
1,2,4,5-Tetrazine derivatives as components and precursors of photo- and electroactive materials
Galina N.
Lipunova
a,
Emiliya V.
Nosova
 *ab,
Grigory V.
Zyryanov
ab,
Valery N.
Charushin
ab and
Oleg N.
Chupakhin
ab
*ab,
Grigory V.
Zyryanov
ab,
Valery N.
Charushin
ab and
Oleg N.
Chupakhin
ab
aPostovsky Institute of Organic Synthesis, Ural Branch of the Russian Academy of Sciences, 22 S. Kovalevskaya st./20 Akademicheskaya st., Ekaterinburg 620137, Russia
bDepartment of Organic and Biomolecular Chemistry, Ural Federal University, 19 Mira st., Ekaterinburg 620002, Russia. E-mail: emilia.nosova@yandex.ru
First published on 3rd June 2021
Abstract
Extensive research on the synthesis and application of tetrazine derivatives for electronic devices, luminescent elements, photoelectric conversion elements, and image sensors has been published recently. This review covers reported data on the modern trends in the design of functionalized tetrazines obtained within the period 2010–2020. Aryl(heteroaryl) and arylvinyl derivatives of tetrazines and their photoluminescence and application for fluorogenic probes are discussed. Examples of photosensitive oligomers and polymer 3,6-dithienyltetrazines are reviewed.
Introduction
Since the discovery of 1,2,4,5-tetrazines (s-tetrazines)1 and over the whole past century, around 300 publications describing s-tetrazine syntheses have been published. Only in the last four decades the properties and applications of various 1,2,4,5-tetrazines derivatives have gained much interest from researcher groups in various fields.2–13 The most important synthetic application of s-tetrazines is associated with their ability to participate as dienes in the inverse electron demand (ID) Diels–Alder cycloaddition reactions to afford multi-substituted pyridazines.2,3,5 Saracoglu8 demonstrated such reactions to be successfully used for the convenient preparation of natural products, bioactive compounds, high energy materials, building blocks, and other heterocyclic compounds. Shawali et al. collected and analysed the literature data on synthesis, biological activity and industrial applications of a wide range of annelated 1,2,4,5-tetrazines, from bicyclo- to heptacyclo ones, for the period from 1981 to 2000.6 Additionally, the application of s-tetrazines as dienes in biorthogonal cycloaddition reactions for their rapid bioconjugation to various molecules was described.14 Finally, the data on the biological activity of s-tetrazines as well as their bi-, tetra- and hexahydro-derivatives were collected in two book chapters by Neunhoeffer15 and Sauer.16 The application of s-tetrazines for the creation of (in)organic functional materials/fluorophores was highlighted in many publications. Thus, in coordination chemistry many s-tetrazines have been reported as ligands for metal complexes.7,17 Among various heterocycles, great potential of s-tetrazine-based push–pull systems for designing oligomers and optoelectronic materials was highlighted in a recent review article;10 however, among various heterocycles, s-tetrazine-based molecules were presented very briefly. Early examples of photophysical properties of s-tetrazines were presented by Neugebauer,12,13 and a summary of photophysical properties of 3,6-diaryl(heteroaryl)-1,2,4,5-tetrazines was presented by Audebert et al.9 All the authors noted such unique properties of s-tetrazines as their deep colour from purple to red in a visible spectrum region (due to the weak n–π*-transitions), as well as fluorescence emission with maxima λem = 550 to 590 nm and quantum yield Φf = 2 × 10−3 to 0.40, which, however, depends strongly on the nature of substituents in the tetrazine core.Among the so far reported stable six-membered cyclic azines, s-tetrazines are the electron-poorest ones with a strongest electron attractor, s-tetrazine ring, and the applications of these heterocycles in the NLO field are highly expected. In addition, the fluorescence of s-tetrazines can be quenched in the presence of various electron donors and s-tetrazines can be reversibly reduced to the radical anions.
In the current review, we wish to analyse the reported over the past decade articles on new or improved synthetic approaches to symmetrical and unsymmetric 3,6-disubstituted-1,2,4,5-tetrazine systems, as well as oligo- and polymers based on 3,6-dithienyl-1,2,4,5-tetrazines, and the photophysical and electrochemical properties of such systems, as well as some important fields of their practical applications. The review consists of three main chapters: synthesis and photophysical properties of 3,6-di(het)aryl-1,2,4,5-tetrazines, synthesis and photophysical properties of alkenyl-substituted 1,2,4,5-tetrazines and, finally, synthesis and photophysical/photovoltaic properties of 3,6-dithienyl-1,2,4,5-tetrazine-based oligomers and polymers.
