 ± 12.9 MPa, 12.4 ± 0.8 GPa, and 466.3 ± 144.7 kJ m−3 which were much lower than those of PEI-GO, PG@PEI-GO (dry), and PG@PEI-GO (wet) films, respectively (Fig. 4 and see Fig. S3† for the detailed characterization of PG@GO (wet) films). Likewise, the fracture behaviour of PG@GO (wet) films was analogous to that of GO films (Fig. S4†). These results further confirmed that the comprehensive reinforcement of PG@PEI-GO (wet) films was driven by the interfacial cross-linking reactions between amine groups of PEI-GO sheets and quinone groups of PG which occurred at the interlayer of PEI-GO films.
 | ||
| Fig. 5 SEM images of the tensile-fractured sites of (a) GO, (b) PEI-GO, (c) PG@PEI-GO (wet), and (d) PG@PEI-GO (dry) films. | ||
Finally, the citrate-capped AgNPs were immobilized onto the surface of PG@PEI-GO (wet) films by a simple dip-coating method through the electrostatic interaction between amine groups of PG@PEI-GO (wet) films and citrate ligands of AgNPs (see Fig. S5† for the detailed characterizations of AgNPs). To verify electrostatic interaction between AgNPs and PG@PEI-GO films, the surface charges of GO, PEI-GO, PG@PEI-GO, AgNPs, and AgNP/PG@PEI-GO were measured with a zeta-potential analyzer under the suspended condition. (Fig. S6a†). The zeta-potential value of GO was negative (−27.0 mV) owing to its oxidized structure, but this value became positive to 35.1 mV after PEI functionalization (Fig. S6a†). This positive surface charge indicated PEI-GO presented abundant amine groups, and after reaction with PG, it decreased to 25.4 mV because Schiff-base reaction with phenol groups of PG lead to consumption of amine groups on PEI-GO (Fig. S6a†). It is worthy to note that the net surface charge of PG@PEI-GO was still positive, and this positive charge was further diminished to 12.8 mV after incorporation of negatively charged AgNPs (−29.8 mV) owing to the charge compensation effect (Fig. S6a†). All these results clearly suggested that AgNPs was immobilized onto the surface of PG@PEI-GO films through electrostatic interaction (the UV-vis spectra of GO, PEI-GO, PG@PEI-GO, AgNPs, and AgNP/PG@PEI-GO suspensions were provided in Fig. S6b†). SEM images of the AgNP/PG@PEI-GO (wet) films showed a high surface coverage of AgNPs with a homogeneous distribution (654 ± 32 μm−2) (Fig. 6a). As a control, GO films were also equally incubated with AgNPs, but there was no immobilization of AgNPs on their surface (Fig. 6b). The AgNP/PG@PEI-GO (wet) films were then employed as a heterogeneous and supported catalyst for the reduction of 4-NP to 4-AP (Fig. 7a).
 | ||
| Fig. 6 SEM images of the surface of (a) AgNP/PG@PEI-GO (wet) and (b) AgNP/GO films with different magnifications. | ||
 | ||
| Fig. 7 (a) The catalytic reduction of 4-NP to 4-AP. (b) Optical images of before and after of catalytic reaction using AgNP/GO and AgNP/PG@PEI-GO (wet) films. (c) UV-vis spectra of the catalytic reaction solution with AgNP/PG@PEI-GO (wet) film as a function of reaction time. (d) Kinetic analysis of the catalytic reaction. (e) Conversion rate of 4-NP to 4-AP as a function of recycle number. | ||
The AgNP/PG@PEI-GO (wet) and AgNP/GO films were incubated in aqueous solutions of 4-NP with stirring for 30 min. The yellow 4-NP solution was successfully de-colored with AgNP/PG@PEI-GO (wet) films, and the film structure was well maintained even after stirring for 30 min (Fig. 7b). In contrast, there was no de-coloration of 4-NP, and AgNP/GO films were dispersed into the 4-NP solution by stirring for 30 min owing to their weak interfacial structure (Fig. 7b). Then, the catalytic reaction using AgNP/PG@PEI-GO (wet) film was monitored by using UV-vis spectroscopy. The characteristic absorption band of 4-NP at 320 nm was shifted to 400 nm by addition of 1.0 M of NaBH4, and this peak was used as indicator for the progress of catalytic reaction.30,43 The absorption band at 400 nm was gradually diminished with appearance of a new absorption band at around 300 nm with the progress of reaction (Fig. 7c). These changes indicated that the 4-NP was gradually converted to 4-AP, and the catalytic reaction on the AgNP/PG@PEI-GO (wet) film was completed after 30 min. Based on the kinetic analysis, it was found that the reaction followed the first order kinetic, described as −ln(A) = −kcatt (where A is the absorbance of 4-NP at time t, and kcat is the rate constant). The fitted curve showed the kcat value of 0.11286 min−1 with a high linearity (coefficient of determination, R2 = 0.99499) (Fig. 7d). The structural stability of AgNP/PG@PEI-GO (wet) films was further validated with a recycle test. After 5 recycle test with washing steps, the structure and catalytic conversion rate of AgNP/PG@PEI-GO (wet) films were not notably changed without loss of AgNPs from their surface (Fig. 7e and S7†).
