
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSERS spectral evolution of azo-reactions mediated by plasmonic Au@Ag core–shell nanorods†
Mengen
Hu
ab,
Zhulin
Huang
 *ab,
Rui
Liu
ac,
Ningning
Zhou
c,
Haibin
Tang
*ab and
Guowen
Meng
ab
*ab,
Rui
Liu
ac,
Ningning
Zhou
c,
Haibin
Tang
*ab and
Guowen
Meng
ab
aKey Laboratory of Materials Physics, Anhui Key Laboratory of Nanomaterials and Nanotechnology, Institute of Solid State Physics, HFIPS, Chinese Academy of Sciences, Hefei, 230031, China. E-mail: zlhuang@issp.ac.cn; hbtang@issp.ac.cn
bUniversity of Science and Technology of China, Hefei, 230026, China
cDepartment of Chemical and Materials Engineering, Hefei University, Hefei 230601, China
First published on 13th September 2022
Abstract
The mechanism and application of localized surface plasmon resonance induced photocatalytic reactions remain an issue of interest. In this work, we used Au@Ag core–shell nanorods as a platform for plasmon-driven photocatalysis, which was in situ investigated by surface-enhanced Raman scattering (SERS) spectroscopy. The para-aminothiophenol (PATP) and para-nitrothiophenol (PNTP) adsorbed on the nanorods were irradiated with different excitation wavelengths (633 nm, 785 nm) and transformed into 4,4′-dimercaptoazobenzene (DMAB) as evidenced by the emerging Raman peaks at 1142 cm−1, 1390 cm−1, 1440 cm−1, and 1477 cm−1, corresponding to hot carrier dominated oxidation of PATP and reduction of PNTP. Preliminary azo-reaction kinetics and in situ SERS measurements were conducted by comparing the relative intensity ratio of SERS peaks at 1440 cm−1 (DMAB stretching of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and 1080 cm−1 (C–S stretching of PATP and PNTP). These results indicate that the catalytic efficiency was dominated by the excitation wavelength as well as the resonance condition between the plasmon band of the nanorods and the excitation line. As a proof of concept, the Au@Ag core–shell nanorods were used to catalyze 4-nitrophenol molecules, and 4-hydroxyazobenzene molecules as the product were confirmed by in situ SERS spectra as well theoretical predictions, showing potential in plasmon driven catalysis and degradation of organic molecules.
N) and 1080 cm−1 (C–S stretching of PATP and PNTP). These results indicate that the catalytic efficiency was dominated by the excitation wavelength as well as the resonance condition between the plasmon band of the nanorods and the excitation line. As a proof of concept, the Au@Ag core–shell nanorods were used to catalyze 4-nitrophenol molecules, and 4-hydroxyazobenzene molecules as the product were confirmed by in situ SERS spectra as well theoretical predictions, showing potential in plasmon driven catalysis and degradation of organic molecules.
Introduction
Noble metal nanomaterials have received widespread attention based on their localized surface plasmon resonance (LSPR) phenomenon.1,2 The LSPR effect originates from the collective oscillation of free electrons in the metal nanostructures under excitation light, wherein the frequency of the incident photon light matches the oscillation frequency of the electrons. This leads to a coherent oscillation in energy and space between the metal nanostructures and incident light.3,4 During the decay of LSPR, a series of effects can be aroused, including light scattering and absorbance, excitation of hot electrons, near-field enhancement, and thermal effect.5 This has led to its wide applications in the fields of catalysis,6 energy conversion,7 chemical and biological sensing,8–10 medical imaging, and surface-enhanced Raman scattering (SERS) based signal amplification.11–13 For example, SERS spectroscopy uses the LSPR effect to sharply increase the local electric field intensity, resulting in enhanced Raman scattering signals of adsorbed molecules,14–16 whereas in plasmon induced catalysis, the transient hot electrons and holes can be excited during the decay of plasmon, and they participate in the reduction and oxidation of species adsorbed at the metal interface. This has aroused great interest as it can enable spatially modified phase transformation and activate otherwise energetically unfavorable sites.17The premise of plasmon-driven photocatalysis is the fabrication of plasmonic nanomaterials with high SERS activity and a distinct LSPR effect. Therefore, various shape-controlled Au and Ag related nanostructures with sharp tips and corners have been fabricated.18 Particularly, Au nanorods have a strong LSPR effect in the visible-near-infrared band, demonstrating remarkable optical, electrical, and catalytic properties,19,20 as proved in SERS-based detection, multi-photon luminescence,21,22 imaging, thermal therapy, and other aspects. The characteristics of LSPR largely depend on the size and morphology of the nanostructures.10,18 In addition, the scattering efficiency can be effectively improved by hybridizing Ag with Au nanostructures, as Ag related nanostructures have sharper resonance properties than the counterparts of Au.23 In this way, the core–shell effect of complexed Au@Ag nanorods can generate a cooperative reinforcement of SERS enhancement compared to pure Au nanorods. Furthermore, Ag has a higher Fermi level and LSPR energy than Au, which could induce a higher energetic level of plasmonic hot electrons, consequently generating larger driving force for the catalytic reactions. Therefore, Au@Ag core–shell nanorods can be preferably employed as a research platform in either SERS-based detection or plasmon-induced catalytic reactions. Despite the extensively investigated sensing applications of Au@Ag core–shell nanorods in SERS-based detection, the plasmon-induced catalytic performances remain unclear.
To investigate the plasmon-driven catalysis, it is important to choose a model reaction. To date, it has been revealed by an increasing number of groups that the plasmon-induced azo-reaction of para-aminothiophenol (PATP) and para-nitrothiophenol (PNTP) molecules could generate 4,4′-dimercaptoazobenzene (DMAB) molecules, featuring three abnormal peaks at 1140 cm−1, 1388 cm−1 and 1432 cm−1 in the SERS spectra,24,25 demonstrating the coupling of two amino or nitro groups into azoxy (–NN–).26 Subsequent studies have shown that this reaction would not occur in an inert gas environment without oxygen, as oxygen could be dissociated by hot electrons and participate in the oxidation of PATP molecules.27–30 Being different from the oxidation reaction of PATP, the conversion of PNTP to DMAB is a reduction process.31 It has been proposed that PNTP was reduced to DMAB by plasmon excited hot electrons, and water as a sacrificial agent was oxidized by the hot holes to produce oxygen. The overall reaction of photocatalysis of PNTP is a deoxygenation process.32,33 Nevertheless, there have been few systematic reports on the relationship between excitation light, the LSPR band and catalytic activity, as well as the proof of application. Even though the reaction product has been qualitatively confirmed, the catalytic mechanism and reaction pathway remain unclear.
In this work, we chose Au and Au@Ag core–shell nanorods to monitor the plasmon-induced catalysis of azo-reactions. The nanorods were selected because they have a tunable longitudinal LSPR band, which can be excited by incident light polarized along the axis, and achieve resonant excitation in the vis-infrared spectral range.34,35 Based on the Au and Au@Ag core–shell nanorods with an aspect ratio of 2.0–4.0, the time-dependent SERS spectra of PATP and PNTP show that regardless of the resonance between the LSPR bands of nanorods and excitation wavelength, DMAB molecules were essentially generated from PATP or PNTP on both Au nanorods and Au@Ag core–shell nanorods via different hot carrier directed reaction pathways, as evidenced by the new Raman peaks of DMAB at 1142 cm−1, 1390 cm−1 and 1440 cm−1. Au nanorod catalyzed PATP exhibited slightly higher DMAB conversion but a lower reaction rate compared to Au@Ag core–shell nanorods. However, Au@Ag core–shell nanorods showed higher conversion efficiency for PNTP than Au nanorods. Since Au@Ag core–shell nanorods have a large extinction cross-section and strong local electric field enhancement, they have good application prospects in the field of catalytic degradation of organics. As a proof of concept, the core–shell nanorods were utilized for the catalysis of 4-nitrophenol (4-NP). Both in situ SERS spectra and theoretical predictions indicate that some of the 4-NP molecules converted to 4-hydroxyazobenzene (4-HAB) instead of 4-aminophenol (4-AP).