Synthesis and photophysical properties of 3,6-diaryl(heteroaryl)tetrazines
Some developed synthetic approaches to 3,6-diaryl(heteroaryl)tetrazines were summarized by Audebert et al.9 Pinner synthesis proved to be the most common method, and 3,6-diphenyl-tetrazine (2) was obtained from hydrazine and benzonitrile and the following mild oxidation (Scheme 1).1 Further modification of this approach with the involvement of sulfur-containing intermediates, performed by Abdel et al., led to dihydrotetrazines 3 in high yields (76–94%) (Scheme 2).18 The abovementioned method proved its effectiveness when aromatic nitriles were involved, whereas it proved to be ineffective in the case of aliphatic nitriles, and the proposed mechanism of the process was presented by Audebert et al.19 | ||
| Scheme 1 Synthesis of 3,6-diphenyl-s-tetrazine 2. | ||
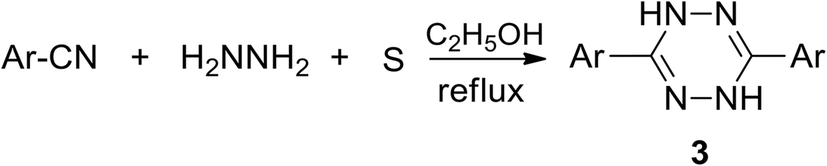 | ||
| Scheme 2 Modification of the Pinner approach. | ||
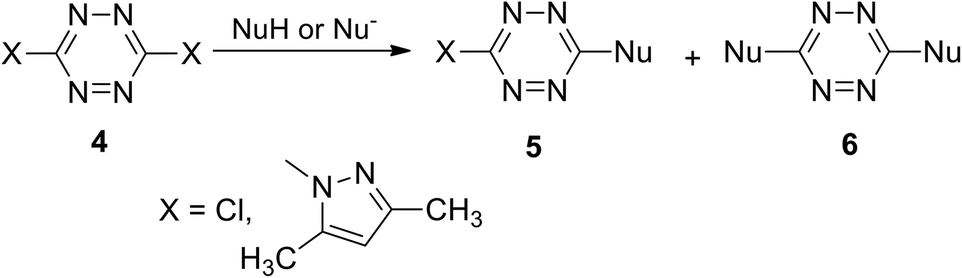
A more common approach to both symmetric and unsymmetric 3,6-diaryl(heteroaryl)tetrazines was based on nucleophilic ipso-substitution reactions in s-tetrazines 4 (Scheme 3).9
 | ||
| Scheme 3 Nucleophilic substitution in the synthesis of tetrazines. | ||
Initially, bis(thiomethyl)tetrazine was used as the starting reagent.20 However, later the convenient approach to 3,6-bis(3,5-dimethyl-1H-pyrazol-1-yl)-s-tetrazine 8 starting from hydrazine and guanidine was developed, and the process included the oxidation step of dihydrotetrazine intermediate 7.21 In the frames of further research,22,23 the two-step conversion of derivative 8 into 3,6-dichloro-s-tetrazine 10 was realised in up to 80% total yield (Scheme 4).
 | ||
| Scheme 4 Synthesis of 3,6-bis(3,5-dimethyl-1H-pyrazol-1-yl)-s-tetrazine 8 and 3,6-dichloro-s-tetrazine 10. | ||
Incorporation of symmetric tetrazines 8 and 10 into the reaction with a wide range of nucleophiles allowed obtaining a huge amount of novel functionalized s-tetrazines. It was shown that nucleophilic mono-substitution proceeds easily at room temperature, whereas further di-substitution requires harsh conditions and/or hard nucleophiles, for example, the participation of anionic nucleophiles. In particular, the reaction of 3-chloro-6-(pyrrolidin-1-yl)-s-tetrazine 11 with lithium alkynes leading to the formation of new derivatives 12 proceeded successfully with acetylenes bearing electron donor aryl substituents (Scheme 5).9
 | ||
| Scheme 5 Interaction of 3-(pyrrolidine-1-yl)-6-chloro-1,2,4,5-tetrazine 11 with lithium alkynes. | ||
Palladium-catalysed cross-coupling reactions are not quite common in s-tetrazines due to the strong electron deficient nature of the 1,2,4,5-tetrazine cycle. However, upon the introduction of electron-donating substituents, such as dimethylamine, pyrrolidine, morpholine, etc. in the tetrazine cycle, the second substituent, such as chlorine or thiomethyl, can be substituted with aryl or heteryl residues via cross-coupling reactions under the common Sonogashira, Negishi or Suzuki conditions9,24,25 as well as by Ag-mediated Pd-catalyzed Liebeskind–Srogl coupling26 (Scheme 6).
 | ||
| Scheme 6 Suzuki and Sonogashira cross-coupling reactions in chloro- and thiomethyl-substituted tetrazines 13, 15. | ||
The growing application potential of substituted s-tetrazines in various fields requires the development of new approaches. In particular, the synthesis of 3,6-disubstituted 1,2,4,5-tetrazines 19 was developed starting from well-available hydrazones 17 in the presence of [hydroxyl(tosyloxy)iodo]benzene (HTIB) (Scheme 7).27 At the first stage, 1,4-dihydrotetrazines 18 formed under mild conditions, while the following de-tosilation/aromatization was carried out by using tetrabutyl ammonium fluoride (TBAF) under reflux in THF. Reaction yields at the first stage were 38–96% and at the second stage were 85–98% except for o-bromoderivatives. The method tolerates a wide scope of substrates to result in a wide variation of substituents at the positions of C3 and C6 in the 1,2,4,5-tetrazines.
 | ||
| Scheme 7 Synthesis of tetrazines 19 from hydrazones 17. | ||
Audebert et al. recently reported an easy and effective approach to 3-monosubstituted unsymmetric 1,2,4,5-tetrazines 20–23 (Scheme 8).28 Thus, mono-substituted 1,2,4,5-tetrazines were obtained in yields up to 75% under mild conditions starting from alkyl(aryl)nitriles and dichloromethane (DCM). When 1,4-dicyanobenzene was used as the nitrile component, both cyano groups were converted sequentially into 1,2,4,5-tetrazine cycles. It should be noted that the approach presented in Scheme 8 demonstrates a very first example of using DCM as a reagent for the synthesis of 1,2,4,5-tetrazines and, according to the authors, this solvent provided its good reactivity and selectivity. The mechanism of this process as well as the elucidation of the role of DCM in the formation of the 1,2,4,5-tetrazine cycle was studied using 13C-labeled DCM. Additionally, optical and electrochemical properties of tetrazines 20 were studied, and as it was expected, all the obtained tetrazines 20 have intensive absorption maxima in the visible region (λabs ∼535 nm). However, only low-intensive fluorescence (λem 575–595 nm) was observed.
