
Cl-Initiated oxidation of methacrolein under NOx-free conditions studied by VUV photoionization mass spectrometry†
Xiaoxiao
Lin‡
 a,
Rongrong
Hu‡
a,
Ziji
Ma
a,
Hao
Yue
a,
Zuoying
Wen
a,
Cuihong
Zhang
ab,
Christa
Fittschen
b,
Weijun
Zhang
*a and
Xiaofeng
Tang
*a
a,
Rongrong
Hu‡
a,
Ziji
Ma
a,
Hao
Yue
a,
Zuoying
Wen
a,
Cuihong
Zhang
ab,
Christa
Fittschen
b,
Weijun
Zhang
*a and
Xiaofeng
Tang
*a
aLaboratory of Atmospheric Physico-Chemistry, Anhui Institute of Optics and Fine Mechanics, HFIPS, Chinese Academy of Sciences, Hefei, 230031 Anhui, China. E-mail: wjzhang@aiofm.ac.cn; tangxf@aiofm.ac.cn
bUniversity Lille, CNRS, UMR 8522, PC2A – Physicochimie des Processus de Combustion et de l’Atmosphère, F-59000 Lille, France
First published on 5th July 2022
Abstract
The Cl-initiated oxidation of methacrolein (MACR, C4H6O) under NOx-free conditions has been investigated in a fast flow tube by using a home-made vacuum ultraviolet (VUV) photoionization mass spectrometer complemented by high-level theoretical calculations. The key species such as intermediates and radicals together with products involved in the oxidation are observed online and confirmed in photoionization mass spectra. The reaction potential energy surfaces of the transient C4H5O and C4H6OCl radicals, formed from the hydrogen-abstraction reaction and the addition reaction of MACR with Cl atoms, with oxygen have been theoretically calculated to illuminate the formation of the peroxy radicals of C4H5OO2 and C4H6OClO2. The photoionization processes of these peroxy radicals, whose cations are not stable, and their individual self-reactions as well as bimolecular reactions with HO2 radical are studied and discussed. In addition, kinetic experiments are also performed to get the time evolution of specific products and compared with theoretical models, providing a detailed insight into the reaction mechanism of the Cl-initiated oxidation of MACR.
1. Introduction
Methacrolein (MACR, C4H6O) is a key reaction intermediate in atmospheric chemistry and plays an essential role in the atmospheric oxidation of isoprene, the most abundant non-methane volatile organic compound (VOC) in the atmosphere.1,2 MACR is also an important VOC in the atmosphere and its global emission can approach ∼45 Tg year−1.3 In particular, due to its essential role involved in the oxidation of isoprene, the atmospheric fate of MACR has attracted a great deal of attention in the past decades.1,4The atmospheric fate of MACR is dominated by its reaction with the hydroxyl radical (OH), and mainly proceeds via two pathways with approximately equal rates: (i) the addition of OH to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of MACR to generate the OH–MACR adduct radical, and (ii) the abstraction of the aldehyde H-atom to produce the acyl radical, C4H5O.5,6 In the atmosphere, these radicals can react with O2 and lead to the production of the hydroxyl peroxy radical and the acyl peroxy radical. In addition, these transient peroxy radicals can perform bimolecular reactions with NOx (NO and NO2), with themselves or other peroxy radicals (RO2), with HO2 or OH radicals depending on the site and time, and contribute to air quality and regional climate.7–12
C double bond of MACR to generate the OH–MACR adduct radical, and (ii) the abstraction of the aldehyde H-atom to produce the acyl radical, C4H5O.5,6 In the atmosphere, these radicals can react with O2 and lead to the production of the hydroxyl peroxy radical and the acyl peroxy radical. In addition, these transient peroxy radicals can perform bimolecular reactions with NOx (NO and NO2), with themselves or other peroxy radicals (RO2), with HO2 or OH radicals depending on the site and time, and contribute to air quality and regional climate.7–12
In recent years, researchers found that halogen radicals, particularly chlorine (Cl) atoms formed from photolysis of chlorine (Cl2), nitrosyl chloride (ClNO), nitryl chloride (ClNO2) and bromine chloride (BrCl), can also have important impacts on the atmospheric degradation of VOCs.13–16 Among them, Cl atoms can efficiently oxidize VOCs, and the reaction rate constants of Cl atoms with VOCs can be ten times larger than those with OH.15–18 For example, the reaction rate constant of MACR with Cl atoms was measured at 3.3 × 10−10 cm3 molecule−1 s−1, much larger than that of MACR with OH (3.4 × 10−11 cm3 molecule−1 s−1).19,20 Therefore, considering the concentrations of OH radicals (∼106 molecules cm−3) and Cl atoms (∼3 × 105 molecules cm−3 during daytime and up to ∼8 × 106 molecules cm−3 in the marine boundary layer),21,22 the Cl-initiated oxidation of MACR can play a significant role in the atmosphere next to that initiated with OH, at least in the marine boundary layer, and has been the subject of experimental and theoretical studies.20,23
Similar to the case with the OH radical, the reaction of MACR with Cl atoms can proceed via two pathways too: the addition of Cl atom to the CC bond of MACR to get the C4H6OCl adduct radical, and the hydrogen abstraction to produce the C4H5O radical.19 Previous theoretical calculations show that the Cl-addition to the external carbon of the CC double bond is the main pathway with a predicted branching ratio of 86%, whereas the branching ratio of the aldehyde-H abstraction is 12% and that of the methyl-H abstraction is 2%.23
In the atmosphere, the C4H6OCl and C4H5O radicals will react with O2 and the transient peroxy radicals, C4H6OClO2 and C4H5OO2, can perform various reactions.11 For example, the peroxy radical C4H6OClO2 reacts with itself to form the C4H6OClO radical, and its decomposition product, chloroacetone (C3H5OCl), was detected with the technique of gas chromatography flame ionization detection (GC-FID).24 The stable secondary products 2,3-dichloro-2-methylpropanal (C4H6OCl2) and methacryloyl chloride (C4H5OCl) formed from the reactions of C4H6OCl and C4H5O radicals with the remaining precursor Cl2 were also detected in experiments with GC-FID.25 But, due to the use of online probe techniques to study these reactions, the available information of the Cl-initiated oxidation of MACR is still scarce and the detailed reaction mechanism as well as the formation of the above theoretically proposed key radicals and intermediates needs to be clarified or confirmed.
In this work, the Cl-initiated oxidation reaction of MACR under NOx-free conditions has been investigated in a fast flow tube reactor by using the online analytical method of vacuum ultraviolet (VUV) photoionization time of flight mass spectrometry complemented by high-level theoretical computations of the structures of radicals and the reaction potential energy surfaces. VUV photoionization mass spectrometry is a powerful analytical method and key species such as radicals and intermediates as well as products involved in the oxidation reactions are directly observed in the experiments.26,27 The adiabatic ionization energies (AIEs) of these key species are also predicted and utilized to assign the photoionization mass spectra. In addition, to confirm the origins of the products and then to reveal the embedded reaction mechanism, kinetic experiments have also been performed and the time behaviors of products have been obtained and compared with modeling results.