Results and discussion
Characterization of Au nanorods and Au@Ag core–shell nanorods
Priorly, we used deionized water to get rid of the adsorbed surfactants before characterization, so that a clearer morphology of the Au nanorods could be observed. Herein, a 1.5 mL tube loaded with Au nanorod solution (1 mL) was centrifuged at a high speed of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min, and the supernatant was removed with a pipette. Then 1 mL deionized water was added into the tube and ultrasonically dispersed for 5 min. The procedure was repeated to remove most of the dummy surfactants bound on the surface of Au nanorods. The typical transmission electron microscope (TEM) image of Au nanorods is displayed in Fig. 1a; as can be seen, the average length of Au nanorods is about 60 nm. As can be seen from the UV-Vis-NIR absorbance spectra (Fig. 1f), the lateral plasmon resonance absorption peak of Au nanorods is around 520 nm, while the longitudinal plasmon resonance absorption peak is 650 nm.
000 rpm for 10 min, and the supernatant was removed with a pipette. Then 1 mL deionized water was added into the tube and ultrasonically dispersed for 5 min. The procedure was repeated to remove most of the dummy surfactants bound on the surface of Au nanorods. The typical transmission electron microscope (TEM) image of Au nanorods is displayed in Fig. 1a; as can be seen, the average length of Au nanorods is about 60 nm. As can be seen from the UV-Vis-NIR absorbance spectra (Fig. 1f), the lateral plasmon resonance absorption peak of Au nanorods is around 520 nm, while the longitudinal plasmon resonance absorption peak is 650 nm.
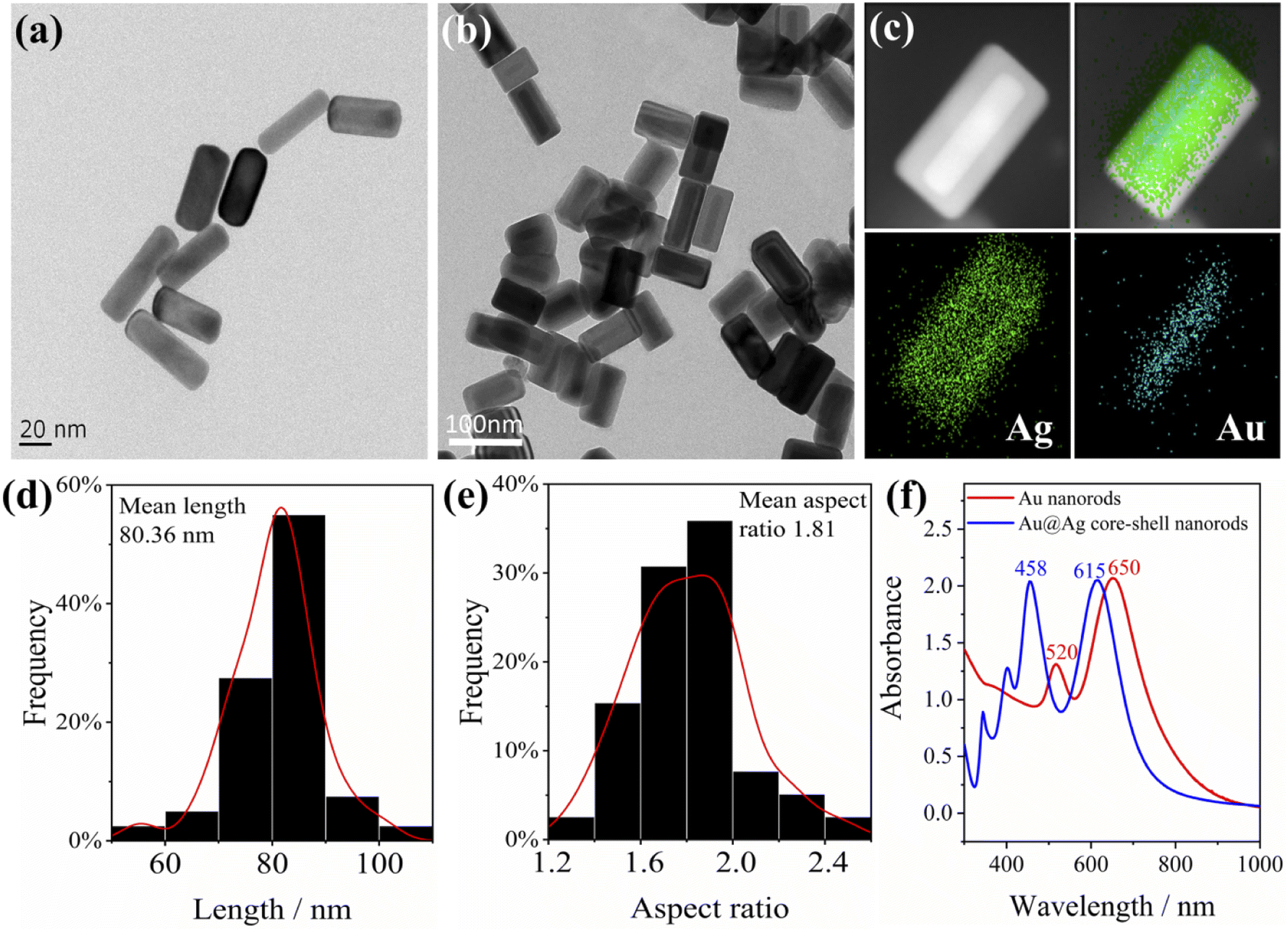 | ||
| Fig. 1 TEM images of (a) Au nanorods, (b) Au@Ag core–shell nanorods, the (c) EDS image of Au@Ag nanorods, (d) length and (e) aspect ratio distribution of Au@Ag core–shell nanorods, and (f) UV-Vis-NIR absorbance spectra of Au nanorods and Au@Ag core–shell nanorods. | ||
To enhance the plasmon effect of Au nanorods, a layer of Ag was coated on the surface of the Au nanorods (the thickness of the coated Ag is about 5–15 nm), and the Au@Ag core–shell nanorods were obtained. The typical TEM image is shown in Fig. 1b. It can be clearly seen from the energy dispersive X-ray spectrometer (EDS) images (Fig. 1c) that the Au nanorods coated with silver have an obvious core–shell structure. The average length of Au@Ag core–shell nanorods is calculated to be 80 nm, and the average aspect ratio is 1.8 (Fig. 1d and e). The UV-Vis-NIR absorption spectrum of Au@Ag core–shell nanorods in Fig. 1f shows a lateral plasmon resonance absorption peak at around 460 nm, indicating the presence of silver. The longitudinal plasmon resonance absorption peak is located at 615 nm, and the overall plasmon modes of Au@Ag core–shell nanorods have a blue shift compared to Au nanorods. Of note, after one year of storage at 4 °C, the plasmon effect maintained while the SERS intensity decreased by 20% (Fig. S1†).