 | ||
| Scheme 8 DCM as a reagent in the synthesis of tetrazines 20–23. | ||
Mao et al.29 noted that the abovementioned reaction requires prolonged microwave irradiation, and this limits its applicability for the synthesis of 1,2,4,5-tetrazines, especially on a large scale. The authors also reported an organocatalytic-based approach to unsymmetric 1,2,4,5-tetrazines based on the commercially available nitriles with various functional groups, and these nitriles can be smoothly activated in the presence of thiol-containing catalysts (L-cysteine, 1,3-propandithiol, 2-aminoethanthiol, thioglycolic acid, and N-acetyl-L-cysteine). By the optimization of reaction conditions, the authors found that in the presence of 3-mercaptopropionic acid the higher yields of target tetrazines were observed. Thus, the reaction between two different nitriles and hydrazine hydrate in ethanol in the presence of this acid at room temperature led to a wide range of unsymmetric tetrazines 24 in up to 71% yields (Scheme 9).
 | ||
| Scheme 9 Synthesis of unsymmetric tetrazines 24. | ||
The authors highlighted that the proposed approach is mild, economical, and compatible with water and does not require dangerous reagents, harsh reaction conditions or sophisticated equipment; moreover, it allows scaling, and, thus, it is suitable for the most chemical and biochemical laboratories. Unfortunately, the properties of the obtained 1,2,4,5-tetrazines were not studied.
Suzuki cross-coupling in chloro/S-alkyl substituted s-tetrazines is another convenient approach for the preparation of unsymmetrically substituted ones. For instance, tetrazines 26 were obtained by Leconte et al. and Bender et al. via the Suzuki cross-coupling protocol using an expanded range of N-alkyl substituted chloro-tetrazines 25 and aryl(heteroaryl) boronic acids (Scheme 10).30,31 Notably, the highest yield was achieved in the reaction of 25 (R = NHCH2CH2CH2CH3) with phenylboronic acid. Products 26 bearing a wide range of aryl/heteryl residues were obtained in the yields up to 89%. According to the authors, the method is promising for the synthesis of new tetrazine derivatives with applications in medical chemistry and materials science.
 | ||
| Scheme 10 Suzuki cross-coupling reaction in the synthesis of 3-amino-6-aryl(heteroaryl)-1,2,4,5-tetrazines 26. | ||
In another work,32 several S-alkyltetrazines 27 were subjected to react with arylboronic acids. It was found that biphenyl-substituted tetrazine 27a was the most effective in this reaction, and a series of desired products 28 was obtained in yields up to 94% (Scheme 11). This approach represents the first example of the Ag-mediated Suzuki cross-coupling reaction. The reaction proceeded under mild conditions and allows introducing the 6-methyltetrazin-3-yl group in a wide range of aromatic and heterocyclic molecules.
 | ||
| Scheme 11 Participation of 3-arylthiomethyl-6-methyltetrazines 27 in the Suzuki cross-coupling reaction. | ||
Fox et al.32 noted that this cross-coupling method is not limited to 6-methyltetrazines. 3-(Methylthio)-6-phenyltetrazine 29 also reacted successfully with arylboronic acids with the yields comparable to the ones observed for the compound 27a (Scheme 12).
 | ||
| Scheme 12 Participation of 3-methylthio-6-phenyltetrazine 29 in the Suzuki cross-coupling reaction. | ||
In a similar manner, the Suzuki cross-coupling reaction between tetrazine 27a and BODIPY-appended phenylboronic acid 31 afforded BODIPY-π-tetrazine conjugate 32 in 78% yield (Scheme 13). It is worth mentioning that this product was reported earlier33 as an “ultra-bright” bioorthogonal probe for fluorogenic bio-visualisation in living cells; however, it was isolated only in 8% yield.
 | ||
| Scheme 13 Synthesis of BODIPY-tetrazine conjugate 32. | ||
Direct TM-catalysed C–H functionalisation of aryl-1,2,4,5-tetrazines was also reported, and in this approach N-atoms act as directing groups.34–36 Thus, palladium-catalysed ortho-halogenation of C3 and C6 phenyl substituents in 3,6-diphenyltetrazine 2 was reported by Hierso et al. (Scheme 14).34 After the optimization of the reaction conditions, mono- and tetrahalogenated 1,2,4,5-tetrazines 33–36 were synthesized. Different palladium salts were applied, and it was shown that microwave irradiation during the fluorination process significantly reduces the reaction time. Notably, these halogenated tetrazine derivatives represent useful precursors for further structure modification. For example, Hierso et al. converted tetrabromoderivative 34a into tetraaryl and tetra(3-thienyl) derivatives through the Suzuki–Miyaura cross-coupling process; the obtained heptaaryls are of great value for further construction of electron-poor, nonplanar phenyl-substituted benzenes, which are complementary to graphene-like fully planar building blocks in materials science.37
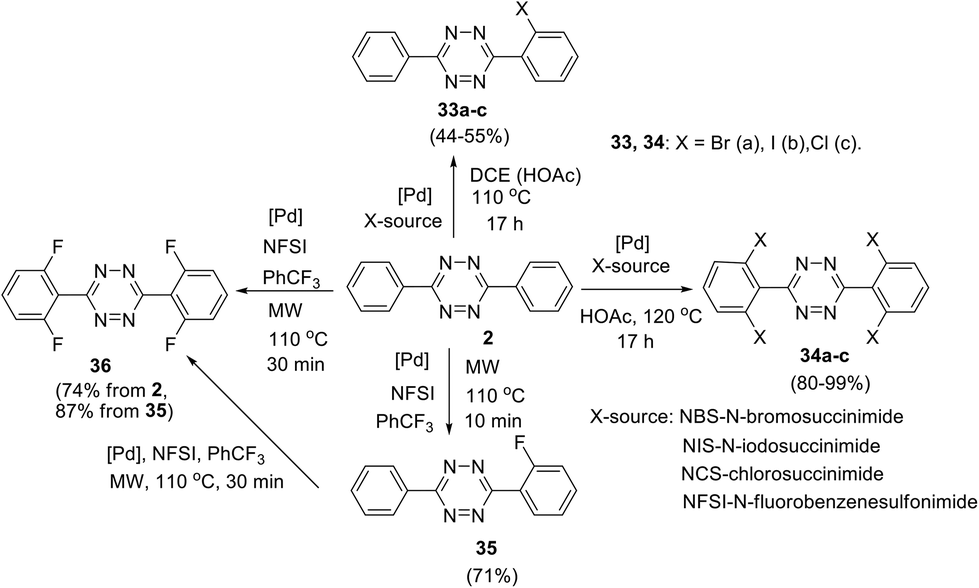 | ||
| Scheme 14 ortho-Halogenation of 3,6-diphenyl-s-tetrazine 2. | ||
