
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel sulfonamide derivatives as multitarget antidiabetic agents: design, synthesis, and biological evaluation†
Mohammed Salah Ayoup *ab,
Nourhan Khaleda,
Hamida Abdel-Hamida,
Doaa A. Ghareebc,
Samah A. Nasrc,
Ahmed Omerde,
Amr Sonousifg,
Asmaa E. Kassab*f and
Abdelazeem S. Eltaweilah
*ab,
Nourhan Khaleda,
Hamida Abdel-Hamida,
Doaa A. Ghareebc,
Samah A. Nasrc,
Ahmed Omerde,
Amr Sonousifg,
Asmaa E. Kassab*f and
Abdelazeem S. Eltaweilah
aDepartment of Chemistry, Faculty of Science, Alexandria University, Alexandria, Egypt. E-mail: mayoup@kfu.edu.sa; mohammedsalahayoup@gmail.com
bDepartment of Chemistry, College of Science, King Faisal University, Al-Ahsa 31982, Saudi Arabia
cBio-screening and Preclinical Trial Lab, Biochemistry Department, Faculty of Science, Alexandria University, Alexandria, Egypt
dPolymer Institute of the Slovak Academy of Sciences, Dúbravská Cesta 9 845 41, Bratislava, Slovakia
ePolymer Materials Research Department, Advanced Technology and New Materials Research Institute (ATNMRI), City of Scientific Research and Technological Applications (SRTA-City), New Borg El-Arab City, Alexandria 21934, Egypt
fDepartment of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Cairo University, P.O. Box 11562 Kasr El-Aini Street, Cairo, Egypt. E-mail: asmaa.kassab@pharma.cu.edu.eg
gUniversity of Hertfordshire hosted by Global Academic Foundation, New Administrative Capital, Cairo, Egypt
hDepartment of Engineering, Faculty of Technology and Engineering, University of Technology and Applied Sciences, Sultanate of Oman
First published on 4th March 2024
Abstract
A series of new sulfonamide derivatives connected through an imine linker to five or seven membered heterocycles were designed and synthesized. All synthesized derivatives were characterized using a variety of spectroscopic methods, including IR, 1HNMR, and 13CNMR. In vitro α-glucosidase and α-amylase inhibition activities, as well as glucose uptake were assessed for each of the synthesized compounds. Four sulfonamide derivatives namely 3a, 3b, 3h and 6 showed excellent inhibitory potential against α-glucosidase with IC50 values of 19.39, 25.12, 25.57 and 22.02 μM, respectively. They were 1.05- to 1.39-fold more potent than acarbose. Sulfonamide derivatives 3g, 3i and 7 (EC50 values of 1.29, 21.38 and 19.03 μM, respectively) exhibited significant glucose uptake activity that were 1.62- to 27-fold more potent than berberine. Both α-glucosidase protein (PDB: 2QMJ) and α-amylase (PDB: 1XCW) complexed with acarbose were adopted for docking investigations for the most active synthesized compounds. The docked compounds were able to inhabit the same space as the acarviosin ring of acarbose. The docking of the most active compounds showed an analogous binding with the active site of α-glucosidase as acarbose. The superior activity of the synthesized compounds against α-glucosidase enzyme than α-amylase enzyme can be rationalized by the weak interaction with the α-amylase. The physiochemical parameters of all synthesized compounds were aligned with Lipinski's rule of five.
1. Introduction
The pandemic of type 2 diabetes mellitus (T2DM) is a multi-factorial chronic health condition and financial burden on modern society.1 It accounts for 90% of all cases of diabetes,2 being distinguished by persistent hyperglycemia brought on by the inability to utilize insulin due to a problem with the pancreatic β-cells.3,4 More than 400 million people globally had T2DM in 2018. The number of diabetic people is increasing at an alarming rate, and by 2030, this figure is expected to increase to nearly 500 million.5,6 In 2019,7 diabetes mellitus was the cause of 4.2 million deaths worldwide. T2DM can lead to serious chronic consequences include end-stage renal disease, blindness, vascular disease, poor wound healing, and limb amputation. It also causes a number of dysfunctions in many organs and tissues.8 Only a small percentage of patients with T2DM are able to maintain long-term glycemic control, despite the development of numerous therapy choices.9,10 Controlling the elevated blood glucose levels is the primary treatment method for this disease. Retarding, controlling, and/or blocking carbohydrate hydrolytic enzymes are methods for regulating blood sugar levels.11 Starch hydrolysis is facilitated by pancreatic enzymes such α-amylase and α-glucosidase, which elevates postprandial hyperglycemia. The key factor causing T2DM to develop is this rise in blood sugar levels. Therapeutically, this postprandial hyperglycemia is managed by blocking these enzymes that break down carbohydrates. The delay in glucose absorption caused by α-amylase and α-glucosidase inhibitors lowers blood sugar levels.12,13 Many α-amylase and α-glucosidase inhibitors, including acarbose, miglitol, and voglibose, are utilized clinically to treat diabetic patients.14,15 The other common groups of drugs for treating hyperglycemia are sulfonylureas (which enhance insulin release from pancreatic islets), biguanides (which decrease hepatic glucose synthesis), and peroxisome proliferator-activated receptor-γ (PPARγ) agonists (which increase insulin action).16 These medications can be taken either on their own or in combination with other hypoglycemic treatments. Severe hypoglycemia, weight gain, decreased therapeutic efficiency as a result of an incorrect or ineffective dosing regimen, low potency and altered adverse effects as a result of drug metabolism, as well as problems with target specificity, solubility, and permeability are the main disadvantages of taking these conventional medications.17 Another crucial tactic that can be used to control blood glucose levels and prevent diabetic hyperglycemia is the stimulation of glucose uptake from the bloodstream.18,19 Most drug design efforts in recent years have focused on creating agents with a wide range of physiological effects, especially for diseases with complicated etiologies like diabetes. The development of these multi-target ligands must prioritize the choice of appropriate targets, which must be thoroughly characterized and are ideally linked to several disease-related pathways, as well as the optimization of the