Plasmon-mediated catalysis of PATP and PNTP
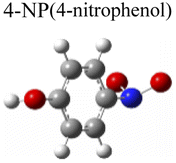
To evaluate the plasmon-driven catalysis, PATP and PNTP probe molecules were utilized, which can evolve into DMAB molecules.36–38 First, the vibrational modes of PATP, PNTP, and DMAB molecules need to be specified, so the DFT theory involved in Gaussian 09W software was used to simulate the optimized structures and Raman spectra of PATP, PNTP, and DMAB molecules. The hybrid density functional B3LYP method with the 6-31G/(d) basis set was used. A scale factor of 0.9613 was used to correct for basis set stacking errors. Table 1 summarizes the optimized molecular structures of PATP, PNTP, and DMAB. The main fingerprints of PATP are located at 1074 cm−1 (C–S stretching), 1188 cm−1 (C–H in-plane bending), and 1600 cm−1 (parallel C–C stretching). The main fingerprints of PNTP are located at 1071 cm−1 (C–S stretching), 1115 cm−1 (C–H in-plane bending), 1340 cm−1 (–NO2 stretching) and 1580 cm−1 (parallel C–C stretching). For the DMAB molecules, the C–S stretching mode shifts slightly to 1069 cm−1, the parallel C–C stretching slightly shifts to 1584 cm−1, and the NN stretching modes appeared at 1382 cm−1, 1424 cm−1 and 1479 cm−1. To be mentioned, these vibration assignments are generally consistent with subsequent actual SERS measurements, given the basis set stacking errors involved in the simulation process.
| Vibrational description | SERS | Calculateda | |
|---|---|---|---|
| a Calculated at B3LYP/6-31G(d) with the vibrational bands above 1000 cm−1 scaled by an optimal factor 0.9613. | |||

|
–NH2 stretching | — | 1642 |
| Ring stretching | 1595 | 1600 | |
| Ring stretching | 1576 | 1562 | |
| C–H wagging | 1190 | 1188 | |
| C–S stretching | 1078 | 1074 | |
| Ring trigonal breathing | 1008 | 1001 | |
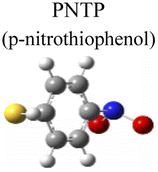
|
Ring stretching | 1575 | 1580 |
| –NO2 stretching | 1345 | 1340 | |
| C–H wagging | 1111 | 1115 | |
| C–S stretching | 1083 | 1071 | |
| Ring trigonal breathing | 1012 | 1010 | |
| S–H wagging | 855 | 866 | |
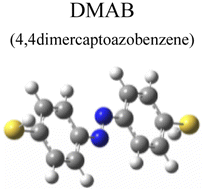
|
Ring stretching | 1580 | 1584 |
| NN stretching |
1477 | 1479 | |
| NN stretching |
1440 | 1424 | |
| NN stretching |
1390 | 1382 | |
| C–N stretching | 1142 | 1140 | |
| C–S stretching | 1078 | 1069 | |
| Ring trigonal breathing | 1007 | 1005 | |
|
Ring stretching | 1592 | 1596 |
| Ring stretching | 1343 | 1348 | |
| –NO2 stretching | 1330 | 1340 | |
| C–O stretching | 1287 | 1272 | |
| C–N stretching | 1114 | 1094 | |
| Ring deforming | 872 | 845 | |
|
Ring stretching | 1619 | 1619 |
| C–N and C–O stretching | 1261 | 1263 | |
| C–H wagging | 1169 | 1160 | |
| Ring deforming | 846 | 832 | |
| –NH2 stretching | 649 | 646 | |
|
NN stretching |
1444 | 1444 |
| NN stretching |
1392 | 1391 | |
| C–O stretching | 1246 | 1250 | |
| C–N stretching | 1132 | 1132 | |
| C–H wagging | 1007 | 1001 | |
| Ring deforming | 760 | 752 | |
Herein, to investigate the catalysis of PATP and PNTP by plasmon-driven Au nanorods and Au@Ag core–shell nanorods, we chose the PATP solution and PNTP solution with a concentration of 10−5 M. The substrates with adsorbed molecules were irradiated with monochromatic light of 633 nm and 785 nm for 10 min. Then, Raman spectroscopy measurements were conducted for the substrates of Au nanorods and Au@Ag core–shell nanorods. As shown in Fig. 2a, three new peaks appear in the SERS spectra of PATP after laser irradiation with centres located at 1142 cm−1, 1390 cm−1, and 1440 cm−1 respectively, indicating that PATP converted to DMAB. The peak position at 1595 cm−1, which represents the stretching of the benzene ring, shifted to 1580 cm−1, because the –NH2 of PATP is relatively light compared to NN, thus the stretching between the benzene ring and NN becomes more difficult. According to Table 1, these newly appeared shifts can be attributed to the NN stretching mode and C–N stretching mode, respectively. However, the main characteristic peak of PATP at 1595 cm−1 still exists in the spectra, suggesting that PATP molecules were not completely converted into DMAB. As shown in Fig. 2b, the SERS spectra of PNTP on the Au nanorods and the Au@Ag core–shell nanorod substrates show the characteristic peaks of DMAB at 1142 cm−1, 1390 cm−1 and 1440 cm−1, indicating that the characteristic peaks of PNTP have been weakened, confirming the conversion of PNTP to DMAB. However, the characteristic peaks of PNTP still exist, and the conversion of PNTP to DMAB was incomplete. A weak PATP characteristic peak can be observed at 1190 cm−1, indicating that a small amount of PATP was produced during the illumination process.
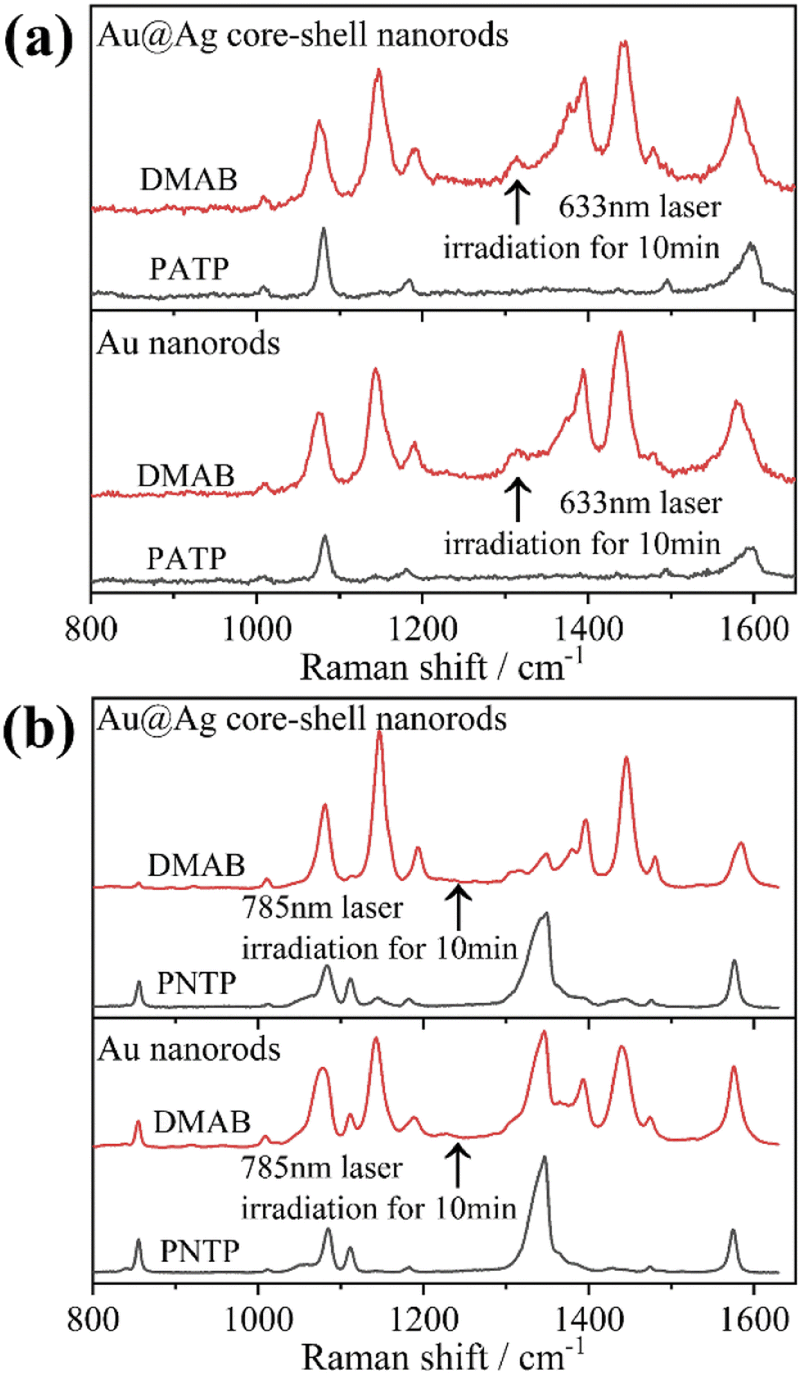 | ||
| Fig. 2 SERS spectral changes after monochromatic light irradiation for 10 min of (a) PATP adsorbed on the surface of Au nanorods and Au@Ag core–shell nanorods and (b) PNTP adsorbed on the surface of Au nanorods and Au@Ag core–shell nanorods. | ||
In addition, we investigated the PATP coupling reaction catalyzed by Au nanorods and Au@Ag core–shell nanorods. The samples adsorbed with PATP were placed in the aqueous solution and irradiated with the laser of a Raman spectrometer for different durations. It was found that with the increase of laser irradiation duration, the peak position of each sample did not change, while the characteristic peak intensity representing DMAB gradually increased (Fig. 3a and e). We compare the peak intensity ratio (RI) at 1440 cm−1 (stretching of NN in DMAB) and 1080 cm−1 (stretching of C–S in PATP) to judge the catalytic activity of plasmon. The RI value calculations and fitting of the relevant time curve formed by the DMAB molecule based on the Poisson equation (I = A + Bexp(−t/τ))39 are shown in Fig. 3c and g. It can be seen that as the irradiation time increases, the RI value first increases and then gradually stabilizes. This also shows that the conversion efficiency of coupling PATP with DMAB increases rapidly with the extension of laser irradiation time and then gradually remains stable.