A general approach to the synthesis of ortho-halogen substituted unsymmetric biphenyl and polyaromatic s-tetrazines was reported later by the same research group.35 The authors used multiple universal Pd-catalysed activation and halogenation of C–H bonds in the phenyl substituent in 1,2,4,5-tetrazine to form C–X bonds (X = I, Br, Cl, F) and to create polyhalogenated unsymmetrically substituted building blocks. Conditions for the accelerating halogenation reaction of aromatic substituents were found by using microwave irradiation. As a result, diverse bi-, tri- and tetra-halogenated aryl-substituted tetrazines bearing different halogen atoms in aryl substituents were obtained, including tetrazine 37 bearing four different halogen atoms (Scheme 15). The synthesized polyhalogenated derivatives can then be used further in the controlled cross-coupling reactions to perform ortho-substitution in the aromatic substituent polycyclic s-tetrazines. Finally, to create new T-shaped (38a) and Z-shaped (38b) tetrazines Hierso et al. used the halogen-selective Suzuki–Miyaura reaction of halogen-functionalized tetrazines 37 (Scheme 15). The same scientific group designed bis-tetrazines 39 by oxidative homocoupling of o-bromo-s-aryltetrazines.38
 | ||
| Scheme 15 The design of biphenyl tetrazines 38 with T-shaped and Z-shaped structure and bis-tetrazines 39. | ||
An effective and selective C–H amidation of s-tetrazines in the presence of the iridium(III) catalyst was reported.36 At first, the reaction conditions were optimized for the reaction of 3-methyl-6-phenyltetrazine 40 with tosylazide. After that, the optimized synthetic protocol process was applied for the diverse sulfonylazides 41, and ortho-disubstituted phenyl moiety tetrazines 42 were obtained in 56–96% yields (Scheme 16). It was observed that mono-substituted tetrazines can be also obtained at lower temperatures. The main features of reaction are the diversity of substrates, tolerance to various groups, atmosphere, and moisture, as well as an easy scaling up of the process. Some examples demonstrated that tri- and tetra-functionalised building blocks can be obtained quickly.
 | ||
| Scheme 16 Selective C–H amidation of s-tetrazines. | ||
Along with the development of approaches for the synthesis of new s-tetrazine derivatives, theoretical and experimental studies of the photophysical and electrochemical properties of 3,6-disubstituted 1,2,4,5-tetrazines are in progress. Thus, the influence of the nature of the substituents on the Red-Ox, spectral and structural characteristic properties of symmetric conjugated aryl- and dithienyl-substituted 1,2,4,5-tetrazines 43 bearing three to five units (Fig. 1) was studied.39 In this study, tricyclic tetrazines were synthesized in high yield by using the Pinner reaction with diethyl azodicarboxylate as an oxidizing agent. Tetrazines 43e–g with five heterocyclic units were prepared by Stille or Suzuki cross-coupling reactions using dibromo-substituted tetrazine precursor 43b.
 | ||
| Fig. 1 Symmetrically conjugated diaryl- and dithienyl s-tetrazines. | ||
In UV-spectra tetrazines 2 and 43a–d exhibited low-intensity longest-wavelength (first) absorption maxima at 531–547 nm, and for the compounds 43e–g, they appear only in the form of a shoulder. Substitution of the phenyl moiety with a thienyl one (switching from compound 2 to compound 43b) led to a hypsochromic shift of the long-wave absorption maximum by 16 nm, while upon the replacement of the phenyl derivative by the 4-pyridinyl-substituted one the position of an absorption band practically did not change (λmax = 547 nm for 2, λmax = 545 nm for 43a). Pron et al.39 noted that according to the quantum-chemical calculations, the first band corresponds to n–π* transition for the 1,2,4,5-tetrazines 2 and 43a, and to π–π* transition for other compounds (43b–g). All derivatives were studied by cyclic voltammetry (CV), and experimental LUMO energy values proved to be comparable with theoretically calculated ones. The single crystal X-ray data for the five cycles containing tetrazine 43e were collected. It was shown that 43e crystallises in the P21/s spatial group, and for this reason the structural arrangement is not favourable for the effective charge transfer inside the crystal. A powder diffraction study for other derivatives showed that their structural organisation is sensitive to the position of the substituents.
In another work40 the dynamics of the excited state of tetrazines 2, 43b and 43h (Fig. 1) was investigated. It was shown that in the most cases the emission from the Sn level is more intense than from the S1 level. Under the excitation of 3,6-diphenyltetrazine 2 at its first absorption maximum (λexc ∼550 nm), weak fluorescence was observed with emission maximum at 600–750 nm (Φf ∼1.5 × 10−4 in cyclohexane). For molecules 43b and 43h the fluorescence quantum yield was significantly higher (in cyclohexane, Φf ∼1.3 × 10−3 for 43h and Φf ∼5.9 × 10−4 for 43b). Time resolved fluorescence and femtosecond transient absorption data led to the conclusion that from the Sn state molecules can undergo either internal conversion to the S1 state or direct emission to the ground state. Relaxation from Sn to S1 represents a rather slow process (time constant ∼20 ps for 43h and ∼30 ps for 43b).