2. Methods
A fast flow tube has been used as a chemical reactor to study the Cl-initiated oxidation reaction of MACR under NOx-free conditions and a home-made VUV photoionization orthogonal acceleration reflectron time-of-flight (TOF) mass spectrometer is employed to probe and analyze reaction intermediates, radicals and products. The configuration of the fast flow tube and the photoionization mass spectrometer has already been introduced in detail before and so only a brief description is presented here.28,29Briefly, the photoionization mass spectrometer is composed of three vacuum chambers: a source chamber, a photoionization chamber and a TOF chamber. The fast flow tube reactor is installed inside the source chamber and is composed of a 45 cm long Pyrex main tube with a 16/12 mm outer/inner diameter and a 60 cm long coaxial movable injector with a 6/4 mm outer/inner diameter. Cl atoms were generated by microwave discharge (GMS-200W, Sairem) of 1% diluted Cl2 gas in helium and introduced into the fast flow tube to initiate the oxidation reactions. MACR was introduced through bubbling its liquid at room temperature, and O2 and helium bath gas were injected via the arms of the main tube or the injector. The total pressure inside the flow tube was monitored by a capacity gauge and fixed at 6 Torr by a closed-loop feedback throttle valve. The initial concentrations of MACR, O2 and Cl atoms in the flow tube were 8 × 1013, 1 × 1016 and ∼1 × 1013 molecule cm−3, respectively. Note that the microwave discharge efficiency is not precisely known and so only an estimated concentration of Cl atoms is provided here. The inner surface of the main tube and the outer surface of the injector were coated with halocarbon wax to reduce radical loss on the walls. The reaction time was adjusted by changing the distance between the injector and the sampling skimmer.
After passing through the skimmer (1 mm diameter), the gas mixture from the fast flow tube entered into the photoionization chamber, which was equipped with a commercial Kr discharge lamp (PKS 106, Heraeus) inside. Then the molecules and radicals absorbed the photons of the lamp with energies of hν = 10.0 and 10.6 eV, and photoionization processes were induced. A TOF mass analyzer with an orthogonal acceleration and reflectron structure was employed to analyze the masses of ions. The total ion flight length of the TOF mass spectrometer is ∼1 m, and its mass resolving power has been measured to be M/ΔM ∼ 2000 (FWHM, the full width at half maximum). During the experiments the pressures of the source chamber, the photoionization chamber and the TOF chamber were 800, 1 × 10−2 and 1 × 10−4 Pa, respectively.
High-level theoretical calculations have also been carried out to get the reaction potential energy surfaces, the structures and the ionization energies of radicals and products. Concretely, the potential energy surfaces were calculated for the reactions of C4H6OCl and C4H5O radicals with O2 to examine the formation channels leading to the peroxy radicals and other products. The potential energy surfaces were computed at the CCSD(T)-F12a/aug-cc-pVTZ//PBE0/aug-cc-pVDZ level of theory. The structural optimizations and the vibrational frequency calculations were performed with the Gaussian 16 program package.30 Then the energies of the reactants, the transition states and the products were calculated with the Molpro package.31 The AIEs of the products were determined from the energy difference of cations and molecules at the PBE0/aug-cc-pVDZ level of theory.
3. Results and discussion
To get detailed information on the reaction system inside the fast flow tube, experiments were performed in sequence and can be divided into two parts.32,33 Firstly, the experiment was performed without adding oxygen into the fast flow tube, and C4H6OCl and C4H5O radicals were formed from the Cl-addition reaction and the hydrogen abstraction reaction of MACR with Cl atoms, respectively. Then, with the addition of oxygen, the transient C4H6OCl and C4H5O radicals reacted with oxygen to produce peroxy radicals and these peroxy radicals could perform self-reactions or react with HO2 radials in the flow tube under NOx-free conditions.113.1 The reaction of MACR with Cl
Without adding oxygen into the fast flow tube, a typical photoionization mass spectrum is presented in Fig. 1(a). Many peaks can be observed in the mass spectrum and the major species have been assigned. The most intense peak at m/z = 70 is ascribed to the reactant MACR itself, whose ionization energy locates at 9.92 eV,34 below the photon energy of the Kr lamp. The mass peaks at m/z = 69 and m/z = 105/107 should be assigned as C4H5O and C4H6OCl radicals, respectively, formed from the hydrogen abstraction reaction and the addition reaction of MACR with Cl atoms.19,23 As far as we known, the transient C4H5O and C4H6OCl radicals are directly detected here for the first time. The ratio of the mass peaks at m/z = 105 and 107 corresponding to the C4H6O35Cl and C4H6O37Cl isotopes is measured at ∼3, agreeing well with the ratio of the natural abundances of the 35Cl and 37Cl isotopes (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :32).35
:32).35
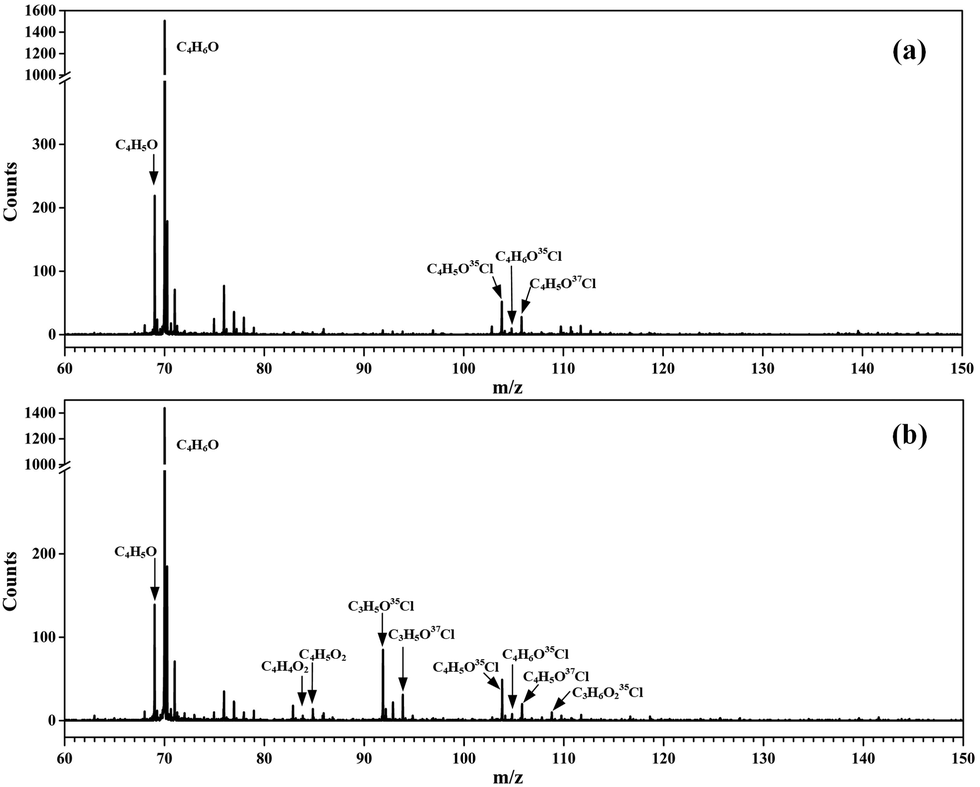 | ||
| Fig. 1 Photoionization mass spectra acquired (a) without O2 and (b) adding O2 into the fast flow tube. | ||
The transient C4H5O and C4H6OCl radicals can react with the residual Cl2 molecules in the fast flow tube and the corresponding products have been observed and identified in the mass spectrum, partially with the aid of the isotopic ratio of 35Cl and 37Cl. For example, the mass peaks at m/z = 104 and 106 are assigned as methacryloyl chloride (C4H5O35Cl and C4H5O37Cl), the product of the reaction of C4H5O radicals with Cl2.25 The small peaks at m/z = 140, 142 and 144 are assigned as 2,3-dichloro-2-methylpropanal (C4H6OCl2) from the reaction of C4H6OCl radicals with Cl2.25 Some products are fragile in the photoionization and their fragment ions have also contributed to the mass spectrum, i.e., at m/z = 76, 78 and 111. The detailed assignment of these peaks and their origins can be found in Table S1 (ESI†).