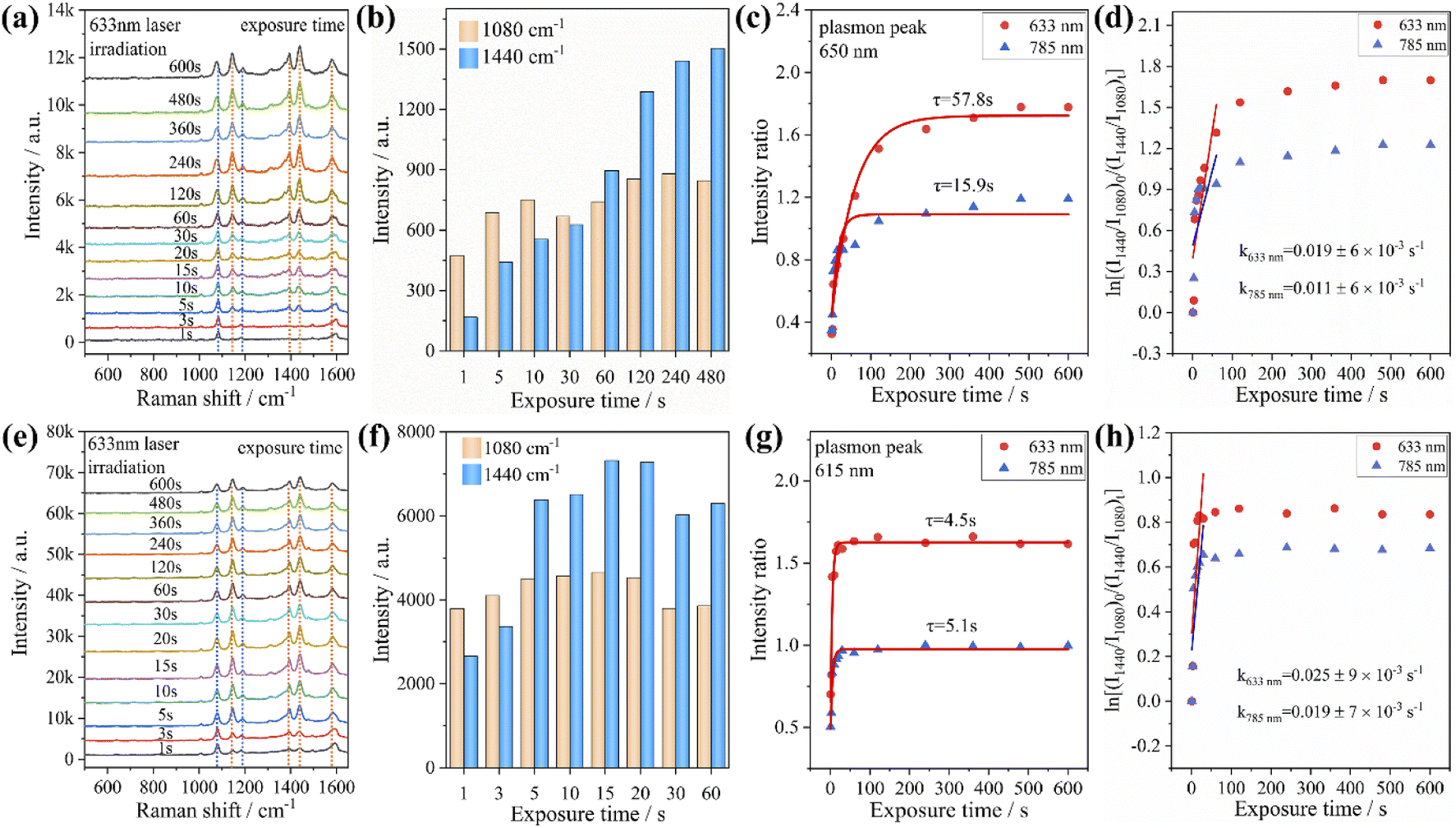 | ||
| Fig. 3 Time-dependent SERS spectra of PATP adsorbed on nanorods and irradiated with 633 nm monochromatic light. (a) The case of Au nanorods; (e) the case of Au@Ag core–shell nanorods. The variations of peak intensities at 1080 cm−1 and 1440 cm−1 with increasing irradiation time for (b) Au nanorods, and (f) Au@Ag core–shell nanorods. The time-dependent relative intensity ratios between the 1440 cm−1 shift of DMAB and the 1080 cm−1 shift of PATP for (c) Au nanorods and (g) Au@Ag core–shell nanorods. Plots of ln[(I1440/I1080)0/(I1440/I1080)t] versus time for the oxidation of PATP with (d) Au nanorods and (h) Au@Ag core–shell nanorods. | ||
According to the RI value-time curve in Fig. 3, for the conversion of PATP to DMAB catalyzed by Au nanorods and Au@Ag core–shell nanorods, the RI value reaches the maximum when the excitation light wavelength was 633 nm, which had the highest catalytic efficiency. The incident power of the excitation light at 633 nm is an order lower than that of the excitation light at 785 nm, but the conversion efficiency and reaction rate are higher than those of the excitation light at 785 nm. The possible reason is that when the wavelength of the excitation light is close to the plasmon band of the Au nanorods, a stronger coupling resonance effect will be generated, which resulted in a higher catalytic efficiency compared to other excitations. Comparing the reaction rate constants, it could be found that the PATP reaction catalyzed by Au@Ag core–shell nanorods is faster than that of Au nanorods. This is because the silver on the surface of the Au@Ag core–shell nanorods has a low work function (4.26 eV), and the photon energy is about 1.96 eV under the excitation light conditions of 633 nm. Compared to gold (5.1 eV), silver has a smaller potential difference with photons and loses electrons more easily. In summary, for the catalytic conversion of PATP to DMAB, the catalytic efficiency of Au nanorods and Au@Ag core–shell nanorods strongly depends on the excitation light wavelength and the plasmon band coupling resonance. The Ag-coated Au nanorods exhibited a faster plasmon response, indicating that the plasmon-driven photocatalytic effect is largely determined by the metal nanostructure and the excitation wavelength. Additionally, we investigated the effect of different aspect ratios of Au nanorods and Au@Ag core–shell nanorods (Fig. S2†) on the conversion of PATP under different wavelengths of excitation light. The results in Fig. S3† also indicate that PATP has the largest conversion rate when the excitation light wavelength is close to the plasmon band of the nanorods.