Incorporation of naphthalenimide fragments into the structure of 3,6-bis(ethynyl)tetrazine 43b using the Pd-catalysed Stille cross-coupling reaction led to the formation of donor–acceptor–donor (D–A–D) tetrazine 46 (Scheme 17).41 For the compound 46, a low-energy band maximum at 454 nm in the absorption spectrum was observed, and this band underwent up to a 75 nm bathochromic shift at the transition from the solution to the films. These films were obtained from the solutions of dithienyltetrazine 46 in dioxane, 1,1,1,2-tetrachloroethane, 1,2-dichlorobenzene or mesitylene/tetralin (1/1) mixture. The films of tetrazines 46 were used for the fabrication of organic field transistors (OFETs) with spin covering. The observed electronic mobility of the transistor was up to 0.15 cm2 V−1 s−1, and for the inkjet-printed OFETs obtained on flexible plastic substrates the electronic mobility reached 0.15 cm2 V−1 s−1.
 | ||
| Scheme 17 Synthesis of bis(thienyl)tetrazine 46 bearing naphthalenimide fragments. | ||
1,2,4,5-Tetrazines 48–50 with the A–D–A structure, containing phenothiazine, 2,7-carbazole or 2,6-benzo[1,2-b:4,5-b′]-dithiophene fragments as donors, were reported.42 All these derivatives were prepared from bromotetrazine 47 by means of the Suzuki reaction in the case of compounds 48 and 49 and the Stille reaction for compound 50 (Scheme 18). It was shown that central donor fragments with different conformations and planarity affect mainly the optical, electrochemical, and solid-state properties of 48–50. It is important that the planarity increases in the following order: phenothiazine < 2,7-carbazole < 2,6-benzo[1,2-b:4,5-b′]dithiophene.
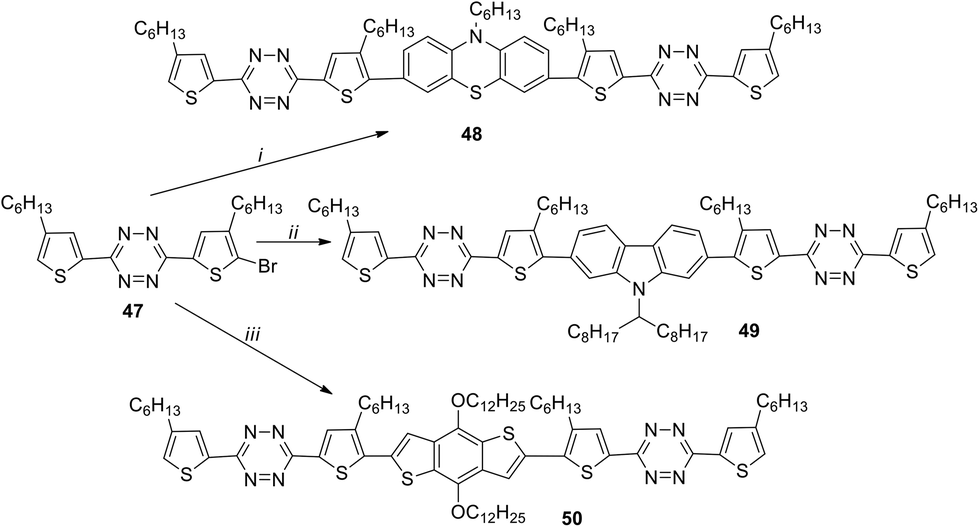 | ||
| Scheme 18 Synthesis of conjugated tetrazine derivatives 48–50 with different planarity of the central donor fragments. Reagents and conditions: i: 10-hexyl-3,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-10H-phenothiazine, Pd(PPh3)4, toluene; ii: 9-(heptadecan-9-yl)-2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9H-carbazole, Pd(PPh3)4, toluene; iii. (4,8-bis(dodecyloxy)benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl)bis(trimethylstannane), Pd(PPh3)4, toluene. | ||
Compounds 48–50 are thermally stable to 270 °C. Their absorption spectra are widened with maxima at 488–553 nm in chloroform solution and at 536–643 nm in thin films. For all the compounds the band gap values obtained from the spectral and CV data are comparable and lie within 1.75–2.31 eV. To study the photovoltaic properties of derivatives 48–50, bulk heterojunction photovoltaic devices were created with a film obtained from solutions of compounds 48–50 and [6.6]-phenyl-C61-butyric acid methyl ester (PC61BM) as an active layer. The component ratio as well as films thickness was varied. The best performance was obtained for the film containing derivative 50 due to its higher coplanarity, highest effect of intramolecular charge transfer (ICT) and excellent crystallinity. All devices showed different performances, and the values of power conversion efficiency (PCE) were changing from 0.1% (with compound 48) to 0.005% (with 49) and 0.78% (with 50). Based on all mentioned above the device based on higher coplanar molecules (50) exhibited the best photovoltaic performance.
Several series of new unsymmetric 3,6-disubstituted-1,2,4,5-tetrazines 51–53 (Fig. 2) were synthesized and investigated by Li et al.43 These compounds were obtained in 71–85% yield by the reaction of one or two different aromatic nitriles with hydrazine hydrate in the presence of sulphur in ethanol upon heating.