relative potency of the drug towards each receptor.20 Drugs that target proteins in several biochemical pathways contributing to the pathogenesis of diabetes mellitus are necessary because of the pathological complexity of this disease. To improve patient compliance, research is also concentrating on identifying multi-targeted ligands.21–23 Several researches described the synthesis of sulfonamides containing molecules that act as antidiabetics by inhibiting α-glucosidase enzyme.24–30 These compounds contain some common structural features namely a p-substituted sulfonamide ring attached to a nitrogen-containing spacer which is consequently attached to phenyl, cyclic, or heterocyclic ring as in compounds A,30 B27 and C28 (Fig. 1). The goal of our work is to synthesize new sulfonamide derivatives with the same essential pharmacophoric features of the reported compounds (Fig. 1). Firstly, a phenyl sulfonamide scaffold substituted at the p-position was used in the design of targeted compounds. The 2nd strategy involved the incorporation of a linker with imine imine-containing group. Both the phenyl sulfonamide motif and the linker were selected to share as H-bond acceptors and/or donors with amino acids in the active sites of α-glucosidase and α-amylase enzymes. Within the 3rd strategy, a five-membered heterocycle (pyrazole or oxazole) or seven-membered heterocycle (diazepine or triazepine ring featuring carbonyl or thioxo group) was grafted. The 4th strategy involved the introduction of variable hydrophobic aromatic moieties on the pyrazole motif. These hydrophobic moieties' substitution design was chosen to ensure a variety of electronic and lipophilic environments, which could affect the activity of the target compounds. In this study, all the synthesized compounds were evaluated for their in vitro α-glucosidase and α-amylase inhibition activities as well as on glucose uptake by yeast cells. Docking studies were done, for the most active synthesized compounds, on both α-glucosidase protein (PDB: 2QMJ) and α-amylase (PDB: 1XCW) complexed with acarbose.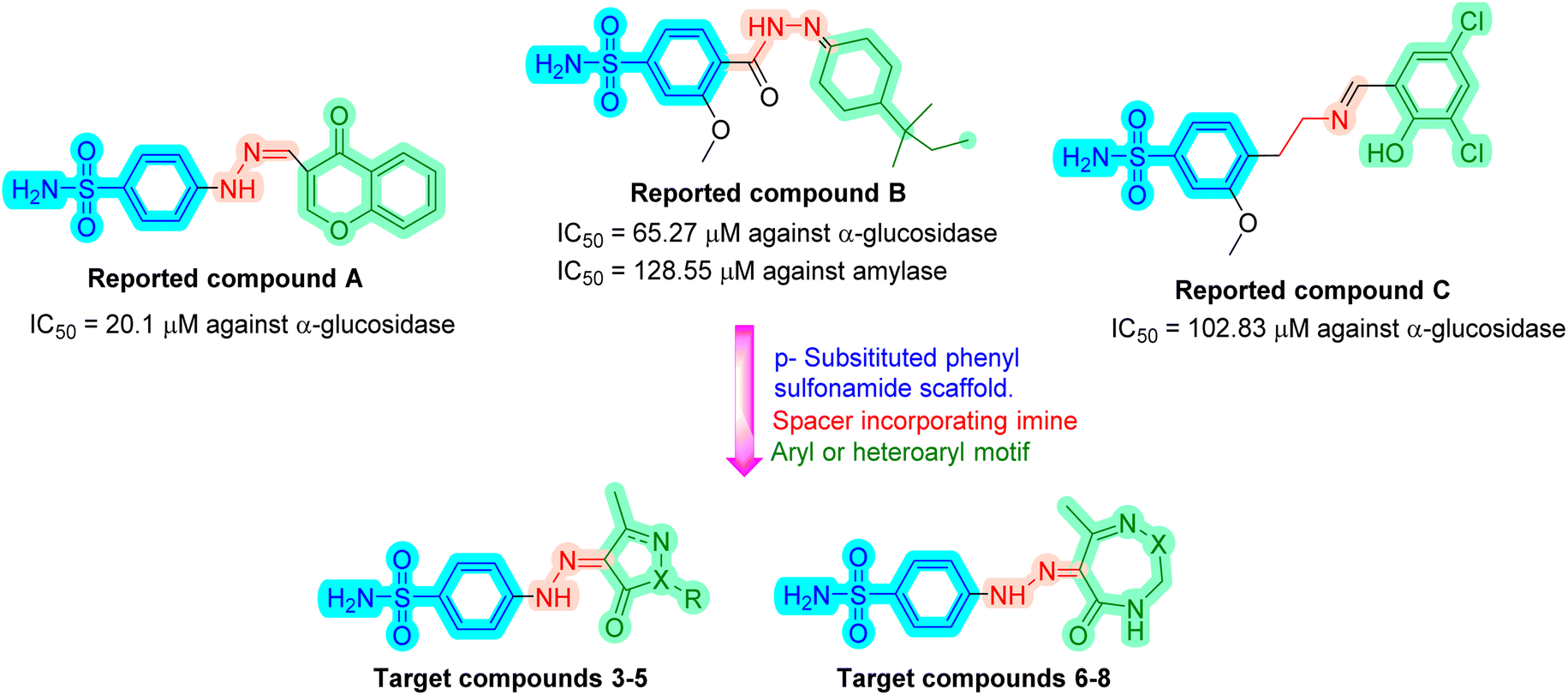 | ||
| Fig. 1 Design strategy for the synthesized sulfonamide derivatives. | ||
2. Results and discussion
2.1. Chemistry
The novel functionalization protocol of the bacteriostatic antibiotic sulfanilamide (Scheme 1) was accomplished initially by diazotization using NaNO2/HCl/AcOH protocol to form the diazonium salt 1. Rapid coupling of 1 with pre-prepared active carbanion of ethyl acetoacetate at 0 °C afforded the corresponding hydrazone ethyl-3-oxo-2-(2-(4-sulfamoylphenyl)-hydrazineylidene) butanoate (2). The structure of 2 was confirmed by IR which showed the strong band at νmax cm−1 3342, 3250, and 3164 correspond to (NH2, NH), and 1687 for conjugated (CO), moreover the 1HNMR spectrum of 2 showed two separate signals of NH and NH2 at δH: 11.54 and 7.25 ppm, respectively. Also, three separated signals in the upfield at δH: 4.28, 2.37, and 1.24 ppm revealed the presence of OCH2, COCH3, and CH3, respectively. Furthermore, the 13CNMR spectrum confirmed the presence of the acetyl and ester groups where two nonhomotopic carbonyls at δC: 194.4 and 162.8 ppm, respectively were present beside the aliphatic carbons at δC: 61.9, 25.8 and 14.3 ppm.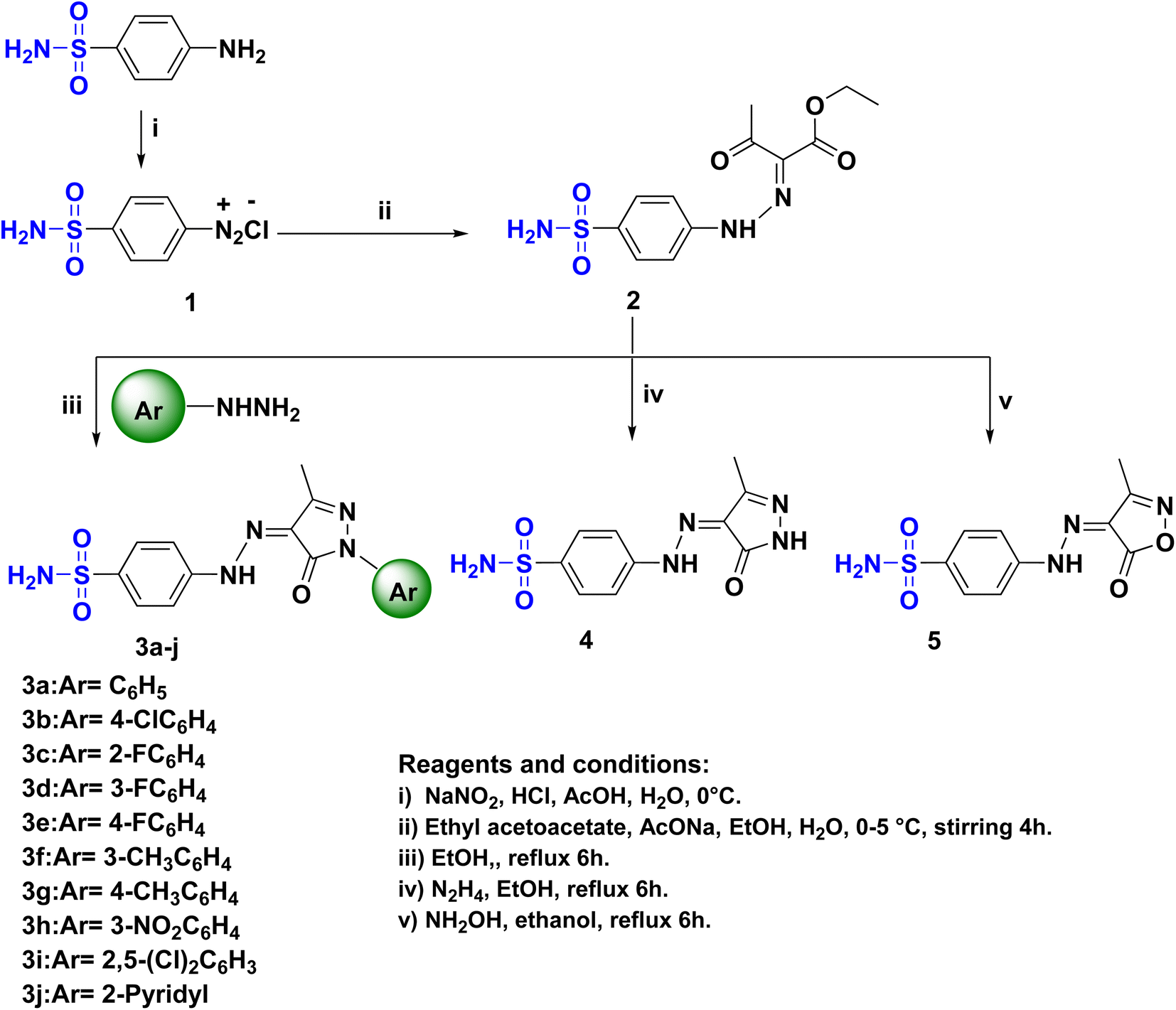 | ||
| Scheme 1 The synthetic pathway and reagents for the preparation of the target compounds 1–5. | ||
A series of sulfonamide hydrazone aryl pyrazole hybrids 3a–j were prepared utilizing the hydrazone sulfonamide ethyl acetoacetate 2 by reflux in ethanol with arylhydrazines namely phenylhydrazine, p-chlorophenylhydrazine, o-fluorophenylhydrazine, m-fluorophenylhydrazine, p-fluorophenylhydrazine, m-methylphenylhydrazine, p-methylphenylhydrazine, m-nitrophenylhydrazine, 2,5-dichlorophenylhydrazine and 2-hydrazinopyridine (Scheme 1). The structure of the pyrazole hybrids were confirmed by NMR spectra where the 1HNMR showed NH's at a range of δH: 13.19–13.03 ppm, the proton of SO2NH2 resonating at δH:7.34–7.36 ppm. The 13CNMR of the hybrids 3a–j showed carbonyl carbon at range δC: 157.0–156.7 ppm and the branched methyl group of pyrazole moiety at δC: 12.2–12.1 ppm. Moreover, the characteristic signal of the C–F bond of the aryl pyrazole 3c–e appeared at δC: 157.3, 162.6, and 159.5 ppm, respectively with 1JCF = 240–249.5 Hz.31,32 sulfanilamide hydrazone/pyrazole or isoxazole hybrids 4 and 5 (Scheme 1) were obtained by treatment of 2 with alcoholic hydrazine or hydroxyl amine, respectively. The IR spectra of 4 and 5 showed a split band revealed to NH2 at νmax 3208 and 3257 cm−1, respectively. The 1HNMR of 4 and 5 showed broad signals corresponding to NH at δH: 11.62 and 12.73 ppm, respectively beside the remarkable signals correspond to H2NSO2 at δH: 7.32 and 7.35 ppm, respectively. Moreover, the 13CNMR confirmed the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the heterocyclic rings in compounds 4 and 5 which was indicated by a signal at δC: 160.4 and 160.6 ppm, respectively.
O of the heterocyclic rings in compounds 4 and 5 which was indicated by a signal at δC: 160.4 and 160.6 ppm, respectively.
Novel sulfonamide hydrazone/seven-membered ring hybrids 6–8 (Scheme 2) were prepared by treatment of 2 with semicarbazide or thiosemicarbazide or ethylenediamine, respectively. The reaction proceeded through condensation followed by ring cyclization. The structures were confirmed by IR spectra, where a strong band in the range of 3307–3107 cm−1 revealed the presence of NH2. The CO band appeared at 1717–1680 cm−1. The 1HNMR confirmed the structure of compounds 6–8, where 6 and 8 were present in the hydrazone form. While 1HNMR of 6 showed four different signals correspond to four different nonhomototopic protons of three N–H′s at δH: 12.96, 7.55, and 7.23 ppm and SO2NH2 at δH: 7.35. Alternatively, hybrid 7 was present as in the azo form rather than hydrazono form in an equilibrium with a tautomer of the azo form 7′ as indicated from the 13CNMR spectrum that revealed the presence of Csp3-H at δC: 61.9 ppm corresponding to C6 of 1,2,4-triazepine moiety. Furthermore 8 is more flexible to exhibit tautomerism where the 13C NMR represent the Csp3 at δC: 50.6, 38.6, 15.5 ppm.