From the previous experimental results, it could be concluded that the Au@Ag core–shell nanorods have a faster plasmon response than Au nanorods. Therefore, we investigated the time-dependent Raman spectra of PNTP catalyzed by Au@Ag core–shell nanorods. It was found that with the increase of laser irradiation time, the intensity of the characteristic peak representing PNTP at 1345 cm−1 (–NO2 stretching) gradually weakens (indicated by the blue dashed line in Fig. 4a and b), while the characteristic peak at 1575 cm−1 (benzene ring stretching) shifts to the right, and the width of the peak increases, which may be caused by the superposition of the characteristic peaks of PNTP and DMAB. The intensity of the characteristic peaks representing DMAB at 1142 cm−1 (C–N stretching), 1390 cm−1 and 1440 cm−1 (NN stretching) gradually increases as indicated by the orange dashed line in Fig. 4a and b, indicating that PNTP was partially converted to DMAB under laser irradiation (Fig. 4a and b). In addition, the decrease of the characteristic peak intensity of PNTP shows that Au@Ag core–shell nanorod catalyzed PNTP conversion is higher than that of Au nanorods, as shown in Fig. 2b. We used the same method to calculate the RI value. For the conversion of PNTP to DMAB catalyzed by Au@Ag core–shell nanorods, the RI value reaches the maximum and takes the shortest time to stabilize under an excitation light of 633 nm, with the shortest stabilization time and the highest reaction rate, which is consistent with the previous catalytic reaction of PATP.
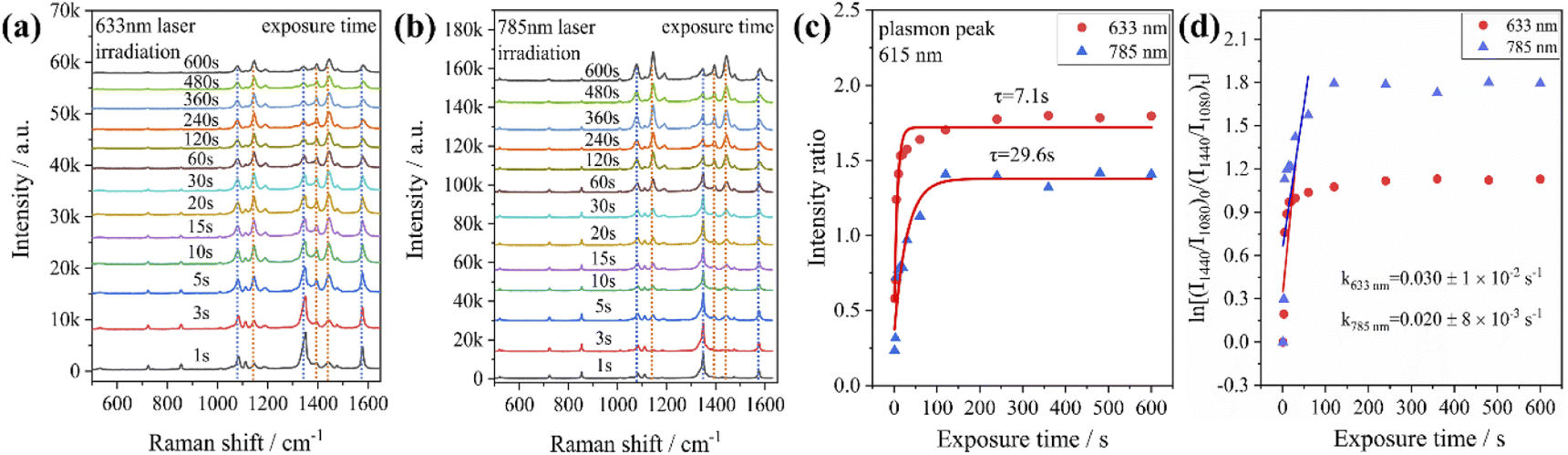 | ||
| Fig. 4 SERS spectral evolution of PNTP adsorbed on Au@Ag core–shell nanorods irradiated with (a) 633 nm and (b) 785 nm laser wavelengths. (c) The time-dependent relative intensity ratios between the 1440 cm−1 shift of DMAB and the 1080 cm−1 shift of PNTP. (d) Plots of ln[(I1440/I1080)0/(I1440/I1080)t] versus time for the reduction of PNTP with Au@Ag core–shell nanorods. | ||
Besides, we took the catalysis of PATP by Au@Ag core–shell nanorods under an excitation light of 633 nm as an example to consider the influence of laser power on the conversion rate. The sample was irradiated for 3 s under different power conditions, and the Raman intensities were averaged from random 5 points. The results of the peak intensity ratio of 1440 cm−1 to 1080 cm−1 in the Raman spectra are shown in Fig. S4.† It can be concluded that the results indicate that the power of the laser does affect the conversion of PATP, but the effect on the conversion is relatively weak when the laser power is below 10%.