3.2 The reactions of C4H5O and C4H6OCl with O2
After adding abundant O2 (∼1016 molecule cm−3) into the fast flow tube, the transient C4H5O and C4H6OCl radicals will mainly react with O2 and the resulting photoionization mass spectrum is presented in Fig. 1(b). To get a detailed understanding of these processes, the potential energy surfaces (PES) of the reactions of C4H5O and C4H6OCl radicals with O2 have been calculated at the CCSD(T)-F12a/aug-cc-pVTZ//PBE0/aug-cc-pVDZ level of theory and are presented in Fig. 2 and 3, respectively, as well as the optimized structures of the species involved in the reactions.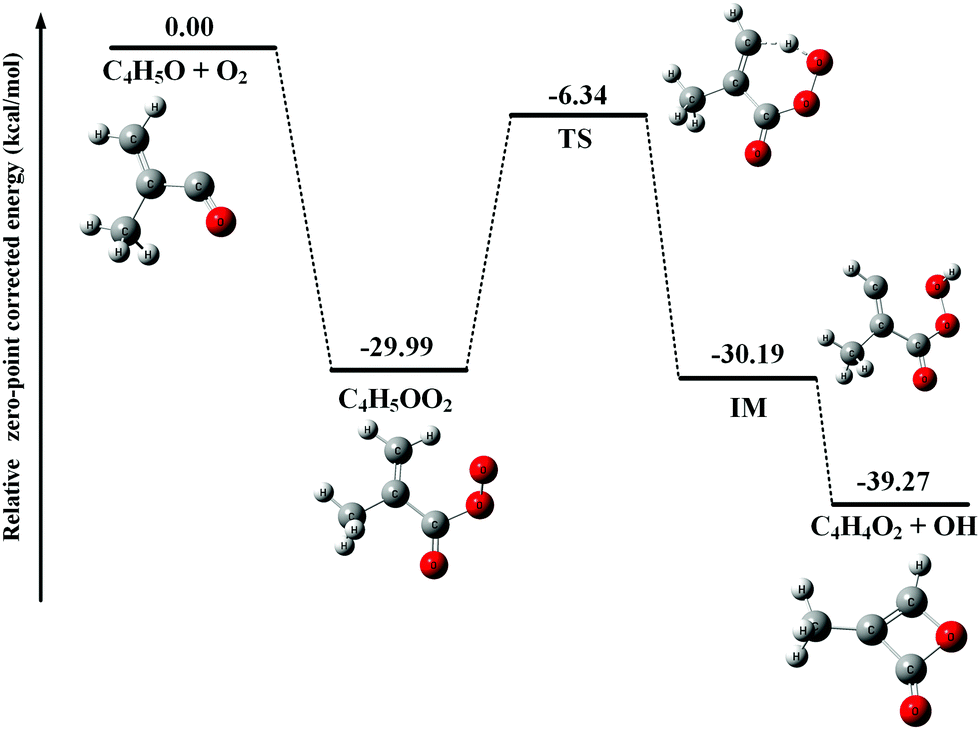 | ||
| Fig. 2 Potential energy surface of the reaction of C4H5O with O2. | ||
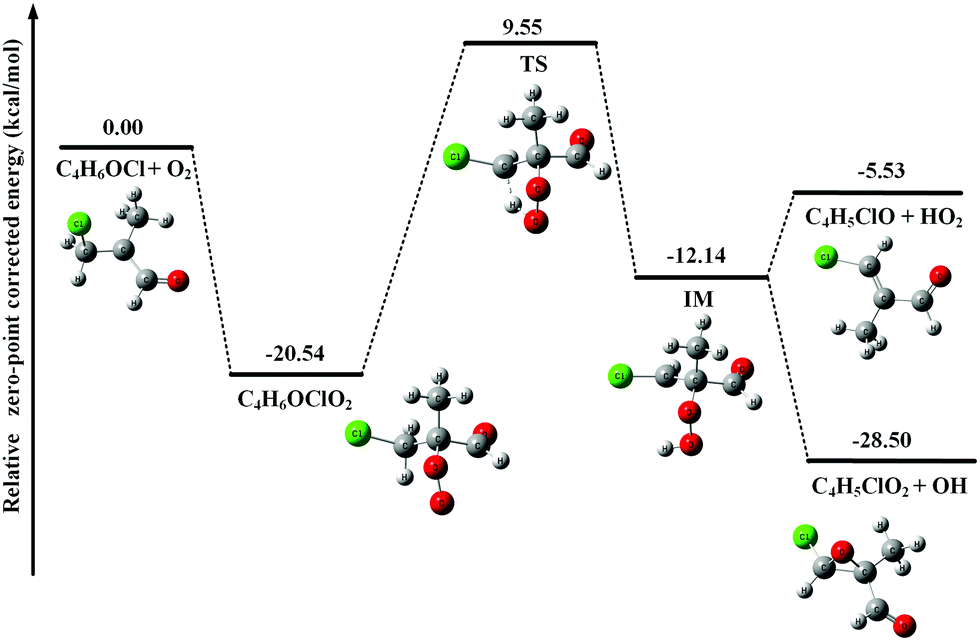 | ||
| Fig. 3 Potential energy surface of the reaction of C4H6OCl with O2. | ||
As shown in Fig. 2, the reaction of C4H5O with O2 is an exothermic reaction and the production of the C4H5OO2 peroxy radical is energetically favorable. In addition, the theoretical calculations show that the C4H5OO2 peroxy radical can perform an H-transfer reaction via a transition state (TS) with a submerged barrier of −6.34 kcal mol−1 and then dissociate to C4H4O2 and OH. The formation of C4H4O2 and OH is exothermic with an energy of −39.27 kcal mol−1, indicating that their formation is energetically feasible. As shown in Fig. 1(b), the product C4H4O2 has been observed and ascribes to the peak of m/z = 84 in the photoionization mass spectrum.