Additionally, the effect of NaBH4 concentration on the catalytic reaction was studied using 0.5 and 2.0 M NaBH4 solutions. Both reaction condition also followed the first order kinetic, and the fitted curve showed the kcat values of 0.0302 and 0.20487 min−1 with 0.5 and 2.0 M NaBH4 solutions and high linearity (coefficient of determination, R2 = 0.98729 and 0.99416, respectively.) (Fig. S8†). These values were 0.3-fold lower and 1.8-fold higher than the original kcat value obtained with 1.0 M NaBH4 solution, respectively. Importantly, the conversion rate of 4-NP to 4-AP was 95.6 and 96.3% with 1.0 and 2.0 M NaBH4 solutions, respectively. However, even after 70 min, only an 87% of conversion rate was achieved with 0.5 M NaBH4 solution (Fig. S8†). Considering these results, the concentration of NaBH4 solution need to be higher than 1.0 M to achieve a high conversion rate over 95% in the present catalytic reaction system.
As control catalytic reactions to verify the origin of catalytic activity from AgNP/PG@PEI-GO (wet) films, the catalytic reaction was carried out with PG@PEI-GO (wet) film and colloidal suspension of AgNPs as a catalyst under the equal reaction condition. In the case of PG@PEI-GO (wet) film, there was no notable change in the UV-vis spectrum of catalytic reaction solution even after 30 min (Fig. S9†). However, the catalytic reaction was completed in 20 min with colloidal AgNPs, and the catalytic rate constant increased 1.9-fold compared to AgNP/PG@PEI-GO (wet) film (Fig. S9†). The enhanced catalytic rate constant with colloidal AgNPs might be derived from the increased available surface area and collision possibility of free-standing AgNPs for the catalytic reaction compared to the surface-immobilized AgNPs on PG@PEI-GO (wet) film. These control experiments clearly supported that the catalytic reaction mainly originated from the incorporated AgNPs onto PG@PEI-GO (wet) films.
Finally, the catalytic rate constant of the AgNP/PG@PEI-GO (wet) film is compared with previously reported literature utilizing noble metal catalysts supported on structural materials. The results confirmed that the catalytic rate constant of AgNP/PG@PEI-GO (wet) film is comparable to those reported in previous literature (Table S1†).
Taken together, it was clearly demonstrated that the present bio-inspired strategy from insect sclerotization is an efficient way to develop a high-performance and functional composite structure using GO derivatives.