The mechanism of plasmon-induced azo-reactions
Although the conversion of PATP to DMAB has been extensively studied, there are relatively few studies on plasmon-driven reduction of PNTP to DMAB. This is because the hot carriers generated by plasmon excitation have extremely short lifetime, so it is difficult to monitor the oxidation or reduction process. In the oxidation process of PATP, the laser irradiated Au@Ag core–shell nanorods generate hot electrons and holes due to the excitation of surface plasmons. The hot electrons activate the oxygen adsorbed on the metal surface and subsequently oxidize PATP to DMAB; the reaction diagram is shown in Fig. 5a. The reaction equations are shown in eqn (1)–(3), and the overall reaction is shown in eqn (4).27,28| 2O2 + 2e− → 2O2− | (1) |
| 2H2O + 4h+ → 4H+ + O2 | (2) |
| 2PATP + 2O2− + 2e− + 4H+ → DMAB + 4H2O | (3) |
| 2PATP + O2 + 4hv → DMAB + 2H2O | (4) |
 | ||
| Fig. 5 Schematic diagram of the reaction mechanism of plasmon-induced conversion of PATP to DMAB (a) and PNTP to DMAB (b). | ||
For PNTP which was reduced to DMAB, the possible reaction is that the plasmon-excited hot electrons of core–shell nanorods combined with oxygen molecules adsorbed on the Au@Ag core–shell nanorod surface and resulted in activated oxygen. Simultaneously, the remaining holes oxidize water to oxygen and hydrogen ions. The hydrogen ions and hot electrons cooperatively reduce PNTP to DMAB, as shown in Fig. 5b. The reaction equations are shown in eqn (5) to (6), and the overall reaction is shown in eqn (7).31
| 4H2O + 8h+ → 8H+ + 2O2 | (5) |
| 2PNTP + 8H+ + 8e− → DMAB + 4H2O | (6) |
| 2PNTP + 8hv → DMAB + 2O2 | (7) |
This could explain the better catalytic effect of Au@Ag core–shell nanorods relative to Au nanorods (shorter equilibration time). Because silver has a stronger ability to adsorb oxygen than gold, there will be more oxygen enrichment on the surface of silver that can participate in the reactions. To qualitatively verify this viewpoint, we sputtered Au and Ag on the surface of the silicon wafer and analyzed the element content on the surface of the sample after sputtering Au and Ag by XPS (as shown in Fig. 6). The oxygen content on the gold surface was calculated to be 9.61%, while the oxygen content on the silver surface was 11.91%, indicating that oxygen content adsorbed on the silver surface is higher than that on the gold surface. The role of oxygen may be as an acceptor of hot electrons, when the laser was irradiated on the Au nanorods and Au@Ag core–shell nanorods, pairs of hot electrons and holes would generate. Hot electrons and holes exist for a very short time and are highly easy to recombine together. The presence of oxygen consumes some of the hot electrons and inhibits the recombination of hot electrons and holes, thereby forming isolated hot holes and consequently promoting catalytic reactions.39
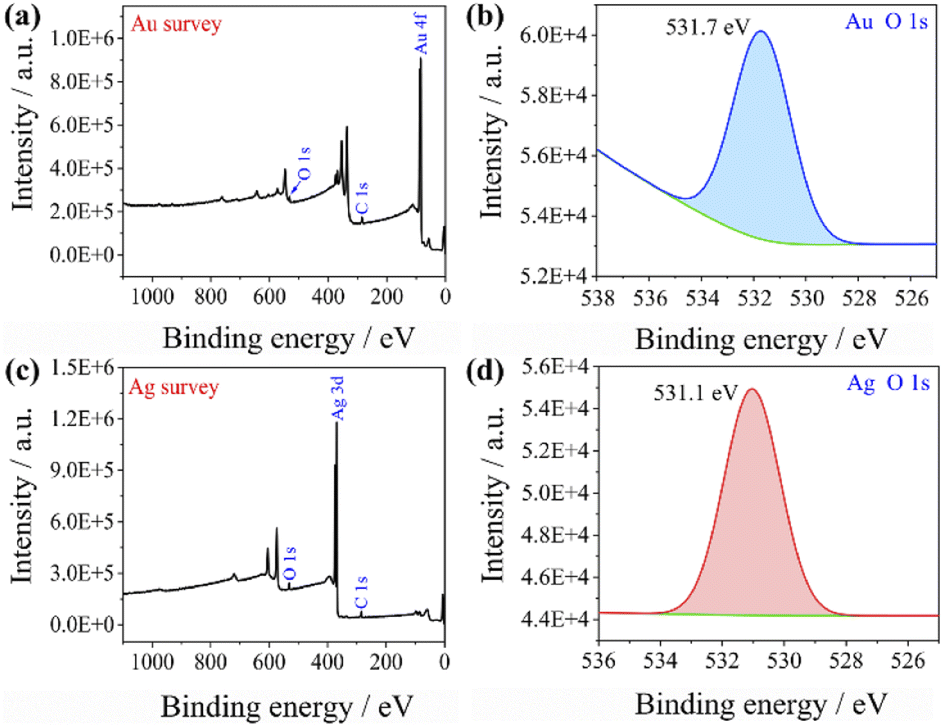 | ||
| Fig. 6 XPS spectra of (a) Au surface wide scan, (b) Au surface oxygen narrow scan, (c) Ag surface wide scan, and (d) Ag surface oxygen narrow scan. | ||
Degradation of 4-NP by Au@Ag core–shell nanorods
To test the effect of plasmon-driven photocatalysis by Au@Ag core–shell nanorods, 4-NP was used as trial analytes. 4-NP is a common organic pollutant with an extremely high risk of carcinogenicity. In addition, 4-NP can easily form solutes in water and is difficult to degrade under natural conditions. The specific operation was to adsorb 4-NP on a silicon substrate dispersed with Au@Ag nanorods, which was immersed in analyte solution and then excited with a 785 nm laser on a Raman spectrometer. As can be seen from Fig. 7a, several new peaks appear in the spectra in a short time, which are located at 760 cm−1, 1132 cm−1, 1246 cm−1, 1392 cm−1, and 1444 cm−1 (red dotted line in Fig. 7a). The intensity of the characteristic peaks representing 4-NP (872 cm−1, 1114 cm−1, 1330 cm−1, and 1592 cm−1) weakens with the prolongation of laser irradiation time (blue dotted line in Fig. 7a), indicating that the plasmonic Au@Ag core–shell nanorods degraded 4-NP molecules. We know that 4-NP is usually converted to 4-AP by the plasmon-induced photocatalytic reaction in the environment of NaBH4,40 but there is no characteristic peak of 4-AP (846 cm−1, 1169 cm−1, 1261 cm−1, and 1619 cm−1) on the Raman spectrum after the reaction. This indicates that the product of the reaction may be a new intermediate product of 4-NP and 4-AP, which is speculated to be 4-hydroxyazobenzene (4-HAB). To verify the product after the catalytic reaction, we used Gaussian 09W software to simulate the Raman spectrum of 4-HAB (Fig. 7c). The corrected results are shown in Table 1. We found that the three strongest peaks (1132 cm−1, 1392 cm−1, and 1444 cm−1) among the new characteristic peaks generated in the produced Raman spectra of 4-NP coincided with the characteristic peaks of 4-HAB.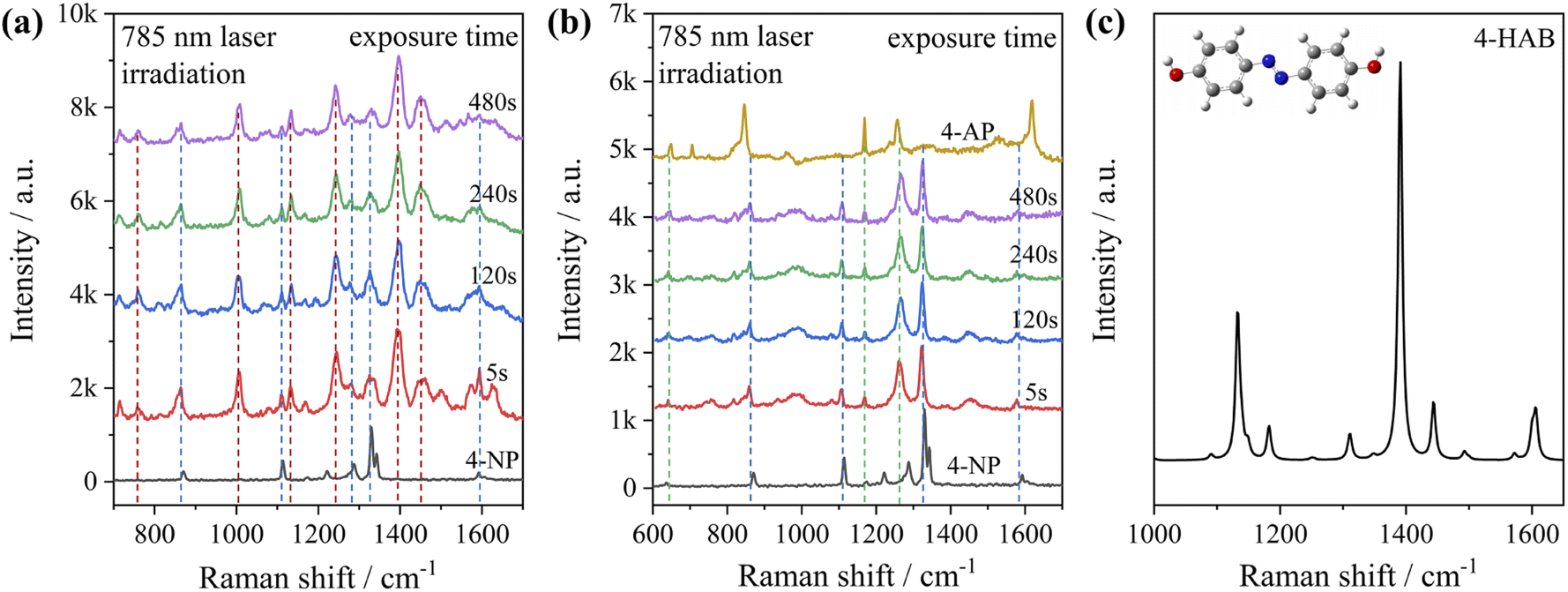 | ||
| Fig. 7 (a) SERS spectral evolution irradiated with 785 nm monochromatic light: (a) 4-NP adsorbed on Au@Ag core–shell nanorods; (b) 4-NP adsorbed on Au@Ag core–shell nanorods after adding NaBH4. (c) Simulated Raman spectrum of 4-HAB molecules and the optimized molecule structures (B3LYP/6-31+G(d)). | ||
Simultaneously, we also conducted a set of control experiments. We added 10 μL of NaBH4 (100 mM) in the solution of 4-NP under the above experimental conditions. As can be seen from Fig. 7b, several new characteristic peaks (649 cm−1, 862 cm−1, 1110 cm−1, 1169 cm−1, 1261 cm−1, and 1324 cm−1) appear shortly after the addition of NaBH4. Compared with the calculation results in Table 1, three new peaks (649 cm−1, 1169 cm−1, and 1261 cm−1) are consistent with the characteristic peaks of 4-AP (green dotted line in Fig. 7b). This suggests that 4-NP can be instantly converted to 4-AP under reducing conditions. However, the electrons and H+ concentration generated by the plasmon-induced photocatalytic reaction are relatively low. Although 4-NP can be reduced, the final reduction product tended to generate azobenzene. This is consistent with the above-mentioned main DMAB product of PNTP catalyzed by Au@Ag core–shell nanorods.