In Fig. 3, the potential energy surface of the reaction of C4H6OCl with O2 shows that the formation of the C4H6OClO2 peroxy radical is also energetically favorable. But, unlike the above case of C4H5OO2, the unimolecular reaction of the C4H6OClO2 peroxy radical to produce C4H5ClO + HO2 and C4H5ClO2 + OH has an energy barrier with a height of 30.09 kcal mol−1, 9.55 kcal mol−1 higher than the total energy of C4H6OCl with O2, which is too high to be overcome at room temperature. Thus the C4H6OClO2 peroxy radical is the only product in the reaction of C4H6OCl with O2.
As discussed above, the C4H5OO2 and C4H6OClO2 peroxy radicals are the main products of the reactions of C4H5O and C4H6OCl radicals with O2 and their corresponding molecular ions should locate at m/z = 101 and m/z = 137/139, respectively, in the photoionization mass spectrum. However, no such mass peaks can be observed in Fig. 1(b).
Previous studies have shown that except the smallest peroxy radical, CH3O2, the cations of most alkyl peroxy radicals are not stable and dissociate to the fragments of alkyl cations and oxygen following the photoionization.32,33,36 Here, to help to assign the photoionization mass spectrum and to explain the dissociative photoionization processes of the C4H5OO2 and C4H6OClO2 peroxy radicals, theoretical calculations have also been performed. Fig. 4 presents the optimized structures of the C4H5OO2 and C4H6OClO2 peroxy radicals and their cations in the ground electronic state. The ground electronic state of these cations is a triplet state after removing an electron from the σ(Cα-OO) orbital of the C4H5OO2 and C4H6OClO2 peroxy radicals. To be specific, the C–OO equilibrium bond length of the neutral C4H6OClO2 is calculated at 1.450 Å, whereas it is substantially elongated to 2.781 Å for the cationic ground electronic state of C4H6OClO2+. Similarly, the C–OO bond length of C4H5OO2+ at the ground electronic state is also elongated from 1.426 to 3.263 Å during photoionization.27,33
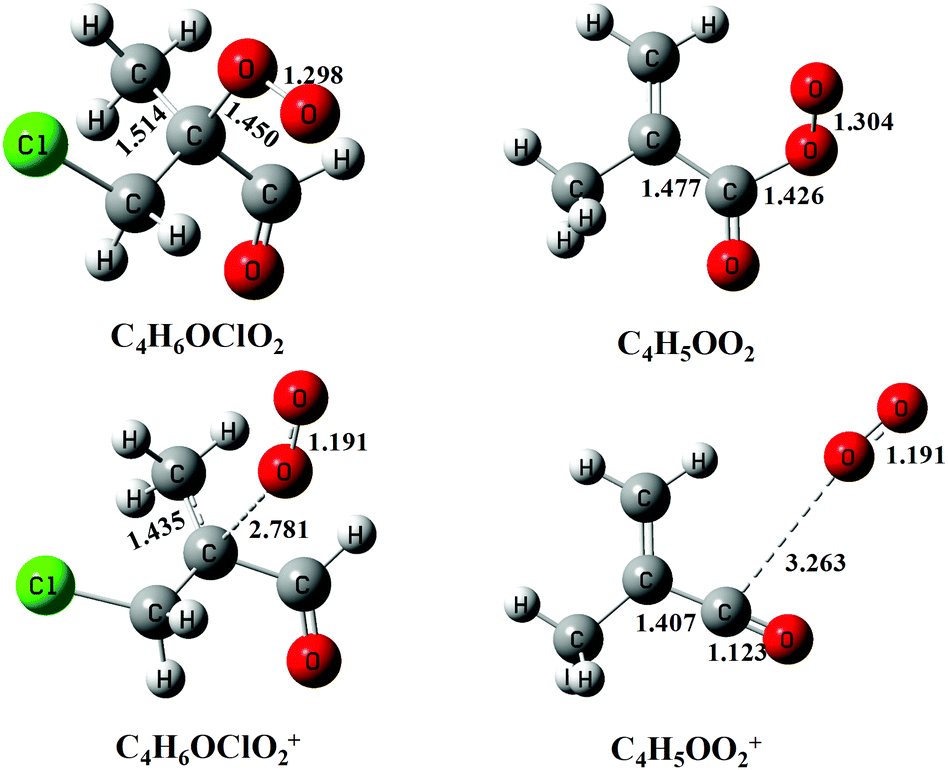 | ||
| Fig. 4 The structures of the C4H6OClO2 and C4H5OO2 peroxy radicals and their cations optimized at the PBE0/aug-cc-pVDZ level of theory. The bond lengths are in Å. | ||
These apparent elongations of the C–OO bond length will reduce the stability of the peroxy radicals’ cations, also making C4H5OO2+ and C4H6OClO2+ only slightly bound and out of the Franck–Condon transition during photoionization.27,33 In addition, the AIEs of the C4H5OO2 and C4H6OClO2 peroxy radicals are calculated and take the values of 7.93 and 9.23 eV, respectively, far below the present photon energy of 10.6 eV. The appearance energies (AEs) of the fragment ions C4H5O+ (m/z = 69) and C4H6OCl+ (m/z = 105/107) in the dissociation of C4H5OO2+ and C4H6OClO2+ are also calculated and locate at 8.00 and 9.08 eV. Therefore, in the photoionization the C4H5OO2+ and C4H6OClO2+ cations will be produced with a high internal energy and dissociate to the C4H5O+ + O2 and C4H6OCl+ + O2 fragments, respectively. Thus, the mass peak at m/z = 69 (C4H5O+) in Fig. 1(b) is attributed to C4H5OO2, and the mass peaks at m/z = 105/107 (C4H6OCl+) is ascribed to the C4H6OClO2 peroxy radical.