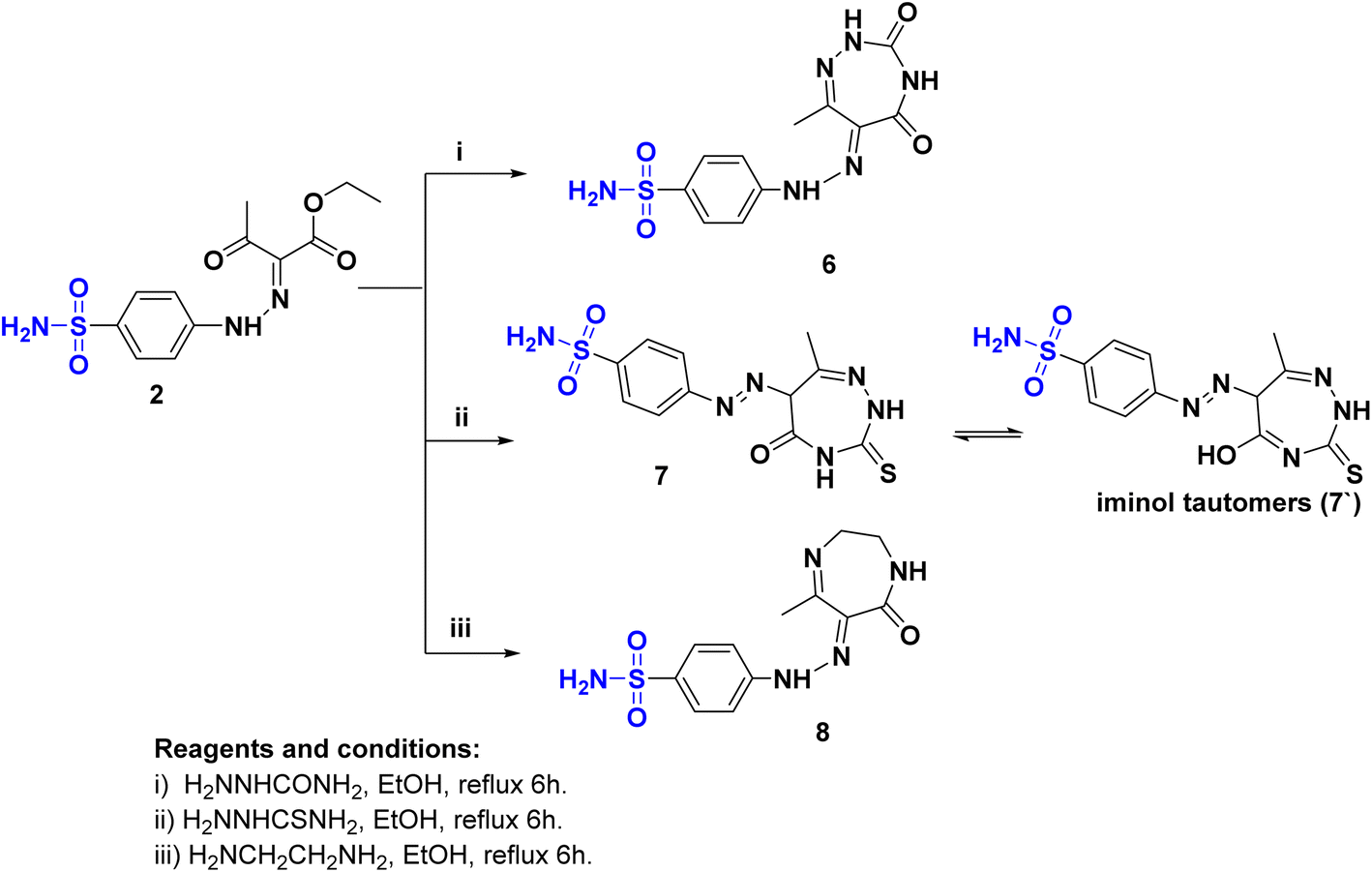 | ||
| Scheme 2 The synthetic pathway and reagents for the preparation of the target compounds 6–8. | ||
2.2. α-Glucosidase inhibitory activity
All the synthesized compounds were evaluated for their in vitro α-glucosidase inhibition activities through the determination of their half maximal inhibitory concentration (IC50) values compared to acarbose as a reference anti-diabetic drug. The results were summarized in Table 1 and represented graphically in Fig. 2. The results showed the most prominent sulfonamide derivatives namely 3a, 3b, 3h, and 6 showed excellent inhibitory potential against α-glucosidase with IC50 values of 19.39, 25.12, 25.57, and 22.02 μM, respectively. In comparison to acarbose, they were 1.05 to 1.39 times more potent. Compounds 3c–f and 3i exhibited moderate α-glucosidase inhibitory activity with IC50 values in the range of 44.82 to 67.68 μM. Sulfonamide derivatives 3g, 3j, 4, 5, and 7 demonstrated weak α-glucosidase inhibitory potential with IC50 values in the range of 76.04 to 99.11 μM. Compound 8 (IC50 = 109.81 μM) showed the least inhibitory activity. The structure–activity relationship (SAR) was investigated. Unsubstituted phenyl ring as in compound 3a showed the best α-glucosidase inhibitory activity. Substitutions on the phenyl ring generally decreased the activity but with 4-Cl (3b) and 3-NO2 (3h) showed better activity than acarbose. Shifting of phenyl ring on the pyrazole scaffold to a hydrogen atom (4) or converting the pyrazole scaffold to isoxazole (5) by replacing the nitrogen atom with an oxygen atom exhibited a delirious effect on the α-glucosidase activity. 1,2,4-Triazepine-3,5-dione (6) increased α-glucosidase activity, changing to 1,2,4-triazepine-3-thione (7) or 1,4-diazepine (8) greatly decreased the activity (Fig. 3).| Compound | α-Glucosidase IC50b (μM ± SD) | α-Amylase IC50b (μM ± SD) | Glucose uptake EC50b (μM ± SD) |
|---|---|---|---|
| a The bold text indicates the compounds that are more potent than the reference standard.b All values are expressed as a mean of three replicates. | |||
| 3a | 19.39 ± 2.7 | 416.46 ± 23.4 | 124.93 ± 9.8 |
| 3b | 25.12 ± 0.7 | 267.19 ± 22.3 | 429.96 ± 33.1 |
| 3c | 67.68 ± 7.1 | 219.29 ± 20.7 | 176.09 ± 11.8 |
| 3d | 61.67 ± 5.6 | 225.55 ± 17.5 | 283.71 ± 19.5 |
| 3e | 44.82 ± 3.4 | 237.41 ± 12.9 | 52.49 ± 4.9 |
| 3f | 55.44 ± 4.8 | 249.10 ± 22.1 | 106.26 ± 9.8 |
| 3g | 99.11 ± 6.8 | 73.93 ± 6.1 | 1.29 ± 0.1 |
| 3h | 25.57 ± 2.1 | 152.23 ± 10.2 | 331.87 ± 22.9 |
| 3i | 50.16 ± 4.8 | 137.95 ± 11.1 | 21.38 ± 1.5 |
| 3j | 82.73 ± 7.4 | 293.86 ± 23.5 | 299.87 ± 15.9 |
| 4 | 76.04 ± 6.1 | 309.19 ± 25.1 | 101.39 ± 8.7 |
| 5 | 78.69 ± 5.9 | 75.72 ± 6.4 | 417.94 ± 22.5 |
| 6 | 22.02 ± 1.9 | 207.91 ± 10.8 | 235.27 ± 11.8 |
| 7 | 85.35 ± 2.3 | 331.96 ± 20.3 | 19.03 ± 1.6 |
| 8 | 109.81 ± 6.7 | 57.36 ± 4.2 | 277.79 ± 18.9 |
| Acarbose | 27.02 ± 1.6 | 13.14 ± 1.1 | — |
| Berberine | — | — | 34.70 ± 2.7 |
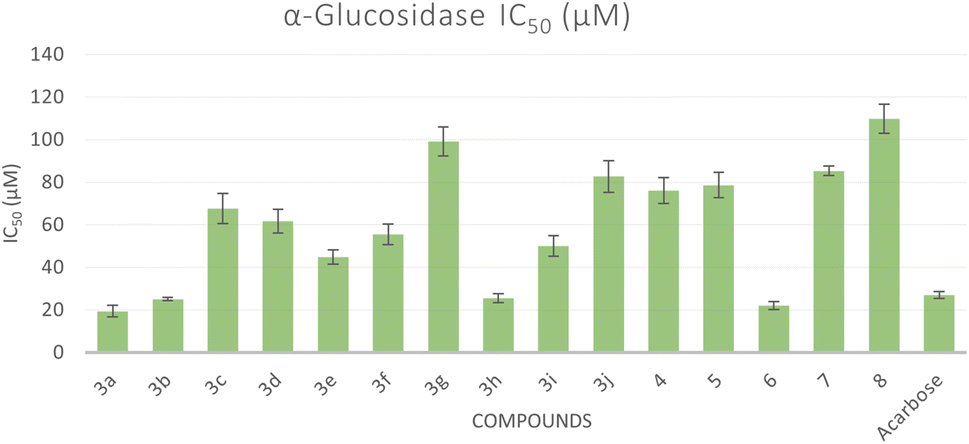 | ||
| Fig. 2 Bar graph representation showing the comparative IC50 values for the α-glucosidase activity of the target compounds and acarbose as a reference drug. | ||
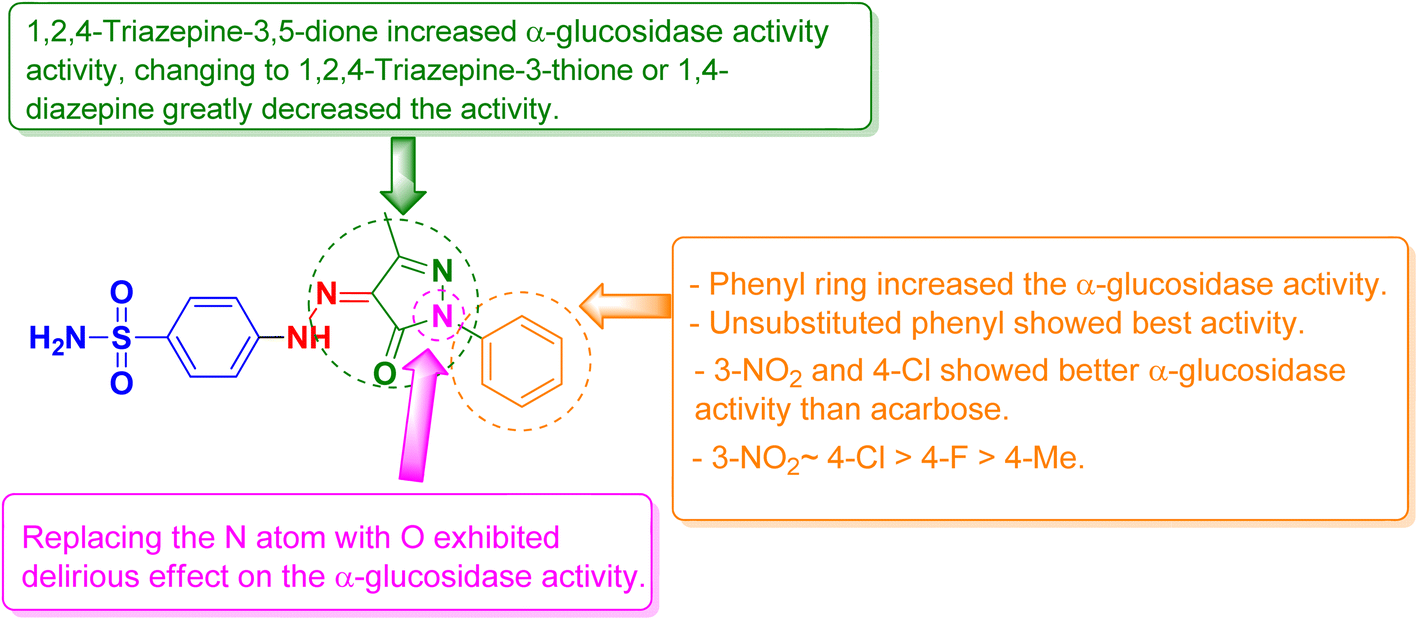 | ||
| Fig. 3 Structure–activity relationship (SAR) of the synthesized sulfonamide derivatives. | ||
2.3. α-Amylase inhibitory activity
The inhibitory activities of all synthesized compounds against α-amylase were evaluated by calculating their half maximum inhibitory concentration (IC50) values in comparison to acarbose, the standard anti-diabetic drug (Table 1). The results indicated that all the synthesized sulfonamide derivatives (IC50 values at the range of 57.36 to 416.46 μM) exhibited weak α-amylase inhibitory potential compared to acarbose.2.4. Glucose uptake assay
The glucose uptake by the yeast cells 33can be used as an in vitro screening tool to evaluate compounds for their hypoglycemic activity.34 Glucose along with different concentrations of the tested compounds were added to yeast cells for a specific amount of time and the amount of glucose remained serves as an indication of the glucose uptake by the yeast cells. Five different concentrations were used for each compound and the EC50 values for all compounds were calculated which correspond to the concentration of the compound at which 50% of glucose uptake occurs. A lower EC50 shows a higher antidiabetic activity (Fig. 4). All compounds showed an increase in glucose uptake with increased concentrations (see ESI†). Compounds 3g, 3i, and 7 showed higher glucose uptake compared to berberine with EC50 of 1.29 μM, 21.38 μM, and 19.03 μM, respectively, representing 1.62- to 27-fold more potency than reference standard berberine (EC50 = 34.70 μM). However, other compounds demonstrated less glucose uptake activity compared to berberine.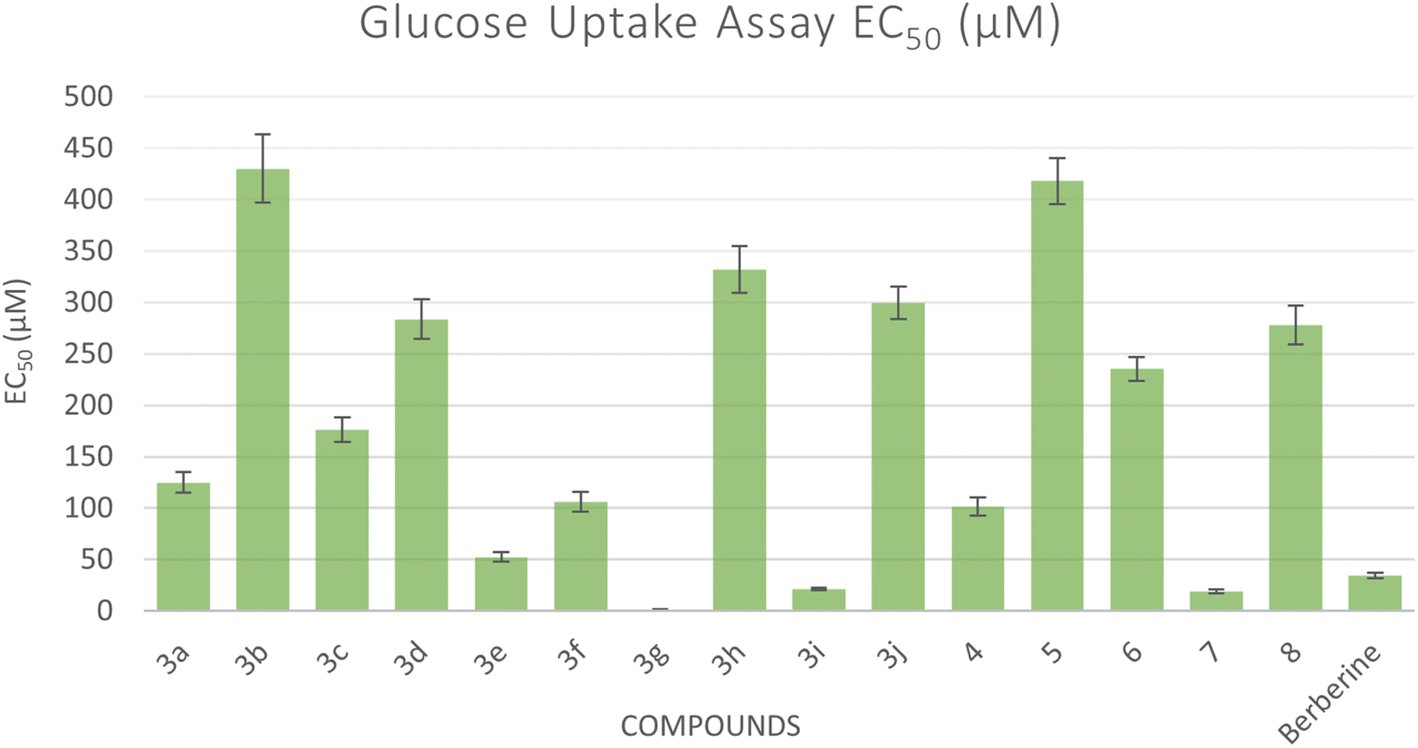 | ||