Conclusions
In summary, Au@Ag core–shell nanorods were utilized as a platform of plasmon-induced photocatalytic reactions. By analyzing the SERS spectra before and after the light illumination, it was found that the molecules PATP, PNTP, and 4-NP can be converted into azobenzene after laser irradiation. At the same time, the transformation effect was investigated by monitoring the SERS fingerprints of DMAB. Plasmon-induced catalytic reactivity was evaluated by comparing the RI values of the SERS spectra of DMAB with PATP and PNTP (ratio of peak intensities at 1440 cm−1 and 1080 cm−1), respectively, and the time required to reach stabilization. From the SERS spectral evolution, both PATP and PNTP adsorbed on Au@Ag core–shell nanorod substrates converted to DMAB molecules and reached equilibrium after plasmon resonance excitation for several to tens of seconds. It was found that the excitation light wavelength close to the plasmon band of the Au@Ag core–shell nanorods resulted in maximum conversion efficiency and the fastest conversion speed. In addition, the plasmonic Au@Ag core–shell nanorods were able to degrade 4-NP molecules, and 4-HAB molecules as a final product was obtained as proved by SERS measurements and theoretical predictions, showing promising applications in catalysis and degradation of organic molecules.Experimental
Materials
Chloroauric acid (HAuCl4) was purchased from Sigma-Aldrich. Cetyltrimethylammonium bromide (CTAB), cetyltrimethylammonium chloride (CTAC), para-aminothiophenol (PATP), and p-nitrothiophenol (PNTP) were obtained from Aladdin. 4-Nitrophenol (4-NP) was purchased from Adamas-beta. The other reagents such as sodium borohydride (NaBH4), silver nitrate (AgNO3), nitric acid (HNO3), hydrochloric acid (HCl), sulfuric acid (H2SO4), hydrogen peroxide (H2O2), acetone (C3H6O), ascorbic acid (AA), and ethanol absolute were bought from Sinopharm Chemical Reagent Co. Ltd. Deionized water (18.2 MΩ) was used for all preparations.Synthesis of Au@Ag core–shell nanorods
Au@Ag core–shell nanorods were synthesized according to our previously reported method.12 The preparation steps of Au@Ag core–shell nanorods are as follows:(1) Preparation of Au nano-seeds: first, HAuCl4 solution (0.01 M, 0.25 mL) was added to CTAB solution (0.1 M, 9.75 mL) and well mixed, followed by injection of ice-cold NaBH4 solution (0.01 M, 0.6 mL). The seed solution was vigorously stirred at 800 rpm for 120 s and further precipitated in a water bath (26 °C) for 2 h. When the color of the solution changed to brown, the gold seeds were formed.
(2) Preparation of Au nanorods: solutions of HAuCl4 (0.01 M, 0.25 mL), AgNO3 (0.01 M, 0.4 mL) and HCl (1.0 M, 0.8 mL) were sequentially added in CTAB solution (0.1 M, 40 mL) and shaken. Then AA solution (0.1 M, 0.32 mL) was injected and shaken for 30 s. Subsequently, Au seed solution (50 μL) was added, shaken for 30 s, and placed in a water bath (28 °C) overnight. In this way, Au nanorods with an aspect ratio of about 2–4 depending on the concentration of Au seeds were obtained.
(3) Synthesis of Au@Ag core–shell nanorods: Au@Ag core–shell nanorods were prepared by wrapping silver shells on Au nanorods. Au nanorods (30 mL) dispersed in CTAB were first sub-surfactant treated with CTAC and then sonicated by adding CTAC (80 mM) after three centrifugation–sonication cycles. Then AgNO3 (0.01 M, 1.5 mL) and AA solution (0.1 M, 2.5 mL) were added. The mixture was shaken and placed in a water bath (70 °C) for 3 h and finally centrifuged and washed repeatedly with deionized water to remove most of the CTAC surfactant.
Characterization
The Au nanorods and Au@Ag nanorods were observed by scanning electron microscopy (SEM, SU8020, Hitachi, Japan) and transmission electron microscopy (TEM, Tecnai G2 F20, American). The UV-Vis-NIR spectra were measured on a Shimadzu UV3600-MPC3100 photometer (Shimadzu, Japan). The Raman spectra of the samples were measured and collected using a confocal Raman microscope (Renishaw inVia Reflex, England) equipped with a monochromatic laser light source (633 nm; 785 nm) and 50× long focus objective. An X-ray photoelectron spectrometer (ESCALAB 250Xi, England) was used to measure the XPS spectrum on the surface of the gold and silver samples.Raman measurements
The measurement of the Raman spectrum includes the following steps. First, the silicon wafer was cut into smaller substrates of 5 × 5 mm2 size and then sonicated in acetone and alcohol for 15 min, respectively. The process was repeated twice. The cleaned silicon wafer was immersed in a mixture of concentrated sulfuric acid and hydrogen peroxide (volume ratio, 1:9) at 60 °C for 6 h. Finally, the silicon wafer was removed and cleaned twice with deionized water and alcohol, respectively. The prepared Au nanorod and Au@Ag core–shell nanorod solutions (10 μL) were dropped onto a clean silicon wafer with a pipette and then dried in an oven at 60 °C to obtain the SERS substrate. The SERS substrate was soaked in analyte solutions for 1 h to make the measured molecules fully adsorbed. For in situ SERS measurements, the substrate was placed in the cap of a centrifugation tube loaded with the measurement solution and covered with a glass slide. The cell was excited by 633 nm (17 mW) and 785 nm (300 mW) laser light, while Raman spectra were acquired continuously with 10% power and an integration time of 2 s.