3.3 The reactions of C4H5OO2 and C4H6OClO2 under NOx free conditions
Under NOx free conditions in the fast flow tube, the C4H5OO2 and C4H6OClO2 peroxy radicals can perform self-reaction or react with HO2 radicals.11 Normally, the self-reaction of peroxy radicals (RO2) has two channels, (i) 2RO2 → 2RO + O2 and (ii) 2RO2 → R–HO + ROH + O2.37 But, based on the specific structures of C4H5OO2 and C4H6OClO2, as shown in Fig. 4, there are no H atoms on the tertiary α-oxyl carbon to be abstracted and thus the self-reactions of C4H5OO2 and C4H6OClO2 do not proceed via channel (ii).38Normally the reaction of the alkoxy radical RO with O2 is one of the major sources to produce HO2 radicals. But, the reaction of C4H5OO with O2 will not produce HO2, as still there is no H atoms on the tertiary α-oxyl carbon to be abstracted. A previous study shows that the reaction of C4H6OClO with O2 can generate HO2, via more than one elementary reaction step,39 and presently this reaction should be the main source of HO2 in the fast flow tube. Note that the AIE of HO2 locates at 11.359 eV, above the present photon energy, and thus no HO2 radicals can be observed in the mass spectra.40
A previous study predicts that the reaction of the C4H5OO2 peroxy radical with HO2 has three channels, (R1a) C4H5OO2 + HO2 → C4H5OO2H + O2, (R1b) C4H5OO2 + HO2 → C4H5OOH + O3, and (R1c) C4H5OO2 + HO2 → C4H5OO + OH + O2.39 The reaction of C4H6OClO2 with HO2 has two channels, (R2a) C4H6OClO2 + HO2 → C4H6OClOOH + O2, (R2b) C4H6OClO2 + HO2 → C4H6OClO + OH + O2, and the branching ratio was predicted at YR2a = 0.2 ± 0.2 and YR2b = 0.8 ± 0.2.39
The alkoxy radical C4H6OClO formed from the self-reaction of C4H6OClO2 or its bimolecular reaction with HO2 can decompose to the stable product of chloroacetone (C3H5O35Cl and C3H5O37Cl) with a fast rate of ∼107 s−1 and has been completely consumed in the fast flow tube within the reaction time of ∼1 ms under the present experimental conditions.39 Therefore, the signal of C4H6OClO (m/z = 121) is not observed in the photoionization mass spectrum of Fig. 1(b), and its decomposition product of chloroacetone (C3H5OCl) contributes to the mass peaks of m/z = 92 and 94 with a ratio of 3:1.
The reaction of C4H6OClO2 with HO2 can produce the hydroperoxide C4H6OClOOH (m/z = 138, 140). The AIE of C4H6OClOOH is calculated at 9.30 eV at the PBE0/aug-cc-pVDZ level of theory, and the appearance energy of its fragment ion C3H5ClO2H+ is predicted at 9.97 eV. Thus in the photoionization the hydroperoxide cation C4H6OClOOH+ is produced with a high internal energy and dissociates into the fragment ion of C3H5ClOOH+, plus the neutral fragment of CHO, contributing to the peaks of m/z = 109 and 111 in the photoionization mass spectrum of Fig. 1(b).
The peroxy radical of C4H5OO2 performs self-reaction and reacts with HO2 to generate the radical C4H5OO, contributing to the mass peak of m/z = 85 in Fig. 1(b). The reaction of C4H5OO2 with HO2 can also produce C4H5OOH (m/z = 86) and C4H5OO2H (m/z = 102). The AIE of C4H5OO2H is calculated at 9.44 eV at the PBE0/aug-cc-pVDZ level of theory. In the photoionization, the C4H5OO2H+ cation was produced with a high internal energy and then dissociated into the C4H5O+ (m/z = 69) and HO2 fragments.
3.4 Reaction mechanism
Kinetic experiments have been performed for the Cl-initiated oxidation reaction of MACR by changing the distance between the injector and the sampling skimmer, to get information inside the fast flow tube and to confirm the above species’ assignments.28,29 The time evolution of ion signals has been measured and some of them, i.e. m/z = 84, 85 and 92 corresponding to C4H4O2, C4H5OO and C3H5OCl, are presented in Fig. 5. In addition, the time evolutions of the species’ concentrations have been theoretically calculated based on the oxidation reactions listed in Table 1 and are presented as solid lines, also seen in Fig. S1 and S2 (ESI†).41,42 Note that the experimental concentration equivalents are less well-defined, as some unknown and conversion factors such as photoionization cross-sections add to the measurement uncertainty. The theoretical results also rely on a number of assumptions, for instance, the initial concentration of Cl atoms and the unknown or estimated rate constants. As shown in Fig. 5, although with some differences, the overall shape of the theoretical modeling results can compare with the time evolution of the ion signals.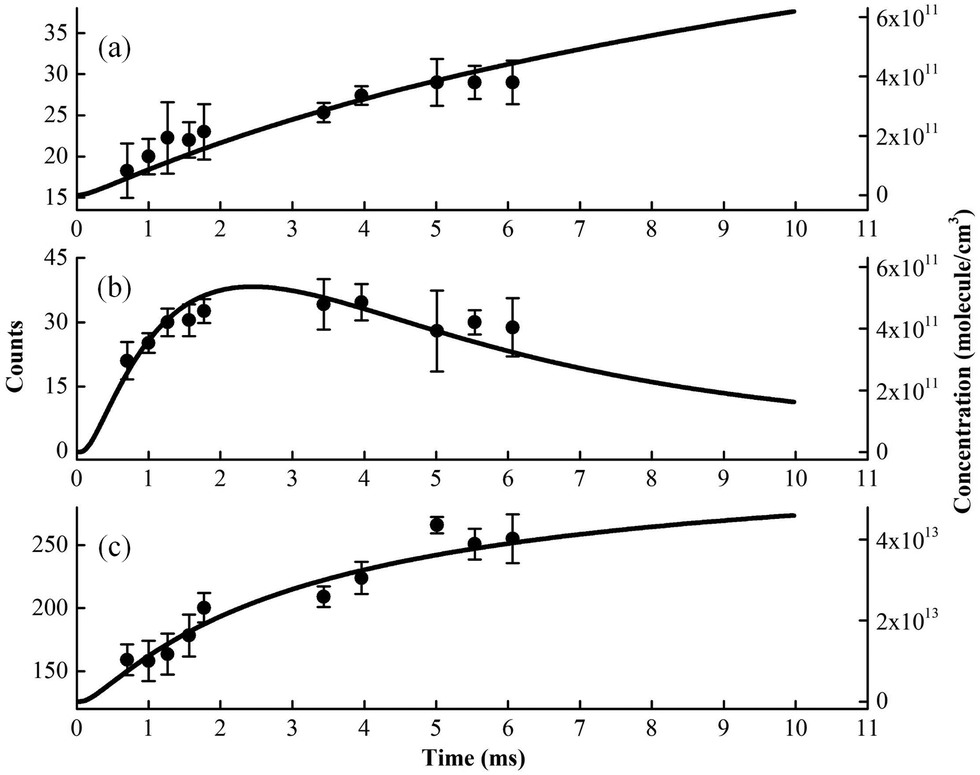 | ||