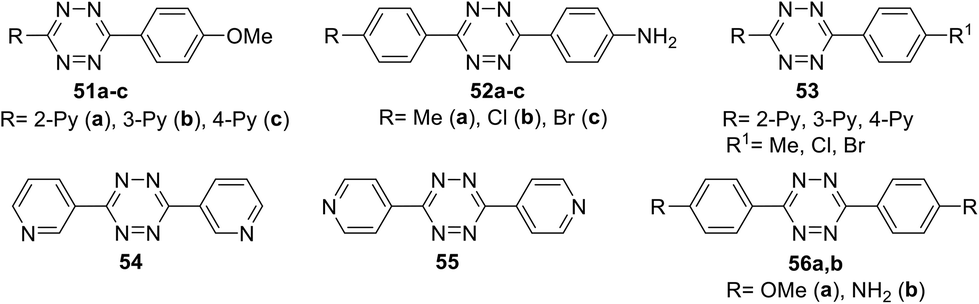 | ||
| Fig. 2 Chemical structure of tetrazine derivatives 51–56. | ||
For all the compounds the absorption maxima are located at 317–346 nm. Experimental and theoretical studies of the spectral properties have shown that among all tetrazines studied the only derivatives 51a–c, 52a–c and 56b could be considered as donor–acceptor systems with an efficient intramolecular charge transfer from methoxyphenyl or aminophenyl donor moieties to pyridyl or aryl acceptor ones. As a result, these tetrazines exhibited intense fluorescence in a visible region with emission maxima at 396–441 nm and the quantum yields ranging from 0.22 to 0.67. The electrochemical behaviour of synthesized tetrazines demonstrates their complete charge reversibility. The effect of pyridinyl substituents on CV characteristics was shown on the example of compounds 51a–c. Thus, 4-pyridyl tetrazine 51c possesses the highest reduction potential (0.314 V) and half-wave potential (0.355 V), whereas its 3-pyridyl counterpart 51b exhibits the lowest ones (Epc = 0.235 V, E1/2 = 0.283 V) (Fig. 3). According to the authors,43 tetrazines 51, 52, and 56 are promising materials for application in organic materials, due to their high electrochemical energy storage capacity and good electrocatalytic reduction effects.
 | ||
| Fig. 3 Cyclic voltammetry (CV) curves for compounds 51a–c, the obtained samples in 2 M KOH electrolyte versus SCE at a scan rate of 20 mV s−1, the voltage window ranged from 0.0–0.5 V. Adapted from ref. 43 with permission from The Royal Society of Chemistry. | ||
Fernández-Gómez et al. reported that 3,5-bis(3,5-dicyanophenyl)-1,2,4,5-tetrazine exhibits more suitable electron injection properties, whereas its fluorinated and brominated counterparts followed by the halogenated derivatives show efficient electron transport properties.44
Several D–A–D type systems containing triphenylamine (TPA) as an electron donor group and an electron acceptor tetrazine ring bound through a π-conjugated linker were reported.45,46 Thus, new electroactive bichromophores 57–60 (Fig. 4) with a 3-methoxy or 3-chlorotetrazine moiety connected to a triarylamine one via simple linkers were prepared in 16–87% yields by using the nucleophilic substitution reaction in 3,6-dichlorotetrazine under the action of oxy- or oxime-containing triarylamines.
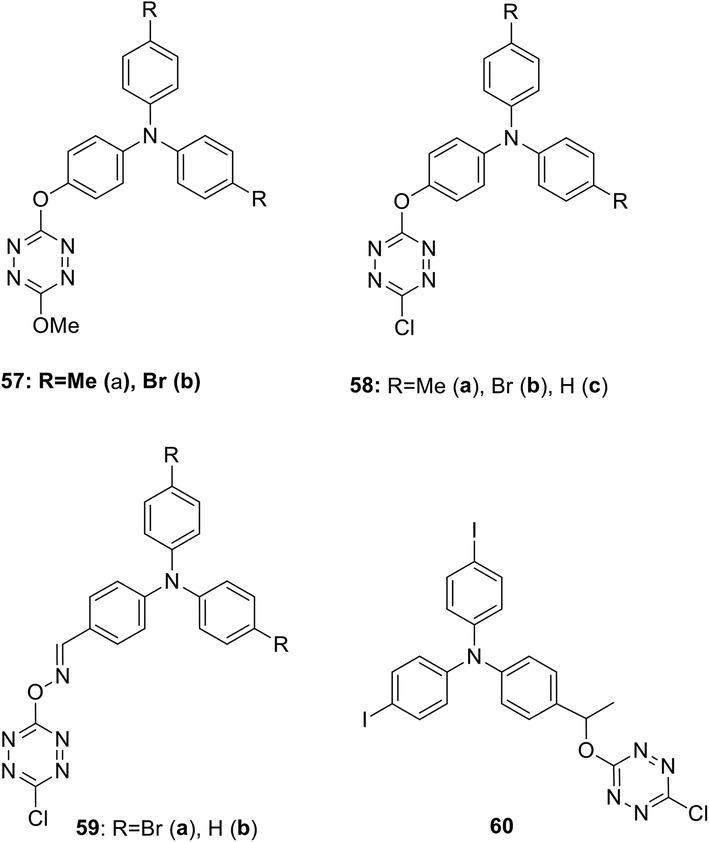 | ||
| Fig. 4 Chemical structure of methoxy and chloro tetrazine derivatives 57–60. | ||
A study of the electrochemical and photophysical properties of derivatives 57 and 58, bearing an oxygen atom as a linker, showed that these compounds practically do not emit in a neutral state (in dichloromethane solution the photoluminescence quantum yield value was significantly lower than 10−3).45 The quenching of fluorescence in these molecules is associated with chromophore proximity and photoinduced electron transfer from triphenylamine to tetrazine. However, the chemical oxidation of TPA into cation-radicals using Cu(ClO4)2 in acetonitrile caused only the appearance of low intensity fluorescence, although the photoluminescence quantum yield value was still low (Φf ∼10−3). In this way the redox-fluorescence switching was achieved, because synthesized bichromophores can exist in the neutral state, in which they are non-fluorescent, and in the fluorescent cation-radical state. Using redox-dependent spectroscopy for compound 58b authors demonstrated that triphenylamine can be oxidised into its stable cation-radical. Upon the chemical oxidation of tetrazine 58b with Cu(ClO4)2, aimed to mimic an electrochemical oxidation process, the appearance of cation-radical absorption bands at 696 nm in the UV–vis spectra upon addition of increasing copper(II) amounts was observed (Fig. 5). In the emission spectra some fluorescence recovery was observed due to the conversion of tetrazine 58b to the cation radical (Fig. 6).