| Fig. 4 Bar graph representation showing the comparative EC50 values for the glucose uptake activity of the target compounds and acarbose as a reference drug. EC50 means the concentration in μM of the compound that can cause 50% of glucose uptake by the yeast cells. | ||
2.5. Molecular docking study
Molecular docking is used as a computational method that can give valuable information about the binding interactions between the synthesized molecules and the active site of the target enzyme. This helps in evaluating the drugs and may propose further development. Docking studies were done, for the most active synthesized compounds, on both α-glucosidase protein (PDB: 2QMJ)35 and α-amylase (PDB: 1XCW)36 complexed with acarbose using MOE (2022.09)37 software. The co-crystallized acarbose was docked first the active site to validate the docking protocol. The docking procedure was able to replicate the pose of acarbose with the relative mean standard deviation (RMSD) between the docked and the modeled acarbose within the cutoff limit (Fig. 5). The structure of acarbose consists of acarviosin moiety (2 rings) and maltose moiety (2 rings). In contrast to the maltose moiety that is solvent exposed and showed no binding with the enzyme, the acarviosin moiety is embedded deeply in the gorge of the active site and is responsible for its interaction with Arg526, Met444, His600, Asp203, Asp542 residues of the enzyme. The same docking protocol was then applied to the synthesized compounds to evaluate their interactions with the α-glucosidase binding site. The Lipinski parameters38 and the docking results of the best derivatives along with the binding interactions are shown in Table 2. The physiochemical parameters of all synthesized compounds were aligned with Lipinski's rule of five. Generally, the docked compounds were able to inhabit the same region as the acarviosin ring of acarbose. The docking of the most active compounds (Fig. 6) showed an similar interaction with the active site as acarbose. The sulfonamide group of compounds 3a was able to form hydrogen bond with His600 and Asp327. In addition, the phenyl ring interacts with Tyr299 and Trp406 via Arene–H interactions which help in the stabilization of the compound conformation in its active site. Compound 3h with the m-nitro substitution showed higher interactions with the nitro group accepting 3 hydrogen bonds from Trp441, Trp539 and Arg526. Further, the pyrazolinone carbonyl oxygen bonded to Arg526 and the phenyl ring interacts with Tyr299 and Phe575 via Arene–H interactions (Fig. 7). In contrast, the docking simulation of the least active compounds 8 and 5 showed less binding energy to the active site (−5.974 kcal mol−1 and −5.516 kcal mol−1, respectively) albeit the same binding pattern (docking poses of compounds are included in ESI†). The synthesized compounds demonstrated superior activity to α-glucosidase enzyme than α-amylase enzyme. For example, compound 3a was 21 times more active towards α-glucosidase enzyme (IC50 = 19.39 μM) than α-amylase enzyme (IC50 = 416.46 μM). This difference in activity can be explained by the weak interaction with the α-amylase. The docking of compound 3a in the active site of α-amylase (PDB: 1XCW) showed that compound 3a has less binding interaction with the α-amylase with only one hydrogen bond with Glu233 (Fig. 8).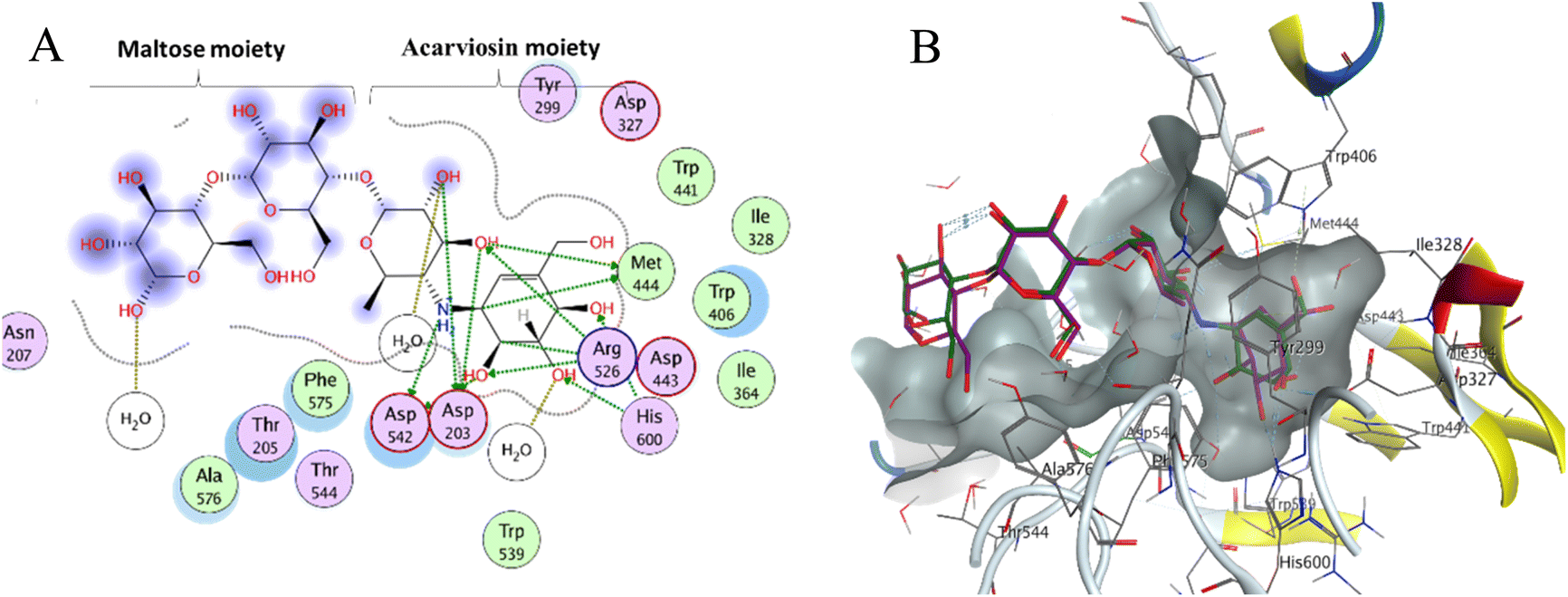 | ||
| Fig. 5 (A) Two-dimensional diagram of acarbose binding with alpha-glucosidase binding pocket; (B) three-dimensional diagram of both the co-crystallized acarbose inhibitor (green) and its re-docked pose (magenta) in the active site of alpha-glucosidase showing RMSD (0.76 Å) within the acceptable range. | ||
| Compound | Docking score (PDB: 2QMJ) (kcal mol−1) | HBA (≤10) | HBD (≤5) | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (−4.0–5) P (−4.0–5) |
PSA (0–150) Å2 | Interactions |
|---|---|---|---|---|---|---|
| 3a | −6.054 | 5 | 2 | 3.495 | 117.22 | HB (Asp327) |
| HB (His600) | ||||||
| Arene–H (Tyr299) | ||||||
| Arene–H (Trp406) | ||||||
| 3b | −6.295 | 5 | 2 | 4.087 | 117.22 | HB (Asp327) |
| HB (His600) | ||||||
| Arene–H (Tyr299) | ||||||