| Fig. 5 The experimental (dots) and theoretical (lines) time behavior of products in the Cl-initiated oxidation of MACR. (a) m/z = 84, (b) m/z = 85 and (c) m/z = 92. | ||
| Reaction | Rate coefficient/cm3 molecule−1 s−1 or s−1 | Ref. | |
|---|---|---|---|
| a Analogic result from ref. 45. b The reaction consists of more than one elementary reaction step, seen the detail in ref. 39. | |||
| 1 | C4H6O + Cl → C4H6OCl | 1.98 × 10−10 | 23 |
| 2 | C4H6O + Cl → C4H5O + HCl | 2.76 × 10−11 | 23 |
| 3 | C4H6OCl + O2 → C4H6OClO2 | 2.87 × 10−12 | 43 |
| 4 | C4H5O + O2 → C4H5OO2 | 2.0 × 10−12 | 44 |
| 5 | C4H5OO2 → C4H4O2 + OH | 12.9a | 45 |
| 6 | 2C4H6OClO2 → 2C4H6OClO + O2 | 2.4 × 10−12 | 39 |
| 7 | 2C4H5OO2 → 2C4H5OO + O2 | 1.0 × 10−11 | 39 |
| 8(a) | C4H6OClO2 + HO2 → C4H6OClO2H + O2 | 2.0 × 10−12 | 39 |
| 8(b) | C4H6OClO2 + HO2 → C4H6OClO + OH + O2 | 8.0 × 10−12 | 39 |
| 9(a) | C4H5OO2 + HO2 → C4H5OO2H + O2 | 7.7 × 10−12 | 39 |
| 9(b) | C4H5OO2 + HO2 → C4H5OOH + O3 | 5.5 × 10−12 | 39 |
| 9(c) | C4H5OO2 + HO2 → C4H5OO + OH + O2 | 8.8 × 10−12 | 39 |
| 10 | C4H6OClO (+O2) → C3H5OCl + HO2 + CO | 1 × 107b |
39 |
| 11 | C4H5OO → C3H5 + CO2 | 7 × 102 | 46 |
| 12 | 2HO2 → H2O2 + O2 | 1.7 × 10−12 | 47 |
| 13 | HO2 → diffusion | 3 | 41 |
We can see that the ion signal of m/z = 84 increases with time within the experimentally covered time region, which is consistent with the theoretically modeled time behavior of C4H4O2 in Fig. 5(a). The signal intensity at m/z = 85 firstly increases and then decreases with time, which is a typical radical behavior of C4H5OO, as shown in Fig. 5(b). The signal of m/z = 92 firstly increases and then stabilizes, which is in agreement with the expected time behavior of chloroacetone. The detailed reaction mechanism of the Cl-initiated oxidation of MACR under NOx free conditions has been summarized in Fig. 6. Note that Fig. 6 only presents the two dominant entrance channels for the reaction of MACR with Cl atoms, the terminal addition of the CC double bond and the aldehyde-H abstraction with their branching ratios of about 86% and 12%, respectively.39 The branching ratio of the minor methyl-H abstraction channel is predicted at only 2%, not shown in Fig. 6, and this isomeric production channel is difficult to be separated only with the fixed-photon-energy photoionization mass spectrometry.
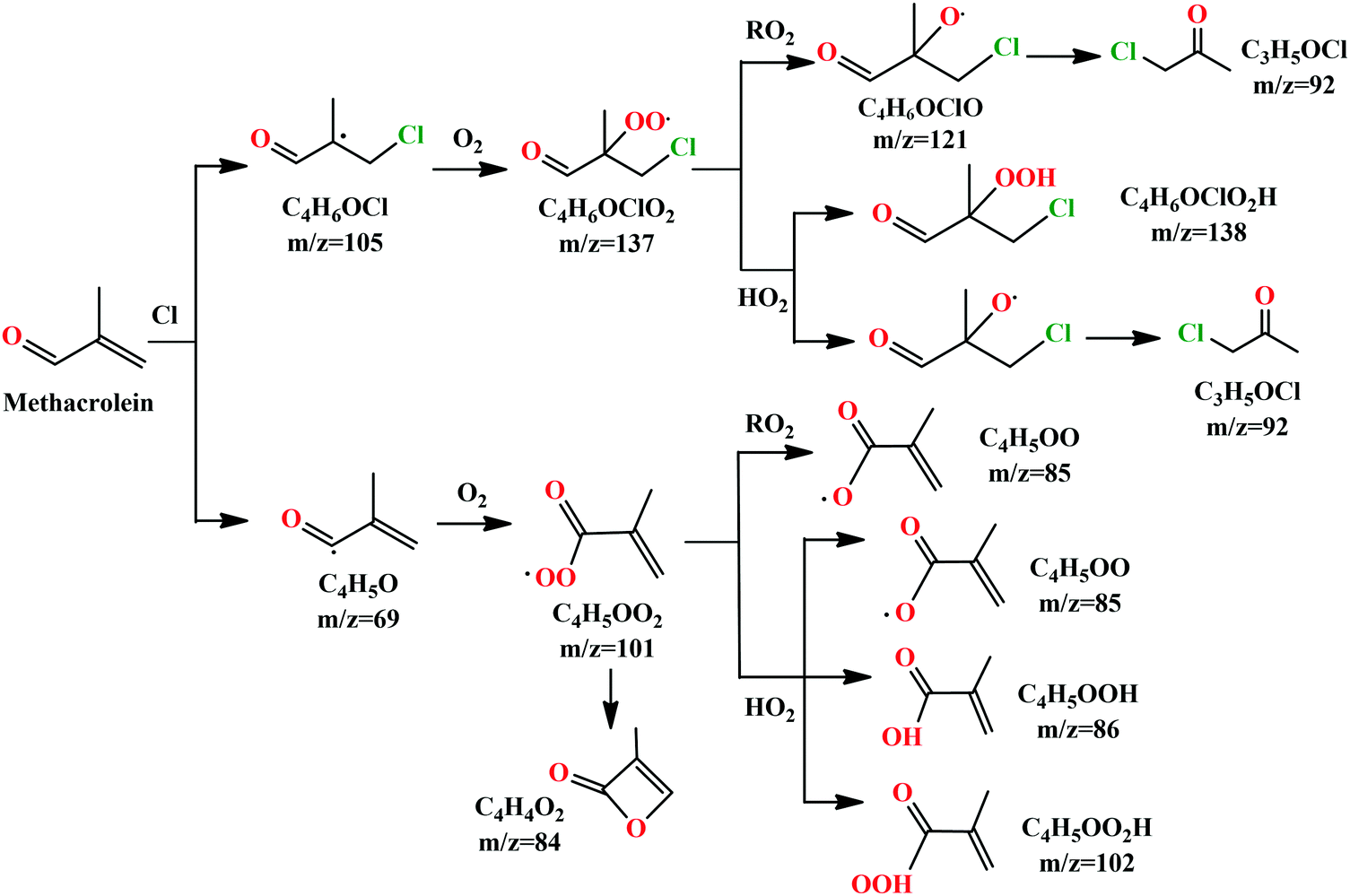 | ||
| Fig. 6 Reaction mechanisms of Cl-initiated oxidation of MACR under NOx free conditions. | ||
4. Conclusions
In summary, we present here a combined experimental and theoretical study on the Cl-initiated oxidation of MACR under NOx-free conditions. A microwave discharge fast flow tube was used to investigate the oxidation reaction of MACR. The reaction products and the key intermediates as well as radicals have been observed online by using a home-made VUV photoionization mass spectrometer. It is shown that the reaction of MACR with Cl atoms can proceed via two pathways, the addition of Cl atom to the CC double bond of MACR to produce the C4H6OCl adduct radical and the abstraction of the aldehyde hydrogen atom to produce the C4H5O radical. These radicals can react with oxygen to produce their corresponding peroxy radicals, whose photoionization processes have also been discussed and utilized to assign the photoionization mass spectra. The transient C4H5O and C4H6OCl radicals, as well as the peroxy radicals C4H5OO2 and C4H6OClO2, are experimentally detected here for the first time, directly confirming the above reaction mechanisms. Under NOx-free conditions, these peroxy radicals will perform self-reaction and react with HO2 radicals. The resulting specific products are clearly identified in the photoionization mass spectra too, with the aid of theoretical calculations. In addition, the time evolutions of products have been measured in the kinetic experiments and compared with theoretically modeled results. The present work provides a detailed insight into the reaction mechanisms of the Cl-initiated oxidation of MACR and will be helpful to understand the atmospheric fate of MACR.