 | ||
| Fig. 5 UV–vis absorption spectra of a 5 × 10−6 mol dm−3 solution of 58b upon gradual addition of copper perchlorate in acetonitrile. Inset, increase of the 696 nm peak with the number of Cu(II) equivalents. Reproduced with permission of Elsevier.46 | ||
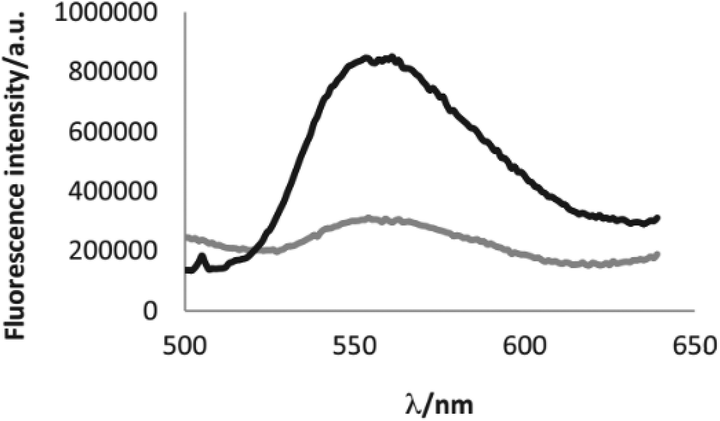 | ||
| Fig. 6 Increase of the fluorescence of molecule 58b upon addition of 2 eq. of copper perchlorate. Bottom curve (grey) fluorescence spectrum of the native molecule; top curve (black) fluorescence spectrum of the oxidized molecule. Reproduced with permission of Elsevier.46 | ||
In their next paper46 Quinton et al. reported some bichromophoric systems 59 and 60, containing an oxime or alkoxy linker (Fig. 4), and the influence of the linker nature on electrochemical behaviour and spectral electrochemistry was discussed. According to the results of the electrochemical studies by means of the CV method, two redox fragments (triarylamino and tetrazine) are capable of electron transfer and their electrochemical behaviour is similar in all bichromophores 57–60. In the absorption spectra of these bichromophores, one or two intensive absorption bands with maxima at 300–362 nm were observed and a low intensity band (because of a forbidden transition) with λmax 490–565 nm featuring the tetrazine moiety absorption (n–π* transition band). The band(s) observed in the UV-region agree with π–π* transitions located both on tetrazine and triphenylamine units with the prevalence of the last ones as they have a higher extinction coefficient compared to free triphenylene and tetrazine. According to the authors, a value of the π–π* transition located on the triphenylamine moiety depends on the nature of the link. For instance, for the compound 58 with the phenoxy link, the π–π* transition band is centred at ca. 310 nm, while for the compound 59 with an oxime link the π–π* transition is centred around 360 nm. This confirms the more attracting effect of an oxime group as a linker. Upon the chemical oxidation of tetrazine 58b with Cu(ClO4)2, aimed to mimic an electrochemical oxidation process, the appearance of cation-radical absorption bands in the UV–vis absorption spectra upon addition of increasing copper(II) amounts was observed. In the emission spectra some fluorescence recovery was observed due to the conversion of tetrazine 58b to the cation radical. The electro-fluorochromic properties of compounds 59a,b were tested in the thin-layer electrochemical cell, wherein the derivative 59b showed no change in oxidation intensity. In the case of derivative 59a, a well reversible and potential-dependent modulation of fluorescence intensity was observed, demonstrating, according to the authors, that this mechanism includes not only the electron transfer process, but probably also energy transfer.
Conjugated systems 61–63 of D–π-A–π-D and D–π-A types with the central tetrazine core modified with one or two TPA groups through the π-spacer (phenylene and 3-decylthiophenylene) were prepared and reported (Fig. 7).47,48 To obtain the derivatives with a phenylene spacer the Stille cross-coupling reaction was used, whereas the synthesis of thiophene-containing derivatives was carried out by the Suzuki–Miyaura cross-coupling reaction.
 | ||
| Fig. 7 Chemical structure of triphenylamine-containing derivatives 61–63. | ||
Next, the optical properties of derivatives 61–63 were studied by experimental and theoretical methods.47 These compounds were characterized by two or three bands of single-photon absorption in dichloromethane solution. The absorption band at 300–307 nm (λ1) was assigned to the electronic transition between the orbitals delocalised on the tetrazine core and linkers. The calculated energy gaps of 3.75–3.92 eV, obtained by the TD-DFT method, were consistent with the experimental λ1 values. A noticeable ICT for compounds 62 and 63 was noted. For compounds 62a and 62b bearing the p-phenylene linker, the absorption band λ2 was characterised by maxima at 412 nm and 401 nm, respectively, whereas in the case of the thiophenylene linker (compound 63) the absorption maximum was located at 441 nm. According to the calculated data, this band corresponds to the electron transfer from the π-orbital, localised on the TPA fragment, to the localised on the core tetrazine π*-orbital and to a linker. An extinction coefficient for compounds 62a and 63a, bearing two TPA moieties, was doubled compared to monosubstituted derivatives 62b and 63b. For compound 61 with a m-phenylene spacer the λ2 band was shifted to the blue region and overlapped with the λ1 band.