4 Conclusions
In conclusion, our study presents a biomimetic strategy inspired by insect exoskeleton sclerotization to fabricate strong and functional GO films. PEI-GO sheets were synthesized and assembled into the nacre-like laminated films through vacuum-assisted filtration. The phenol-amine reactions between PEI-GO and PG were optimized by comparing the dry and wet treatments in terms of the internal structure, cross-linking, and mechanical reinforcement. As a result, the wet method was found as the optimal process, and the resulting PG@PEI-GO (wet) films exhibited the considerably enhanced tensile strength, modulus, and toughness to 216.0 ± 12.9 MPa, 17.0 ± 1.1 GPa, and 2192 ± 538.5 kJ m−3 in comparison with those of GO films (79.5 ± 5.5 MPa, 6.1 ± 0.4 GPa, and 937.2 ± 76.8 kJ m−3), respectively. In addition, the PG@PEI-GO (wet) films afforded immobilization of AgNPs on their surface due to the abundant amine groups. The AgNP/PG@PEI-GO (wet) films showed a high catalytic activity in the conversion of 4-NP to 4-AP with the remarkable structure stability during 5 recycles. Although mechanical properties of the resulting PG@PEI-GO (wet) films are still inferior to synthetic engineering plastics such as polyimide and aramid fiber, the overall and significant mechanical reinforcement shows the strong potential of a rational design of interfacial structures in the GO composite films. Based on the rational design with knowledge of interfacial interactions and structure in natural strong materials, we believe there will be more efficient and practical ways to develop a high-performance composite film by using functional GO derivatives and natural materials. Considering all those features, this study can provide a novel perspective and insight to fabricate the mechanically robust and functional GO based composite films.Author contributions
Yoo-Bin Kwon: conceptualization, data curation, investigation, methodology, formal analysis, writing – original draft, and writing – review & editing. Seongwon Cho: data curation and investigation. Dal-Hee Min: funding acquisition, supervision, and writing – review & editing. Young-Kwan Kim: conceptualization, data curation, methodology, formal analysis, investigation, funding acquisition, supervision, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT) (No. 2021R1A2B5B03086506, 2022R1C1C1008388). This research was also supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2022R1A6A1A03053343).References
- Z. M. Han, D. H. Li, H. Bin Yang, Y. X. Zhao, C. H. Yin, K. P. Yang, H. C. Liu, W. Bin Sun, Z. C. Ling, Q. F. Guan and S. H. Yu, Adv. Funct. Mater., 2022, 32, 2202221 CrossRef CAS.
- W. Chen, P. Zhang, R. Zang, J. Fan, S. Wang, B. Wang and J. Meng, Adv. Mater., 2020, 32, 1907413 CrossRef CAS PubMed.
- J. J. Martin, B. E. Fiore and R. M. Erb, Nat. Commun., 2015, 6, 8641 CrossRef PubMed.
- A. R. Studart, Adv. Funct. Mater., 2013, 23, 4423–4436 CrossRef CAS.
- G. X. Gu, M. Takaffoli and M. J. Buehler, Adv. Mater., 2017, 29, 1700060 CrossRef PubMed.
- Y. Zhang, S. Gong, Q. Zhang, P. Ming, S. Wan, J. Peng, L. Jiang and Q. Cheng, Chem. Soc. Rev., 2016, 45, 2378–2395 RSC.
- A. Ghazlan, T. Ngo, P. Tan, Y. M. Xie, P. Tran and M. Donough, Composites, Part B, 2021, 205, 108513 CrossRef CAS.
- Q. Cheng, M. Wu, M. Li, L. Jiang and Z. Tang, Angew. Chem., Int. Ed., 2013, 125, 3838–3843 CrossRef.
- M. Wu, H. Shuai, Q. Cheng and L. Jiang, Angew. Chem., Int. Ed., 2014, 53, 3358–3361 CrossRef CAS PubMed.
- Q. Cheng, L. Jiang and Z. Tang, Acc. Chem. Res., 2014, 47, 1256–1266 CrossRef CAS PubMed.
- M. Suzuki, K. Saruwatari, T. Kogure, Y. Yamamoto, T. Nishimura, T. Kato and H. Nagasawa, Science, 2009, 325, 1388–1390 CrossRef CAS PubMed.
- J. B. Thompson, G. T. Paloczi, J. H. Kindt, M. Michenfelder, B. L. Smith, G. Stucky, D. E. Morse and P. K. Hansma, Biophys. J., 2000, 79, 3307–3312 CrossRef CAS PubMed.
- A. Razaq, F. Bibi, X. Zheng, R. Papadakis, S. H. M. Jafri and H. Li, Materials, 2022, 15, 1012 CrossRef CAS PubMed.
- W. Yu, L. Sisi, Y. Haiyan and L. Jie, RSC Adv., 2020, 10, 15328–15345 RSC.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- J. Sun and B. Bhushan, RSC Adv., 2012, 2, 7617–7632 RSC.
- H. Zhao, Z. Yang and L. Guo, NPG Asia Mater., 2018, 10, 1–22 CrossRef CAS.
- D. Madhav, B. Buffel, P. Moldenaers, F. Desplentere and V. Vandeginste, Prog. Mater. Sci., 2023, 139, 101168 CrossRef CAS.
- Z. An, O. C. Compton, K. W. Putz, L. C. Brinson and S. T. Nguyen, Adv. Mater., 2011, 23, 3842–3846 CrossRef CAS PubMed.