| Arene–H (Trp406) | ||||||
| 3c | −6.394 | 5 | 2 | 2.364 | 117.22 | HB (Asp327) |
| HB (His600) | ||||||
| HB (Asp443) | ||||||
| 3d | −6.598 | 5 | 2 | 2.403 | 117.22 | HB (Asp327) |
| HB (His600) | ||||||
| 3e | −6.385 | 5 | 2 | 3.648 | 117.22 | HB (Asp327) |
| HB (His600) | ||||||
| HB (Asp443) | ||||||
| 3f | −6.501 | 5 | 2 | 3.830 | 117.22 | HB (Asp327) |
| HB (His600) | ||||||
| HB (Asp443) | ||||||
| 3g | −6.212 | 5 | 2 | 3.793 | 117.22 | HB (Asp327) |
| HB (His600) | ||||||
| HB (Asp443) | ||||||
| 3h | −6.756 | 5 | 2 | 3.467 | 163.04 | HB (Trp441) |
| HB (Trp539) | ||||||
| HB (Arg526) | ||||||
| Arene–H (Tyr299) | ||||||
| Arene–H (Phe575) | ||||||
| 3i | −6.740 | 5 | 2 | 4.714 | 117.22 | HB (Asp327) |
| HB (His600) | ||||||
| HB (Asp443) | ||||||
| 3j | −6.193 | 6 | 2 | 2.668 | 130.11 | HB (Asp327) |
| HB (His600) | ||||||
| HB (Asp443) | ||||||
| 4 | −5.644 | 5 | 3 | 1.644 | 126.01 | HB (Asp203) |
| HB (His600) | ||||||
| HB (Met444) | ||||||
| HB (Phe450) | ||||||
| HB (Trp539) | ||||||
| Arene–H (Tyr299) | ||||||
| Arene–H (Trp406) | ||||||
| 5 | −5.516 | 5 | 2 | 2.112 | 123.21 | HB (Met444) |
| HB (Arg526) | ||||||
| HB (Trp539) | ||||||
| Arene–H (His600) | ||||||
| 6 | −5.695 | 6 | 4 | 1.265 | 155.11 | HB (Asp327) |
| HB (His600) | ||||||
| HB (Asp443) | ||||||
| Arene–H (Tyr299) | ||||||
| 7 | −5.694 | 6 | 4 | 3.216 | 170.13 | HB (Asp327) |
| HB (His600) | ||||||
| HB (Asp443) | ||||||
| Arene–H (Tyr299) | ||||||
| Arene–H (Trp406) | ||||||
| 8 | −5.974 | 5 | 3 | 0.750 | 126.01 | HB (Asp327) |
| HB (His600) | ||||||
| HB (Asp443) | ||||||
| Arene–H (Tyr299) | ||||||
| Acarbose | −7.8552 | 18 | 13 | −7.508 | 325.75 | HB (Arg526) |
| HB (Met444) | ||||||
| HB (His600) | ||||||
| HB (Asp203) | ||||||
| HB (Asp542) |
 | ||
| Fig. 6 (A) Two-dimensional diagram and (B) three-dimensional representation of molecular docking of compound 3a (magenta) in the binding pocket (PDB: 2QMJ). | ||
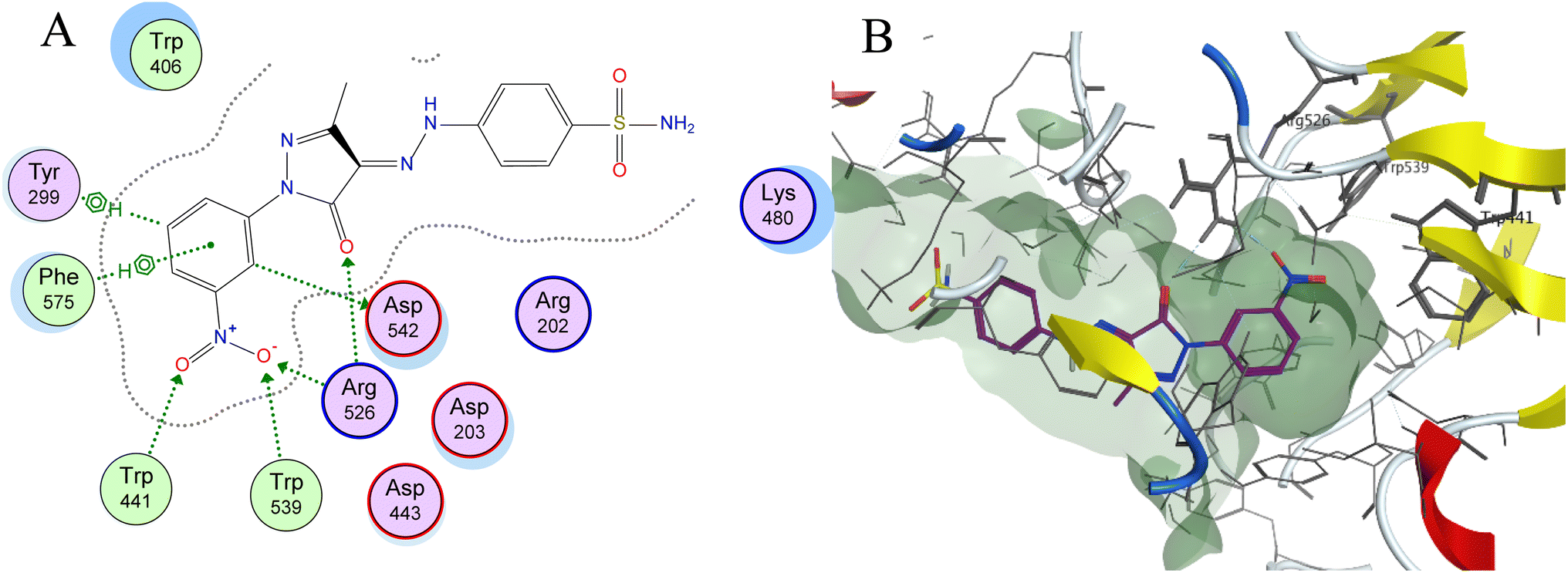 | ||
| Fig. 7 (A) Two-dimensional diagram and (B) three-dimensional representation of molecular docking of compound 3h (magenta) in the binding pocket (PDB: 2QMJ). | ||
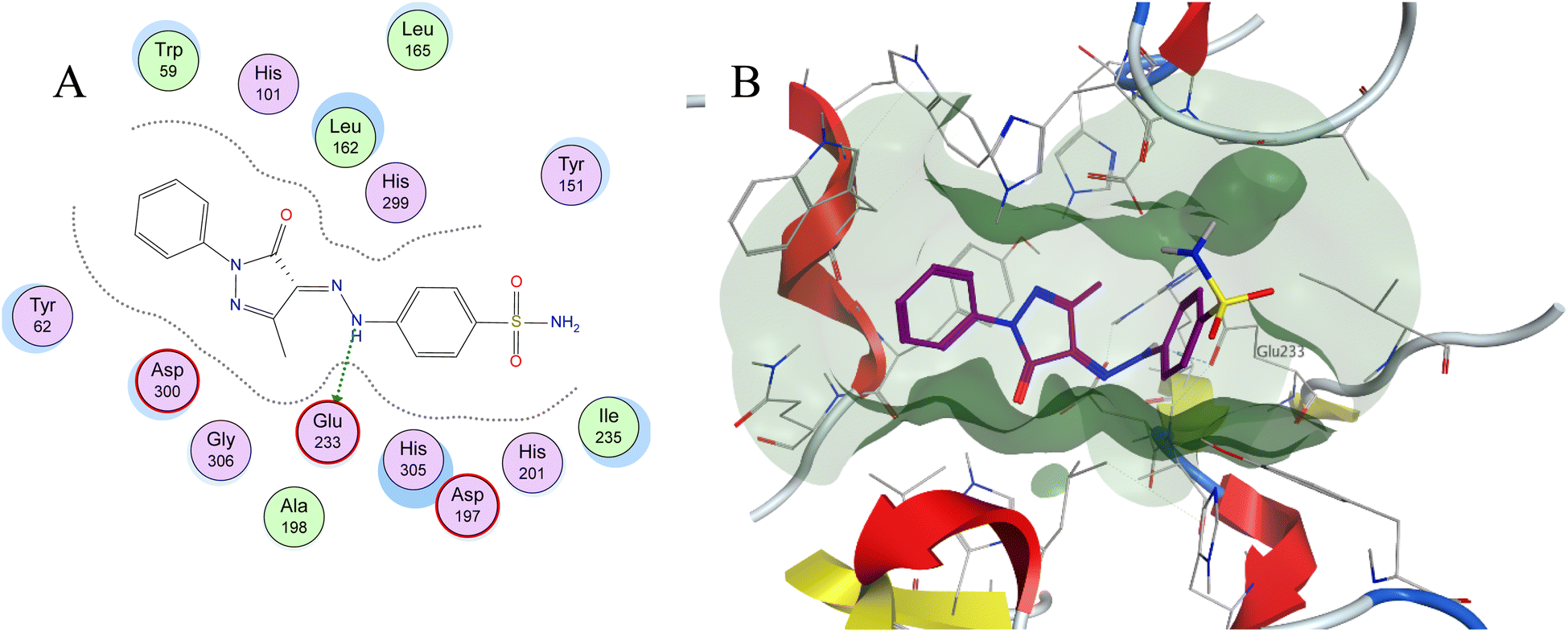 | ||
| Fig. 8 (A) Two-dimensional diagram and (B) three-dimensional representation of molecular docking of compound 3a (magenta) in the binding pocket (PDB: 1XCW). | ||
3. Conclusion
In conclusion, a series of novel sulfonamides were synthesized and evaluated for their α-glucosidase, and α-amylase enzyme activity as well as their glucose uptake activity. Out of these fifteen novel compounds, four compounds showed better α-glucosidase inhibitory compared to the acarbose reference such as 3a (19.39 μM for α-glucosidase), 3b (25.12 μM for α-glucosidase), 3h (25.57 μM for α-glucosidase) and 6 (22.02 μM for α-glucosidase). All compounds showed less α-amylase inhibition activity compared to acarbose. This low amylase activity can be rationalized computationally by the weak interactions of the compounds with the gorge of the α-amylase active site. Compounds 3g, 3i, and 7 showed better glucose uptake activity with EC50 compared to berberine (EC50 = 34.70 μM) values of 1.29, 21.38 and 19.03 μM, respectively.4. Experimental
4.1. Chemistry
:n-hexane, 2:1); IR: νmax cm−1 3342, 3250 (NH2), 3164 (NH), 2994 (Csp3-H), 1687 (CO); 1H NMR (500 MHz, DMSO-d6) δH:11.54 (s, 1H, N–H), 7.76 (d, J = 9.0 Hz, 2H, Ar–H), 7.50 (d, J = 9.0 Hz, 2H, Ar–H), 7.25 (s, 2H, NH2SO2), 4.28 (q, J = 7.5 Hz, 2H, OCH2), 2.37 (s, 3H, CH3), 1.24 (t, J = 7.0 Hz, 3H, CH2CH3), 13C NMR (125 MHz, DMSO-d6) δC: 194.4, 162.8, 145.7, 138.8, 133.9, 127.8, 115.3 (Ar–C), 61.9, 25.8, 14.3 (aliphatic-C). Anal. calcd for C12H15N3O5S (313.07): C, 46.00; H, 4.83; N, 13.41. Found: C, 46.21; H, 5.04; N, 13.12.4.1.2.1. 4-(2-(3-Methyl-5-oxo-1-phenyl-1,5-dihydro-4H-pyrazol-4-ylidene) hydrazinyl)benzenesulfonamide (3a). As reddish orange crystal; 72% yield; m.p. 260–263 °C; Rf = 0.62 (EtOAc
:n-hexane, 2:1); IR: νmax cm−1 3350, 3327 (NH2), 3249 (NH), 2924 (Csp3-H), 1668 (CO). 1H NMR (500 MHz, DMSO-d6) δH: 13.19 (s, 1H, N–H), 7.87 (d, J = 8.0 Hz, 2H, Ar–H), 7.83 (d, J = 8.5 Hz, 2H, Ar–H), 7.72 (d, J = 8.0 Hz, 2H, Ar–H), 7.42 (t, J = 7.5 Hz, 2H, Ar–H), 7.35 (s, 2H, H2NSO2), 7.18 (t, J = 8.0 Hz, 1H, Ar–H), 2.27 (s, 3H, CH3). 13C NMR (125 MHz, DMSO-d6) δC: 156.7, 149.2, 144.5, 140.8, 138.3, 129.9, 129.6, 127.8, 125.4, 118.2, 116.6 (Ar–C), 12.1 (aliphatic-C). Anal. calcd for C16H15N5O3S (357.09): C, 53.77; H, 4.23; N, 19.60. Found: C, 53.52; H, 4.34; N, 19.81.