Author contributions
Xiaoxiao Lin: conceptualization, formal analysis, investigation, writing – original draft. Rongrong Hu: investigation, writing – original draft. Ziji Ma: investigation. Hao Yue: investigation. Zuoying Wen: investigation. Cuihong Zhang: investigation. Christa Fittschen: writing – review & editing. Weijun Zhang: investigation. Xiaofeng Tang: conceptualization, methodology, investigation, funding acquisition, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 42075113, 91961123, 42120104007), the International Partnership Program of Chinese Academy of Sciences (No. 116134KYSB20170048) and the Key Program of Research and Development of Hefei Science Center, CAS (No. 2020HSCKPRD001).References
- P. O. Wennberg, K. H. Bates, J. D. Crounse, L. G. Dodson and R. C. McVay, et al., Gas-phase reactions of isoprene and its major oxidation products, Chem. Rev., 2018, 118, 3337–3390 CrossRef CAS PubMed.
- X. Wang, J. Sun, L. Bao, Q. Mei and B. Wei, et al., Mechanisms and kinetic parameters for the gas-phase reactions of 3-methyl-3-buten-2-one and 3-methyl-3-penten-2-one with ozone, J. Phys. Chem. A, 2019, 123, 2745–2755 CrossRef CAS PubMed.
- J. D. Crounse, F. Paulot, H. G. Kjaergaard and P. O. Wennberg, Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 2011, 13, 13607–13613 RSC.
- T. Gierczak, J. B. Burkholder, R. K. Talukdar, A. Mellouki and S. Barone, et al., Atmospheric fate of methyl vinyl ketone and methacrolein, J. Photochem. Photobiol., A, 1997, 110, 1–10 CrossRef CAS.
- H. G. Kjaergaard, H. C. Knap, K. B. Ørnsø, S. Jørgensen and J. D. Crounse, et al., Atmospheric fate of methacrolein. 2. Formation of lactone and implications for organic aerosol production, J. Phys. Chem. A, 2012, 116, 5763–5768 CrossRef CAS PubMed.
- J. J. Orlando, G. S. Tyndall and S. E. Paulson, Mechanism of the OH-initiated oxidation of methacrolein, Geophys. Res. Lett., 1999, 26, 2191–2194 CrossRef CAS.
- M. Claeys, W. Wang, A. C. Ion, I. Kourtchev and A. Gelencsér, et al., Formation of secondary organic aerosols from isoprene and its gas-phase oxidation products through reaction with hydrogen peroxide, Atmos. Environ., 2004, 38, 4093–4098 CrossRef CAS.
- Z. Chen, H. Wang, L. Zhu, C. Wang and C. Jie, et al., Aqueous-phase ozonolysis of methacrolein and methyl vinyl ketone: A potentially important source of atmospheric aqueous oxidants, Atmos. Chem. Phys., 2008, 8, 2255–2265 CrossRef CAS.
- Y. Liu, F. Siekmann, P. Renard, A. El Zein and G. Salque, et al., Oligomer and SOA formation through aqueous phase photooxidation of methacrolein and methyl vinyl ketone, Atmos. Environ., 2012, 49, 123–129 CrossRef CAS.
- X. Zhang, Z. Chen and Y. Zhao, Laboratory simulation for the aqueous OH-oxidation of methyl vinyl ketone and methacrolein: Significance to the in-cloud SOA production, Atmos. Chem. Phys., 2010, 10, 9551–9561 CrossRef CAS.
- J. J. Orlando and G. S. Tyndall, Laboratory studies of organic peroxy radical chemistry: An overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC.
- C. Fittschen, The reaction of peroxy radicals with OH radicals, Chem. Phys. Lett., 2019, 725, 102–108 CrossRef CAS.
- T. Sherwen, M. J. Evans, R. Sommariva, L. D. Hollis and S. M. Ball, et al., Effects of halogens on European air-quality, Faraday Discuss., 2017, 200, 75–100 RSC.
- W. R. Simpson, S. S. Brown, A. Saiz-Lopez, J. A. Thornton and R. von Glasow, Tropospheric halogen chemistry: Sources, cycling, and impacts, Chem. Rev., 2015, 115, 4035–4062 CrossRef CAS PubMed.
- R. Gao, L. Zhu, Q. Zhang and W. Wang, Atmospheric oxidation mechanism and kinetic studies for OH and NO3 radical-initiated reaction of methyl methacrylate, Int. J. Mol. Sci., 2014, 15, 5032–5044 CrossRef PubMed.
- M. Lawler, R. Sander, L. Carpenter, J. Lee and R. v Glasow, et al., HOCl and Cl2 observations in marine air, Atmos. Chem. Phys., 2011, 11, 7617–7628 CrossRef CAS.
- Y. Sun, Q. Zhang, J. Hu, J. Chen and W. Wang, Theoretical study for OH radical-initiated atmospheric oxidation of ethyl acrylate, Chemosphere, 2015, 119, 626–633 CrossRef CAS PubMed.
- X. Liu, H. Qu, L. G. Huey, Y. Wang and S. Sjostedt, et al., High levels of daytime molecular chlorine and nitryl chloride at a rural site on the north China plain, Environ. Sci. Technol., 2017, 51, 9588–9595 CrossRef CAS PubMed.
- C. E. Canosa-Mas, E. S. N. Cotter, J. Duffy, K. C. Thompson and R. P. Wayne, The reactions of atomic chlorine with acrolein, methacrolein and methyl vinyl ketone, Phys. Chem. Chem. Phys., 2001, 3, 3075–3084 RSC.
- W. H. Wang, M. J. Ezell, A. A. Ezell, G. Soskin and B. J. Finlayson, Rate constants for the reactions of chlorine atoms with a series of unsaturated aldehydes and ketones at 298 K: Structure and reactivity, Phys. Chem. Chem. Phys., 2002, 4, 1824–1831 RSC.
- C.-T. Chang, T.-H. Liu and F.-T. Jeng, Atmospheric concentrations of the Cl atom, CIO radical, and HO radical in the coastal marine boundary layer, Environ. Res., 2004, 94, 67–74 CrossRef CAS PubMed.