- W. Cui, M. Li, J. Liu, B. Wang, C. Zhang, L. JiANG and Q. Cheng, ACS Nano, 2014, 9511–9517 CrossRef CAS PubMed.
- K. Chen, S. Zhang, A. Li, X. Tang, L. Li and L. Guo, ACS Nano, 2018, 12, 4269–4279 CrossRef CAS PubMed.
- Y. Lee, K. Jun, K. Lee, Y. C. Seo, C. Jeong, M. Kim, I. K. Oh and H. Lee, Angew. Chem., Int. Ed., 2020, 59, 3864–3870 CrossRef CAS PubMed.
- C. Zuo, L. Wang, Y. Tong, L. Shi, W. Ding and W. Li, Sep. Purif. Technol., 2021, 267, 118660 CrossRef CAS.
- Y. Wang, E. J. Jeon, J. Lee, H. Hwang, S. W. Cho and H. Lee, Adv. Mater., 2020, 32, 2002118 CrossRef CAS PubMed.
- H. K. Park, D. Lee, H. Lee and S. Hong, Mater. Horiz., 2020, 7, 1387–1396 RSC.
- K. Chen, X. Tang, B. Jia, C. Chao, Y. Wei, J. Hou, L. Dong, X. Deng, T. Xiao, K. Goda and L. Guo, Nat. Mater., 2022, 21, 1121–1130 CrossRef CAS PubMed.
- Y. L. Hong, S. Ryu, H. S. Jeong and Y. K. Kim, Appl. Surf. Sci., 2019, 480, 514–522 CrossRef CAS.
- X. Zou and S. Dong, J. Phys. Chem. B, 2006, 110, 21545–21550 CrossRef CAS PubMed.
- J. Jeon, M. Choi, S. B. Kim, T. H. Seo, B. C. Ku, S. Ryu, J. H. Park and Y. K. Kim, J. Ind. Eng. Chem., 2021, 102, 233–240 CrossRef CAS.
- F. Jiang, W. Zhao, Y. Wu, Y. Wu, G. Liu, J. Dong and K. Zhou, Appl. Surf. Sci., 2019, 479, 963–973 CrossRef CAS.
- Y. B. Kwon, S. H. Go, C. Choi, T. H. Seo, B. Yang, M. W. Lee and Y. K. Kim, Diamond Relat. Mater., 2021, 119, 108565 CrossRef CAS.
- Y. B. Kwon, S. R. Lee, T. H. Seo and Y. K. Kim, Macromol. Res., 2022, 30, 279–284 CrossRef CAS.
- Y. B. Kwon, S. Y. Cho, H. Jang, J. H. Kim and Y. K. Kim, Langmuir, 2021, 37, 14205–14213 CrossRef CAS PubMed.
- J. Xi, L. Zhang, W. Zheng, Q. Zeng, Y. He, Z. He and J. Chen, J. Mater. Sci., 2021, 56, 18061–18077 CrossRef CAS.
- Y. Chen, C. Zhong, J. Wu, J. Ma, X. Yu and Y. Liu, Langmuir, 2022, 38, 14192–14199 CrossRef CAS PubMed.
- Z. Fan, K. H. L. Po, K. K. Wong, S. Chen and S. P. Lau, ACS Appl. Nano Mater., 2018, 1, 1811–1818 CrossRef CAS.
- J. Jeon and Y. K. Kim, Appl. Surf. Sci., 2023, 611, 155566 CrossRef CAS.
- X. Li, J. Agric. Food Chem., 2012, 60, 6418–6424 CrossRef CAS PubMed.
- C. Yin, X. Du, Z. Ding, Q. Zeng, X. Li, C. He, B. Xiong, J. Li and Y. Zhou, RSC Adv., 2022, 12, 6561–6572 RSC.
- X. Zhang, L. Hou, F. Richard and P. Samorì, Chem.–Eur. J., 2018, 24, 18518–18528 CrossRef CAS PubMed.
- L. Chen, Z. Li, G. Wu, Y. Wang, T. Wang, Y. Ma and B. Fei, Composites, Part A, 2018, 115, 341–347 CrossRef CAS.
- Y. K. Kim, Y. J. Kim, J. Park, S. W. Han and S. M. Kim, Carbon, 2021, 173, 376–383 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08932k |
| This journal is © The Royal Society of Chemistry 2024 |