4.1.2.2. 4-(2-(1-(4-Chlorophenyl)-3-methyl-5-oxo-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazinyl)benzenesulfonamide (3b). Reddish orange crystal; 64% yield; m.p. = 247–249 °C; Rf = 0.51 (EtOAc
:n-hexane, 1:1); IR: νmax cm−1 3339, 3250 (NH2), 3104 (NH), 2992 (Csp3-H), 1654 (CO); 1H NMR (500 MHz, DMSO-d6) δH: 13.13 (s, 1H, N–H), 7.87 (d, J = 8.5 Hz, 2H, Ar–H), 7.82 (d, J = 8.5 Hz, 2H, Ar–H), 7.71 (d, J = 8.5 Hz, 2H, Ar–H), 7.46 (d, J = 8.5 Hz, 2H, Ar–H), 7.35 (s, 2H, NH2SO2), 2.24 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δC: 156.6, 149.6, 144.4, 140.9, 137.1, 129.6, 129.5, 129.1, 127.8, 119.5, 116.7 (Ar–C), 12.2 (aliphatic-C). Anal. calcd for C16H14ClN5O3S (391.05): C, 49.05; H, 3.60; N, 17.87. Found: C, 49.31; H, 3.76; N, 17.96.
4.1.2.3. 4-((2-(2-Fluorophenyl)-5-methyl-3-oxo-2,3-dihydro-1H-pyrazol-4-ylidene)hydrazinyl)benzenesulfonamide (3c). As an orange powder; in 56% yield; m.p. 224–226 °C; Rf = 0.54 (EtOAc
:n-hexane, 1:1); IR: νmax cm−1 3358, 3264 (NH2), 3175 (NH), 2933 (Csp3-H), 1678 (CO); 1H NMR (500 MHz, DMSO-d6) δH: 13.08 (s, 1H, N–H), 7.82 (d, J = 9.5 Hz, 2H, Ar–H), 7.72 (d, J = 8.5 Hz, 2H, Ar–H), 7.50 (t, J = 7.5 Hz, 1H, Ar–H), 7.45–7.38 (m, 2H, Ar–H), 7.34 (s, 2H, H2NSO2), 7.29 (t, J = 7.5 Hz, 1H, Ar–H), 2.25 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δC: 157.3 (d, 1JCF = 249.5 Hz, C–F), 156.7, 149.4, 144.5, 140.8, 130.3 (d, 3JCF = 7.2 Hz, Ar–C), 128.9, 127.8 (d, 2JCF = 12 Hz), 125.4 (d, 4JCF = 2.5 Hz), 124.8 (d, 2JCF = 12 Hz, Ar–C), 117.2, 117.1, 116.7 (Ar–C) 12.2 (aliphatic-C). Anal. calcd for C16H14FN5O3S (375.08): C, 51.20; H, 3.76; N, 18.66. Found: C, 51.43; H, 3.96; N, 18.85.
4.1.2.4. 4-((2-(3-Fluorophenyl)-5-methyl-3-oxo-2,3-dihydro-1H-pyrazol-4-yl)-diazenyl)benzenesulfonamide (3d). As yellowish orange powder; in 64% yield; m.p. 265–267 °C, Rf = 0.45 (EtOAc
:n-hexane, 1:1); IR: νmax cm−1 3378, 3315 (NH2), 3238 (NH), 1672 (CO); 1H NMR (500 MHz, DMSO-d6) δH: 13.12 (s, 1H, N–H), 7.82 (d, J = 8.5 Hz, 2H, Ar–H), 7.73–7.67 (m, 4H, Ar–H), 7.47–7.46 (m, 1H, Ar–H), 7.35 (s, 2H, H2NSO2), 7.0 (t, J = 7.0 Hz, 1H, Ar–H), 2.25 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δC: 162.6 (d, 1JCF = 240 Hz, C–F), 156.8, 149.8, 144.4, 140.9, 139.7 (d, 3JCF = 10.7, CF), 131.4 (d, 4JCF = 8.3, CF), 127.8, 116.7, 113.6, 111.8 (d, 2JCF = 21.5 Hz, CF), 104.8 (d, 2JCF = 27.5 Hz, CF) (Ar–C), 12.2 (aliphatic-C). Anal. calcd for C16H14FN5O3S (375.08): C, 51.20; H, 3.76; N, 18.66. Found: C, 51.38; H, 3.64; N, 18.76.
4.1.2.5. 4-(2-(1-(4-Fluorophenyl)-3-methyl-5-oxo-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazinyl)benzenesulfonamide (3e). As orange powder; in 59% yield; m.p. 277–279 °C; Rf = 0.66 (EtOAc
:n-hexane, 1:1); IR: νmax cm−1 3359, 3324 (NH2), 3247 (NH), 2925 (Csp3-H), 1666 (CO). 1H NMR (500 MHz, DMSO-d6) δH: 13.15 (s, 1H, N–H), 7.88–7.81 (m, 4H, Ar–H), 7.72 (d, J = 8.0 Hz, 2H, Ar–H), 7.35 (s, 2H, H2NSO2), 7.27 (t, J = 8.5 Hz, 2H, Ar–H), 2.25 (s, 3H, CH3). 13C NMR (125 MHz, DMSO-d6) δC: 159.5 (d, 1JCF = 240 Hz, C-F), 156.7, 149.3, 144.4, 140.8, 134.7, 129.7, 127.8, 120.2 (d, 3JCF = 8.3 Hz, Ar–C), 116.7, 116.3 (d, 2JCF = 22.7 Hz, Ar–C), 117.2, 117.1, 116.7 (Ar–C) 12.2 (aliphatic-C). Anal. calcd for C16H14FN5O3S (375.08): C, 51.20; H, 3.76; N, 18.66. Found: C, 51.31; H, 3.85; N, 18.42.
4.1.2.6. 4-(2-(3-Methyl-5-oxo-1-(m-tolyl)-1,5-dihydro-4H-pyrazol-4-ylidene)-hydrazinyl)benzenesulfonamide (3f). As pale orange solid; in 69% yield; m.p. 242–245 °C; Rf = 0.68 (EtOAc
:n-hexane, 1:1); IR: νmax cm−1 3319, 3227 (NH2), 3122 (NH), 2925 (Csp3-H), 1645 (CO); 1H NMR (500 MHz, DMSO-d6) δH: 13.18 (s, 1H, N–H), 7.82 (d, J = 8.5 Hz, 2H, Ar–H), 7.70–7.66 (m, 4H, Ar–H), 7.35 (s, 2H, H2NSO2), 7.27 (t, J = 7.5 Hz, 1H, Ar–H), 6.98 (d, J = 8.0 Hz, 1H, Ar–H), 2.29 (s, 3H, CH3), 2.23 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δC: 156.7, 149.1, 144.4, 140.7, 138.9, 138.2, 129.9, 129.3, 127.8, 126.0, 118.6, 116.5, 115.3 (Ar–C), 21.7, 12.1 (aliphatic-C). Anal. calcd for C17H17N5O3S (371.11): C, 54.98; H, 4.61; N, 18.86. Found: C, 55.12; H, 4.81; N, 18.91.
4.1.2.7. 4-(2-(3-Methyl-5-oxo-1-(p-tolyl)-1,5-dihydro-4H-pyrazol-4-ylidene)-hydrazinyl)benzenesulfonamide (3g). As orange powder; in 70% yield; m.p. 272–275 °C; Rf = 0.65 (EtOAc
:n-hexane, 1:1); IR: νmax cm−1 3331, 3246 (NH2), 3088 (NH), 2921 (Csp3-H), 1653 (CO)·1H NMR (500 MHz, DMSO-d6) δH: 13.18 (s, 1H, N–H), 7.82 (d, J = 8.5 Hz, 2H, Ar–H), 7.74–7.71 (m, 4H, Ar–H), 7.35 (s, 2H, H2NSO2), 7.21 (d, J = 8.0 Hz, 2H, Ar–H), 2.26 (s, 3H, CH3), 2.24 (s, 3H, CH3);13C NMR (125 MHz, DMSO-d6) δC: 156.5, 149.0, 144.4, 140.7, 135.9, 134.6, 129.9, 127.8, 118.2, 116.5 (Ar–C), 21.0, 12.1 (aliphatic-C). Anal. calcd for C17H17N5O3S (371.11): C, 54.98; H, 4.61; N, 18.86. Found: C, 55.08; H, 4.77; N, 18.72.