- C. Mallik, L. Tomsche, E. Bourtsoukidis, J. N. Crowley and B. Derstroff, et al., Oxidation processes in the eastern Mediterranean atmosphere: Evidence from the modelling of HOx measurements over Cyprus, Atmos. Chem. Phys., 2018, 18, 10825–10847 CrossRef CAS.
- C. Sun, B. Xu and S. Zhang, Atmospheric reaction of Cl+ methacrolein: A theoretical study on the mechanism, and pressure-and temperature-dependent rate constants, J. Phys. Chem. A, 2014, 118, 3541–3551 CrossRef CAS PubMed.
- J. J. Orlando, G. S. Tyndall, E. C. Apel, D. D. Riemer and S. E. Paulson, Rate coefficients and mechanisms of the reaction of Cl-atoms with a series of unsaturated hydrocarbons under atmospheric conditions, Int. J. Chem. Kinet., 2003, 35, 334–353 CrossRef CAS.
- E. W. Kaiser, I. R. Pala and T. J. Wallington, Kinetics and mechanism of the reaction of methacrolein with chlorine atoms in 1–950 torr of N2 or N2/O2 diluent at 297 K, J. Phys. Chem. A, 2010, 114, 6850–6860 CrossRef CAS PubMed.
- X. Lin, X. Tang, Z. Wen, B. Long and C. Fittschen, et al., Direct observation of the particle-phase bicyclic products from OH-initiated oxidation of 1,3,5-trimethylbenzene under NOx-free conditions, Atmos. Environ., 2022, 271, 118914 CrossRef CAS.
- X. Tang, X. Lin, G. A. Garcia, J.-C. Loison and Z. Gouid, et al., Identifying isomers of peroxy radicals in the gas phase: 1-C3H7O2vs. 2-C3H7O2., Chem. Commun., 2020, 56, 15525–15528 RSC.
- Z. Wen, X. Tang, C. Fittschen, C. Zhang and T. Wang, et al., Online analysis of gas-phase radical reactions using vacuum ultraviolet lamp photoionization and time-of-flight mass spectrometry, Rev. Sci. Instrum., 2020, 91, 043201 CrossRef CAS PubMed.
- Z. Wen, X. Tang, C. Wang, C. Fittschen and T. Wang, et al., A vacuum ultraviolet photoionization time-of-flight mass spectrometer with high sensitivity for study of gas-phase radical reaction in a flow tube, Int. J. Chem. Kinet., 2019, 51, 178–188 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria and M. Robb, et al., Gaussian 16 revision a.03. 2016, Gaussian Inc., 2016 Search PubMed.
- H. J. Werner and P. J. Knowles, Getting started with Molpro version 2015.1, 2015 Search PubMed.
- X. Tang, X. Gu, X. Lin, W. Zhang and G. A. Garcia, et al., Vacuum ultraviolet photodynamics of the methyl peroxy radical studied by double imaging photoelectron photoion coincidences, J. Chem. Phys., 2020, 152, 104301 CrossRef CAS PubMed.
- Z. Wen, X. Lin, X. Tang, B. Long and C. Wang, et al., Vacuum ultraviolet photochemistry of the conformers of the ethyl peroxy radical, Phys. Chem. Chem. Phys., 2021, 23, 22096–22102 RSC.
- NIST Chemistry WebBook, 2022, https://webbook.nist.gov/chemistry/.
- K. J. R. Rosman and P. D. P. Taylor, Isotopic compositions of the elements 1997, J. Anal. At. Spectrom., 1999, 14, 5N–24N Search PubMed.
- G. Meloni, P. Zou, S. J. Klippenstein, M. Ahmed and S. R. Leone, et al., Energy-resolved photoionization of alkylperoxy radicals and the stability of their cations, J. Am. Chem. Soc., 2006, 128, 13559–13567 CrossRef CAS PubMed.
- D. M. Rowley, P. D. Lightfoot, R. Lesclaux and T. J. Wallington, Ultraviolet absorption spectrum and self-reaction of cyclopentylperoxy radicals, J. Chem. Soc., Faraday Trans., 1992, 88, 1369–1376 RSC.
- G. Hasan, R. R. Valiev, V.-T. Salo and T. Kurtén, Computational investigation of the formation of peroxide (ROOR) accretion products in the OH- and NO3-initiated oxidation of α-pinene, J. Phys. Chem. A, 2021, 125, 10632–10639 CAS.
- A. S. Hasson, G. S. Tyndall, J. J. Orlando, S. Singh and S. Q. Hernandez, et al., Branching ratios for the reaction of selected carbonyl-containing peroxy radicals with hydroperoxy radicals, J. Phys. Chem. A, 2012, 116, 6264–6281 CrossRef CAS PubMed.
- X. Tang, X. Lin, G. A. Garcia, J.-C. Loison and C. Fittschen, et al., Threshold photoelectron spectroscopy of the HO2 radical, J. Chem. Phys., 2020, 153, 124306 CrossRef CAS PubMed.
- C. Zhang, M. Shamas, M. Assali, X. Tang and W. Zhang,
et al., Absolute absorption cross-section of the à ←
![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) electronic transition of the ethyl peroxy radical and rate constant of its cross reaction with HO2, Photonics, 2021, 8, 296 CrossRef CAS.
electronic transition of the ethyl peroxy radical and rate constant of its cross reaction with HO2, Photonics, 2021, 8, 296 CrossRef CAS. - M. Shamas, M. Assali, C. Zhang, X. Tang and W. Zhang, et al., Rate constant and branching ratio for the reactions of the ethyl peroxy radical with ttself and with the ethoxy radical, ACS Earth Space Chem., 2021, 6, 181–188 CrossRef.
- J. D. DeSain, S. J. Klippenstein, J. A. Miller and C. A. Taatjes, Measurements, theory, and modeling of OH formation in ethyl + O2 and propyl + O2 reactions, J. Phys. Chem. A, 2003, 107, 4415–4427 CrossRef CAS.
- C. E. Mcdade, T. M. Lenhardt and K. D. Bayers, The rate of reaction of acetyl and benzoyl radicals with O2, J. Photochem., 1982, 20, 1–7 CrossRef CAS.
- F. Zhang and T. S. Dibble, Effects of olefin group and its position on the kinetics for intramolecular H-shift and HO2 elimination of alkenyl peroxy radicals, J. Phys. Chem. A, 2011, 115, 655–663 CrossRef CAS PubMed.
- H. Ning, J. Wu, L. Ma, W. Ren and D. F. Davidson, et al., Combined ab initio, kinetic modeling, and shock tube study of the thermal decomposition of ethyl formate, J. Phys. Chem. A, 2017, 121, 6568–6579 CrossRef CAS PubMed.
- W. DeMoore, S. Sander and D. Golden, JPL Publication 97-4, Institute of Technology, 1997 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp02101c |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2022 |