Author contributions
Zhulin Huang conceived and designed the study. Mengen Hu and Rui Liu synthesized and characterized the materials. Mengen Hu and Zhulin Huang wrote and edited the manuscript. Haibin Tang and Ningning Zhou revised the manuscript. Guowen Meng provided very helpful suggestions for this work. All authors agreed on the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grants 52222208, 52072373, 21673245, and 51972308), the HFIPS Director's Fund (Grant No. BJPY2021B04 and YZJJZX202019), Natural Science Foundation of Anhui Province (Grants No. 2008085ME171 and 201903a07020021), and the Key Research Program of Frontier Sciences, Chinese Academy of Sciences (Grant No. QYZDJSSW-SLH046).Notes and references
- T. G. Habteyes, S. Dhuey, S. Cabrini, P. J. Schuck and S. R. Leone, Nano Lett., 2011, 11, 1819–1825 CrossRef CAS PubMed.
- C. X. Kan, J. J. Zhu and X. G. Zhu, J. Phys. D: Appl. Phys., 2008, 41, 155304 CrossRef.
- L. W. Chou, N. Shin, S. V. Sivaram and M. A. Filler, J. Am. Chem. Soc., 2012, 134, 16155–16158 CrossRef CAS PubMed.
- S. W. Li, P. Miao, Y. Y. Zhang, J. Wu, B. Zhang, Y. C. Du, X. J. Han, J. M. Sun and P. Xu, Adv. Mater., 2021, 33, 2000086 CrossRef CAS PubMed.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- Y. Horiguchi, T. Kanda, K. Torigoe, H. Sakai and M. Abe, Langmuir, 2014, 30, 922–928 CrossRef CAS PubMed.
- C. C. Zhang, Y. Y. Zhang and W. Xie, J. Energy Chem., 2021, 63, 40–53 CrossRef CAS.
- C. Gutierrez-Sanchez, M. Pita, C. Vaz-Dominguez, S. Shleev and A. L. De Lacey, J. Am. Chem. Soc., 2012, 134, 17212–17220 CrossRef CAS PubMed.
- P. K. Jain, X. H. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS PubMed.
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828–3857 CrossRef CAS PubMed.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Y. Li, C. H. Moran, Q. Zhang, D. Qin and Y. N. Xia, Chem. Rev., 2011, 111, 3669–3712 CrossRef CAS PubMed.
- Z. L. Huang, X. Lei, Y. Liu, Z. W. Wang, X. J. Wang, Z. M. Wang, Q. H. Mao and G. W. Meng, ACS Appl. Mater. Interfaces, 2015, 7, 17247–17254 CrossRef CAS PubMed.
- Z. Q. Tian, B. Ren, J. F. Li and Z. L. Yang, Chem. Commun., 2007, 3514–3534 RSC.
- H. Y. Liang, H. Wei, D. Pan and H. X. Xu, Nanotechnol. Rev., 2015, 4, 289–302 Search PubMed.
- B. Wiley, Y. G. Sun, B. Mayers and Y. N. Xia, Chem.–Eur. J., 2005, 11, 454–463 CrossRef CAS PubMed.
- H. Y. Liang, W. Z. Wang, Y. Z. Huang, S. P. Zhang, H. Wei and H. X. Xu, J. Phys. Chem. C, 2010, 114, 7427–7431 CrossRef CAS.
- K. Sytwu, M. Vadai, F. Hayee, D. K. Angell, A. Dai, J. Dixon and J. A. Dionne, Science, 2021, 371, 280–283 CrossRef CAS PubMed.
- Y. N. Xia, Y. J. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- E. B. Guidez and C. M. Aikens, J. Phys. Chem. C, 2013, 117, 12325–12336 CrossRef CAS.
- R. A. Sperling, P. Rivera gil, F. Zhang, M. Zanella and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896–1908 RSC.
- X. Li, F. J. Kao, C. C. Chuang and S. L. He, Opt. Express, 2010, 18, 11335–11346 CrossRef CAS PubMed.
- H. F. Wang, T. B. Huff, D. A. Zweifel, W. He, P. S. Low, A. Wei and J. X. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15752–15756 CrossRef CAS PubMed.
- Z. Yin, Y. Wang, C. Q. Song, L. H. Zheng, N. Ma, X. Liu, S. W. Li, L. L. Lin, M. Z. Li, Y. Xu, W. Z. Li, G. Hu, Z. Y. Fang and D. Ma, J. Am. Chem. Soc., 2018, 140, 864–867 CrossRef CAS PubMed.
- D. Y. Wu, X. M. Liu, Y. F. Huang, B. Ren, X. Xu and Z. Q. Tian, J. Phys. Chem. C, 2009, 113, 18212–18222 CrossRef CAS.
- Y. R. Fang, Y. Z. Li, H. X. Xu and M. T. Sun, Langmuir, 2010, 26, 7737–7746 CrossRef CAS PubMed.
- M. T. Sun and H. X. Xu, Small, 2012, 8, 2777–2786 CrossRef CAS PubMed.
- P. Xu, L. L. Kang, N. H. Mack, K. S. Schanze, X. J. Han and H. L. Wang, Sci. Rep., 2013, 3, 2997 CrossRef PubMed.
- Y. F. Huang, M. Zhang, L. B. Zhao, J. M. Feng, D. Y. Wu, B. Ren and Z. Q. Tian, Angew. Chem., Int. Ed., 2014, 53, 2353–2357 CrossRef CAS PubMed.
- U. Aslam, V. G. Rao, S. Chavez and S. Linic, Nat. Catal., 2018, 1, 656–665 CrossRef.
- P. Christopher, H. L. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CrossRef CAS PubMed.
- G. Xu, Y. Sun, Y. Zhang and L. Xia, Spectrochim. Acta A Mol. Biomol. Spectrosc., 2021, 264, 120282 CrossRef PubMed.
- L. B. Zhao, M. Zhang, Y. F. Huang, C. T. Williams, D. Y. Wu, B. Ren and Z. Q. Tian, J. Phys. Chem. Lett., 2014, 5, 1259–1266 CrossRef CAS PubMed.
- M. Zhang, L. B. Zhao, W. L. Luo, R. Pang, C. Zong, J. Z. Zhou, B. Ren, Z. Q. Tian and D. Y. Wu, J. Phys. Chem. C, 2016, 120, 11956–11965 CrossRef CAS.
- H. Z. Wang, Y. Y. Gao, J. Liu, X. Y. Li, M. W. Ji, E. H. Zhang, X. Y. Cheng, M. Xu, J. J. Liu, H. P. Rong, W. X. Chen, F. T. Fan, C. Li and J. T. Zhang, Adv. Energy Mater., 2019, 9, 1803889 CrossRef.
- Z. L. Huang, G. W. Meng, X. Y. Hu, Q. J. Pan, D. X. Huo, H. J. Zhou, Y. Ke and N. Q. Wu, Nano Res., 2019, 12, 449–455 CrossRef CAS.
- Y. Ke, B. Chen, M. Hu, N. Zhou, Z. Huang and G. Meng, Nanomaterials, 2022, 12, 1156 CrossRef CAS PubMed.
- J. T. Huang, W. X. Niu, C. L. Li, C. L. Tan, P. F. Yin, H. F. Cheng, Z. N. Hu, N. L. Yang, Q. Y. He, G. H. Nam and H. Zhang, ACS Mater. Lett., 2020, 2, 409–414 CrossRef CAS.
- J. L. Wang, R. A. Ando and P. H. C. Camargo, Angew. Chem., Int. Ed., 2015, 54, 6909–6912 CrossRef CAS PubMed.
- Y. Sun, Y. Zhang, Y. Wang and L. X. Xia, Microchim. Acta, 2020, 187, 462 CrossRef CAS PubMed.
- Q. Cao, K. P. Yuan, Q. H. Liu, C. Y. Liang, X. Wang, Y. F. Cheng, Q. Q. Li, M. Wang and R. C. Che, ACS Appl. Mater. Interfaces, 2015, 7, 18491–18500 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2na00486k |
| This journal is © The Royal Society of Chemistry 2022 |