4.1.2.8. 4-(2-(3-Methyl-1-(3-nitrophenyl)-5-oxo-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazinyl)benzenesulfonamide (3h). As orange crystals; in 74% yield; m.p. 278–280 °C; Rf = 0.55 (EtOAc
:n-hexane, 1:1); IR: νmax cm−1 3381 (NH), 3192, 3114 (NH2), 1673 (CO); 1H NMR (500 MHz, DMSO-d6) δH: 13.09 (s, 1H, N–H), 8.74 (s, 1H, Ar–H), 8.22 (d, J = 6.0 Hz, 1H, Ar–H), 7.98 (d, J = 6.5 Hz, 2H, Ar–H), 7.98–7.68 (m, 6H, Ar–H), 7.36 (s, 2H, H2NSO2), 2.26 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δC: 156.8, 150.3, 148.5, 144.5, 141.0, 139.0, 131.2, 129.2, 127.8, 123.4, 119.5, 116.9, 111.7 (Ar–C), 12.2 (aliphatic-C). Anal. calcd for C16H14N6O5S (402.07): C, 47.76; H, 3.51; N, 20.89. Found: C, 47.52; H, 3.74; N, 20.72.
4.1.2.9. 4-(2-(1-(2,5-Dichlorophenyl)-3-methyl-5-oxo-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazinyl)benzenesulfonamide (3i). As a yellowish Orange powder; in 76% yield; m.p. 227–229 °C; Rf = 0.63 (EtOAc
:n-hexane, 1:1); IR: νmax cm−1 3239 (NH), 3134, 3088 (NH2), 1674 (CO). 1H NMR (500 MHz, DMSO-d6) δH: 13.03 (s, 1H, N–H), 7.82 (d, J = 8.5 Hz, 2H, Ar–H), 7.74 (d, J = 8.5 Hz, 2H, Ar–H), 7.66 (m, 2H, Ar–H), 7.54 (dd, J = 8.5, 2.0 Hz, 1H, Ar–H), 7.34 (s, 2H, H2NSO2), 2.25 (s, 3H, CH3)·13C NMR (125 MHz, DMSO-d6) δC: 156.8, 149.4, 140.8, 136.0, 132.4, 132.1, 130.5, 130.0, 129.5, 128.6, 127.8, 116.8 (Ar–C), 12.2 (aliphatic-C). Anal. calcd for C16H13Cl2N5O3S (425.01): C, 45.08; H, 3.07; N, 16.43. Found: C, 45.22; H, 3.14; N, 16.51.
4.1.2.10. 4-(2-(3-Methyl-5-oxo-1-(pyridin-2-yl)-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazinyl)benzenesulfonamide (3j). As an orange powder, in 70% yield; m.p. 274–276 °C, Rf = 0.69 (EtOAc
:n-hexane, 2:1); IR: νmax cm−1 3294, 3227 (NH2), 3109 (NH), 1723 (CO). 1H NMR (500 MHz, DMSO-d6) δH: 13.14 (s, 1H, N–H), 8.44 (s, 1H, Ar–H), 7.90–7.71 (m, 6H, Ar–H), 7.35 (s, 2H, H2NSO2), 7.24 (t, J = 6.0 Hz, 1H, Ar–H), 2.27 (s, 3H, CH3). 13C NMR (125 MHz, DMSO-d6) δC: 157.0, 149.9, 149.4, 148.9, 144.6, 140.7, 138.9, 129.5, 127.8, 121.7, 116.7, 114.4 (Ar–C), 12.2 (aliphatic-C). Anal. calcd for C15H14N6O3S (358.08): C, 50.27; H, 3.94; N, 23.45. Found: C, 50.03; H, 4.17; N, 23.22.
4.1.3.1. 4-(2-(3-Methyl-5-oxo-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazinyl)-benzenesulfonamide (4). As reddish orange powder; in 58% yield; m.p. 227–229 °C; Rf = 0.58 (EtOAc
:n-hexane, 2:1); IR: νmax cm−1 3208 (split band, NH2), 1677 (CO). 1H NMR (500 MHz, DMSO-d6) δH: 13.12 (bs, 1H, N–H), 11.62 (s, 1H, N–H), 7.79 (d, J = 9.0 Hz, 2H, Ar–H), 7.63 (d, J = 9.0 Hz, 2H, Ar–H), 7.32 (s, 2H, H2NSO2), 2.12 (s, 3H, CH3)·13C NMR (125 MHz, DMSO-d6) δC: 160.4, 147.5, 144.6, 140.2, 130.2, 127.8, 116.0 (Ar–C), 12.1 (CH3). Anal. calcd for C10H11N5O3S (281.06): C, 42.70; H, 3.94; N, 24.90. Found: C, 42.54; H, 4.12; N, 24.81.
4.1.3.2. 4-(2-(3-Methyl-5-oxoisoxazol-4(5H)-ylidene)hydrazinyl)benzene-sulfonamide (5). As pale yellow powder, in 54% yield; m.p. 227–229 °C; Rf = 0.52 (EtOAc
:n-hexane, 2:1); IR: νmax cm−1 3437 (split band, NH2), 3257 (NH), 1720 (CO); 1H NMR (500 MHz, DMSO-d6) δH: 12.73 (s, 1H, N–H), 7.81 (d, J = 8.5 Hz, 2H, Ar–H), 7.76 (d, J = 9.0 Hz, 2H, Ar–H), 7.35 (s, 2H, H2NSO2), 2.22 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δC: 162.4, 160.6, 144.5, 141.2, 127.7, 122.7, 117.3 (Ar–C), 10.6 (aliphatic-C). Anal. calcd for C10H10N4O4S (282.04): C, 42.55; H, 3.57; N, 19.85. Found: C, 42.71; H, 3.62; N, 19.72.
4.1.4.1. 4-(2-(5-Methyl-3,7-dioxo-1,2,3,7-tetrahydro-6H-1,2,4-triazepin-6-ylidene)hydrazinyl)benzenesulfonamide (6). As pale yellow powder, in 51% yield; m.p. 244–247 °C, Rf = 0.56 (EtOAc
:n-hexane, 2:1); IR: νmax cm−1 3419 (NH), 3334, 3307 (NH2), 3193 (NH), 2930 (Csp3-H), 1717 (CO); 1H NMR (500 MHz, DMSO-d6) δH: 12.96 (s, 1H, N–H), 7.82 (d, J = 7.0 Hz, 2H, Ar–H), 7.73 (d, J = 7.0 Hz, 2H, Ar–H), 7.55 (s, 1H, N–H), 7.35 (s, 2H, H2NSO2), 7.23 (s, 1H, N–H), 2.20 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δC: 157.8, 149.8, 149.5, 144.5, 141.0, 128.7, 127.8, 116.9, (Ar–C), 12.2 (aliphatic-C). Anal. calcd for C11H12N6O4S (324.06): C, 40.74; H, 3.73; N, 25.91. Found: C, 40.85; H, 3.81; N, 25.71.
4.1.4.2. (E)-4-((7-methyl-5-oxo-3-thioxo-3,4,5,6-tetrahydro-2H-1,2,4-triazepin-6-yl)diazenyl)benzenesulfonamide (7). As yellowish green powder, in 58% yield; m.p. 233–235 °C, Rf = 0.71 (EtOAc
:n-hexane, 2:1); IR: νmax cm−1 3431 (NH), 3244, 3207 (NH2), 1682 (CO); 1H NMR (500 MHz, DMSO-d6) δH: 9.44 (s, 0.44H, N–H), 8.89 (s, 0.36H, N–H), 8.61 (m, 6H (Ar–H), (NH2SO2)), CH3 appeared at 2.37 and 2.23; 13C NMR (125 MHz, DMSO-d6) δC: 181.6, 141.1, 127.8, 127.7, 117.2, 115.3 (Ar–C) 61.9, 14.1, 12.2 (aliphatic-C). Anal. calcd for C11H12N6O3S2. (340.04) C, 38.82; H, 3.55; N, 24.69. Found: C, 38.95; H, 3.62; N, 24.87.
4.1.4.3. 4-(2-(5-Methyl-7-oxo-1,2,3,7-tetrahydro-6H-1,4-diazepin-6-ylidene)hydrazinyl)benzenesulfonamide (8). As a pale-yellow powder, in 60% yield; m.p. 283–285 °C; Rf = 0.67 (EtOAc
:n-hexane, 2:1); IR: νmax cm−1 broad peak 3312–3103 (OH, NH, and NH2), 2945 (Csp3-H), 1638 (CO); 1H NMR (500 MHz, DMSO-d6) δH: 13.90 (s, 1H, N–H), 11.30 (s, 1H, N–H), 7.74–7.34 (m, 6H, Ar–H, H2NSO2), 3.72–3.54 (m, 4H, CH2CH2), 2.20 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δC: 168.8, 164.3, 145.9, 138.0, 130.1, 127.9, 114.4 (Ar–C), 50.6, 38.6, 15.5 (aliphatic-C). Anal. calcd for C12H15N5O3S (309.09): C, 46.59; H, 4.89; N, 22.64. Found: C, 46.45; H, 4.81; N, 22.44.
4.2. Molecular modeling studies
The X-ray structure of the α-glucosidase enzyme (PDB: 2QMJ) and α-amylase (PDB: 1XCW) were downloaded from the protein databank (PDB) website (https://www.rcsb.org/), at a resolution of 1.90 Å and 2.00 Å respectively. All the molecular modeling and docking studies were carried out using MOE 2020.09 (Chemical Computing Group, Canada) as the computational software. First, all the hydrogen atoms were added using the Protonate 3D algorithm where the protonation states of the amino acid residues were assigned, and the partial charges of atoms were added. In addition, the compounds were drawn using the builder tool and energy was minimized using the MMFF94× force field. MOE induced-fit Dock tool used to dock the synthesized compounds into the active site. The selection of the final docked ligand–enzyme poses was according to the criteria of binding energy score and combined with ligand–receptor interactions.4.3. Biological evaluation
The biological assays were carried out according to the previously reported procedures and have been provided in the ESI;† alpha-glucosidase inhibitory assay,39 alpha-amylase inhibitory assay,40 and glucose uptake assay.33Conflicts of interest
The authors have no conflict of interest concerning this work.Acknowledgements
Nourhan Khaled (M. Sc. Student, faculty of science, Alexandria University), a Scholarship student of Scientists for next generation (SNG) scholarship program (Call 8), N. K. would like to thank the Academy of Scientific Research and Technology, Cairo, Egypt for their financial contribution to this research.References
- M. H. Tschöp, B. Finan and C. Clemmensen, et al., Unimolecular Polypharmacy for Treatment of Diabetes and Obesity, Cell Metab., 2016, 24(1), 51–62, DOI:10.1016/j.cmet.2016.06.021.
- M. Laakso and J. Kuusisto, Insulin resistance and hyperglycaemia in cardiovascular disease development, Nat. Rev. Endocrinol., 2014, 10(5), 293–302, DOI:10.1038/nrendo.2014.29.
- H. Kolb and D. L. Eizirik, Resistance to type 2 diabetes mellitus: a matter of hormesis?, Nat. Rev. Endocrinol., 2012, 8(3), 183–192, DOI:10.1038/nrendo.2011.158.
- S. E. Kahn, M. E. Cooper and S. Del Prato, Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future, Lancet, 2014, 383(9922), 1068–1083, DOI:10.1016/S0140-6736(13)62154-6.
- S. Wild, G. Roglic, A. Green, R. Sicree and H. King, Global Prevalence of Diabetes, Diabetes Care, 2004, 27(5), 1047–1053, DOI:10.2337/diacare.27.5.1047.
- S. Basu, J. S. Yudkin and S. Kehlenbrink, et al., Estimation of global insulin use for type 2 diabetes, 2018–30: a microsimulation analysis, Lancet Diabetes Endocrinol., 2019, 7(1), 25–33, DOI:10.1016/S2213-8587(18)30303-6.
- I. D. Federation, IDF Diabetes Atlas, 9th edn, 2019 Search PubMed.
- Y. Lin and Z. Sun, Current views on type 2 diabetes, J. Endocrinol., 2010, 204(1), 1–11, DOI:10.1677/JOE-09-0260.
- J. S. Skyler, Diabetes Mellitus: Pathogenesis and Treatment Strategies, J. Med. Chem., 2004, 47(17), 4113–4117, DOI:10.1021/jm0306273.
- B. Ahrén, Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes, Nat. Rev. Drug Discovery, 2009, 8(5), 369–385, DOI:10.1038/nrd2782.
- J. B. Xiao and P. Hogger, Dietary polyphenols and type 2 diabetes: current insights and future perspectives, Curr. Med. Chem., 2015, 22(1), 23–38, DOI:10.2174/0929867321666140706130807.
- H. Laoufi, N. Benariba, S. Adjdir and R. Djaziri, In vitro α-amylase and α-glucosidase inhibitory activity of Ononis angustissima extracts, J. Appl. Pharm. Sci., 2017, 7(2), 191–198, DOI:10.7324/JAPS.2017.70227.
- P. Agarwal and R. Gupta, Alpha-amylase inhibition can treat diabetes mellitus, Res. Rev.: J. Med. Health Sci., 2016, 5(4), 1–8 Search PubMed.
- M. TOELLER, α-Glucosidase inhibitors in diabetes: efficacy in NIDDM subjects, Eur. J. Clin. Invest., 2010, 24(S3), 31–35, DOI:10.1111/j.1365-2362.1994.tb02253.x.
- H. Bischoff, Pharmacology of alpha-glucosidase inhibition, Eur. J. Clin. Invest., 1994, 24, 3–10 CAS.
- A. Chaudhury, C. Duvoor and V. S. Reddy Dendi, et al., Clinical review of antidiabetic drugs: implications for type 2 diabetes mellitus management, Front. Endocrinol., 2017, 8, 6, DOI:10.3389/fendo.2017.0006.
- K. R. Feingold, B. Anawalt, A. Boyce, et al., Oral and Injectable (Non-insulin) Pharmacological Agents for Type 2 Diabetes, Endotext South Dartmouth, MA, 2020 Search PubMed.
- Y. Zheng, S. H. Ley and F. B. Hu, Global aetiology and epidemiology of type 2 diabetes mellitus and its complications, Nat. Rev. Endocrinol., 2018, 14(2), 88–98, DOI:10.1038/nrendo.2017.151.
- C. Hu and W. Jia, Therapeutic medications against diabetes: what we have and what we expect, Adv. Drug Delivery Rev., 2019, 139, 3–15, DOI:10.1016/j.addr.2018.11.008.
- A. Artasensi, A. Pedretti, G. Vistoli and L. Fumagalli, Type 2 diabetes mellitus: A review of multi-target drugs, Molecules, 2020, 25(8), 1–20, DOI:10.3390/molecules25081987.
- R. Ottanà, R. Maccari and M. Giglio, et al., Identification of 5-arylidene-4-thiazolidinone derivatives endowed with dual activity as aldose reductase inhibitors and antioxidant agents for the treatment of diabetic complications, Eur. J. Med. Chem., 2011, 46(7), 2797–2806, DOI:10.1016/j.ejmech.2011.03.068.
- R. Ottanà, P. Paoli and M. Cappiello, et al., Search for Multi-Target Ligands as Potential Agents for Diabetes Mellitus and Its Complications—A Structure-Activity Relationship Study on Inhibitors of Aldose Reductase and Protein Tyrosine Phosphatase 1B, Molecules, 2021, 26(2), 330, DOI:10.3390/molecules26020330.
- M. Stefek, V. Snirc and P.-O. Djoubissie, et al., Carboxymethylated pyridoindole antioxidants as aldose reductase inhibitors: Synthesis, activity, partitioning, and molecular modeling, Bioorg. Med. Chem., 2008, 16(9), 4908–4920, DOI:10.1016/j.bmc.2008.03.039.
- H. Ullah, M. Waseem, F. Rahim, A. Hussain and M. Perviaz, molecular docking study of oxadiazole-sulphonamide hybrid analogues, Chem. Data Collect., 2023, 45, 1–12 Search PubMed.
- F. A. Saddique, M. Ahmad, U. A. Ashfaq, M. Muddassar, S. Sultan and M. E. A. Zaki, Identification of Cyclic Sulfonamides with an N-Arylacetamide Group as α-Glucosidase and α-Amylase Inhibitors: Biological Evaluation and Molecular Modeling, Pharmaceuticals, 2022, 15(1), 1–22, DOI:10.3390/ph15010106.
- L. Rasheed, W. Rehman and F. Rahim, et al., Molecular Modeling and Synthesis of Indoline-2,3-dione-Based Benzene Sulfonamide Derivatives and Their Inhibitory Activity against α-Glucosidase and α-Amylase Enzymes, ACS Omega, 2023, 8(17), 15660–15672, DOI:10.1021/acsomega.3c01130.
- Ç. B. Apaydın, G. Hasbal Çelikok, T. Yılmaz Özden and G. Cihan Üstündağ, Design, synthesis and biological evaluation of novel sulfonamide hydrazones as α-glucosidase and α-amylase inhibitors, İstanbul J Pharm, 2022, 52(2), 108–113, DOI:10.26650/istanbuljpharm.2022.1018698.
- P. Taslimi, M. Işık and F. Türkan, et al., Benzenesulfonamide derivatives as potent acetylcholinesterase, α-glycosidase, and glutathione S-transferase inhibitors: biological evaluation and molecular docking studies, J. Biomol. Struct. Dyn., 2020, 1–12, DOI:10.1080/07391102.2020.1790422.
- M. Askarzadeh, H. Azizian and M. Adib, et al., Design, synthesis, in vitro α-glucosidase inhibition, docking, and molecular dynamics of new phthalimide-benzenesulfonamide hybrids for targeting type 2 diabetes, Sci. Rep., 2022, 12(1), 1–16, DOI:10.1038/s41598-022-14896-2.
- G. Wang, M. Chen and J. Wang, et al., Synthesis, biological evaluation and molecular docking studies of chromone hydrazone derivatives as α-glucosidase inhibitors, Bioorg. Med. Chem. Lett., 2017, 27(13), 2957–2961, DOI:10.1016/j.bmcl.2017.05.007.
- M. S. Ayoup, D. B. Cordes, A. M. Z. Slawin and D. O'Hagan, Selectively fluorinated cyclohexane building blocks: Derivatives of carbonylated all- cis- 3-phenyl-1,2,4,5-tetrafluorocyclohexane, Beilstein J. Org. Chem., 2015, 11(1), 2671–2676, DOI:10.3762/bjoc.11.287.
- M. S. Ayoup, D. B. Cordes, A. M. Z. Slawin and D. O'Hagan, Total Synthesis of a Reported Fluorometabolite from Streptomyces sp. TC1 Indicates an Incorrect Assignment. The Isolated Compound Did Not Contain Fluorine, J. Nat. Prod., 2014, 77(6), 1249–1251, DOI:10.1021/np500260z.
- V. P. Cirillo, Mechanism of glucose transport across the yeast cell membrane, J. Bacteriol., 1962, 84(3), 485–491, DOI:10.1128/jb.84.3.485-491.1962.
- M. Khan, R. Ahmad and G. Rehman, et al., Synthesis of Pyridinyl-benzo[d]imidazole/Pyridinyl-benzo[d]thiazole Derivatives and their Yeast Glucose Uptake Activity In Vitro, Lett. Drug Des. Discovery, 2019, 16(9), 984–993, DOI:10.2174/1570180815666181004102209.
- L. Sim, R. Quezada-Calvillo, E. E. Sterchi, B. L. Nichols and D. R. Rose, Human Intestinal Maltase–Glucoamylase: Crystal Structure of the N-Terminal Catalytic Subunit and Basis of Inhibition and Substrate Specificity, J. Mol. Biol., 2008, 375(3), 782–792, DOI:10.1016/j.jmb.2007.10.069.
- C. Li, A. Begum, S. Numao, K. H. Park, S. G. Withers and G. D. Brayer, Acarbose Rearrangement Mechanism Implied by the Kinetic and Structural Analysis of Human Pancreatic α-Amylase in Complex with Analogues and Their Elongated Counterparts, Biochemistry, 2005, 44(9), 3347–3357, DOI:10.1021/bi048334e.
- Molecular Operating Environment (MOE), 2020.09 Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2022 Search PubMed.
- C. A. Lipinski, Lead- and drug-like compounds: the rule-of-five revolution, Drug Discovery Today: Technol., 2004, 1(4), 337–341 CrossRef CAS PubMed.
- T. Matsui, C. Yoshimoto, K. Osajima, T. Oki and Y. Osajima, Vitro Survey of α -Glucosidase Inhibitory Food Components, Biosci., Biotechnol., Biochem., 1996, 60(12), 2019–2022, DOI:10.1271/bbb.60.2019.
- K. Lalitha and V. k. Kumar, In vitro study on α-amylase inhibitory activity of an Ayurvedic medicinal plant, Anacyclus pyrethrum DC root, Indian J. Pharmacol., 2014, 46(3), 350, DOI:10.4103/0253-7613.132204.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01060d |
| This journal is © The Royal Society of Chemistry 2024 |