Additionally, an intensive two-photon absorption was observed for compound 62a at λmax 795 nm with a cross-section value of 860 GM, and for compound 63a at λmax 820 nm with a cross-section value of 650 GM (Fig. 8). According to Quinton et al.,47 compound 62a is promising for the construction of two-photon absorbing materials, and its characteristics are comparable to widespread initiators of stilbene nature.
 | ||
| Fig. 8 Linear (black curves, scale upwards) and two-photon (red curves, scale downwards) absorption spectra of 62a, 63a in DCM. Adapted from ref. 47 with permission from The Royal Society of Chemistry. | ||
The effect of linker nature in symmetric triphenylamino-s-tetrazine derivatives 61a–63a (Fig. 4) on their electrochemical and spectral properties, as well as the ability of these derivatives to undergo electrochemical polymerisation, was investigated.48 These compounds were studied by the CV method, and ionization potentials and electron affinity were calculated. All of them were found to be bipolar, and all of them underwent a quasi-reversible reduction and oxidation. The study of the reduction process using absorption and EPR spectroscopy showed that it leads to the generation of anion radicals. Adjusting the conjugation degree between the donor and acceptor part molecules resulted in different activity during electrochemical polymerization/oligomerization. Oxidation of derivatives 61a and 62a proved to be a single-stage process in which monomers (in 2 mM solution) undergo electro-polymerisation to form a stable electroactive layer on the electrode working surface. Oxidation of the thiophene derivative 60a was found to be a three-stage process, which, however, did not lead to the formation of an electroactive layer on the electrode working surface. The bication-radical formed in this process was more stable due to the charge delocalization also on the linker, compared to the cation-radicals formed from compounds 61a and 62a. The study of polymer layers revealed an interesting fact: thin polymer films exhibited a charge capture phenomenon along with storage properties.
Symmetrically and unsymmetrically substituted tetrazine-triarylamine fluorophores 61–66 were studied as molecular donors for organic solar cells (Fig. 7 and 9).49 Apart from compounds 61 and 62 bearing a phenyl spacer and derivatives 63 with thienyl one, molecules 64 with an alkynyl spacer and compounds 65 and 66, in which donor and acceptor moieties are directly bound to each other, were described. The synthesis was carried out by different methods described previously. The influence of the linker nature on electrochemical and photophysical properties of derivatives 61–66 was studied.
 | ||
| Fig. 9 Chemical structure of triphenylamine-containing derivatives 64–66. | ||
According to cyclic voltammetry data, two well-defined redox systems can be identified in all compounds, corresponding to the oxidation of the triphenylamine group into cation-radicals and the reduction of tetrazine into anion-radicals. It was noted that TPA oxidation peaks are reversible, while tetrazine electrochemistry is not completely reversible. The reduction potential of compounds 61–64 was about 1.45 V, while for compounds 65 and 66 it proved to be 0.15 V higher. The oxidation potential of compounds 61–64 is about 0.45 V, it was slightly lower for derivative 65 (0.30 V) and higher for derivatives 66a andb (0.65 V and 0.57 V, respectively). Audebert et al. concluded that the reduction potential of tetrazine is weakly dependent on the substituent nature, but, on the other hand, tetrazine causes a significant increase in the oxidative potential of triphenylamine when they are directly bound (compounds 65, 66).
In the absorption spectra of compounds 61–66 several bands were observed depending on the delocalisation between the tetrazine and TPA groups. All the compounds exhibited one or two intense bands in the UV region corresponding to the π–π* transitions with extinction coefficients ranging from 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 76000 L mol−1 cm−1 depending on the nature of substituents in tetrazine. Derivatives 62a and 63a demonstrated the highest values of two bands, which is quite suitable for the application in organic solar cells. A low-intensity long-wavelength band at 545 nm corresponds to the n–π* transition in compounds 61 and 62b; for the derivatives 62a and 65 it was observed as a shoulder, while for the rest of the compounds such a band was missing. Solvatochromism of several derivatives was studied, and it was shown that the solvent polarity affects the value of absorption bands at 360–440 nm. Based on the observed absorption with high ICT and a notable solvatochromic behaviour, some important applications of these compounds were suggested by Audebert et al., such as organic solar cell devices or new fluorescent two-photon absorption chromophores.
000 to 76000 L mol−1 cm−1 depending on the nature of substituents in tetrazine. Derivatives 62a and 63a demonstrated the highest values of two bands, which is quite suitable for the application in organic solar cells. A low-intensity long-wavelength band at 545 nm corresponds to the n–π* transition in compounds 61 and 62b; for the derivatives 62a and 65 it was observed as a shoulder, while for the rest of the compounds such a band was missing. Solvatochromism of several derivatives was studied, and it was shown that the solvent polarity affects the value of absorption bands at 360–440 nm. Based on the observed absorption with high ICT and a notable solvatochromic behaviour, some important applications of these compounds were suggested by Audebert et al., such as organic solar cell devices or new fluorescent two-photon absorption chromophores.
Even though the organic photovoltaic solar cell fabricated from derivative 62a demonstrated low performance (PCE = 0.21%) according to the authors the application of more advanced ways for the manufacturing of photovoltaic solar cell devices will lead to their higher productivity.
Semiconductor organic dyes 67–69 of D–π-A type having tetrazine moieties as acceptors (Fig. 10) were designed to sensitize titanium dioxide in the photochemical hydrogen evolution reaction (HER).50,51
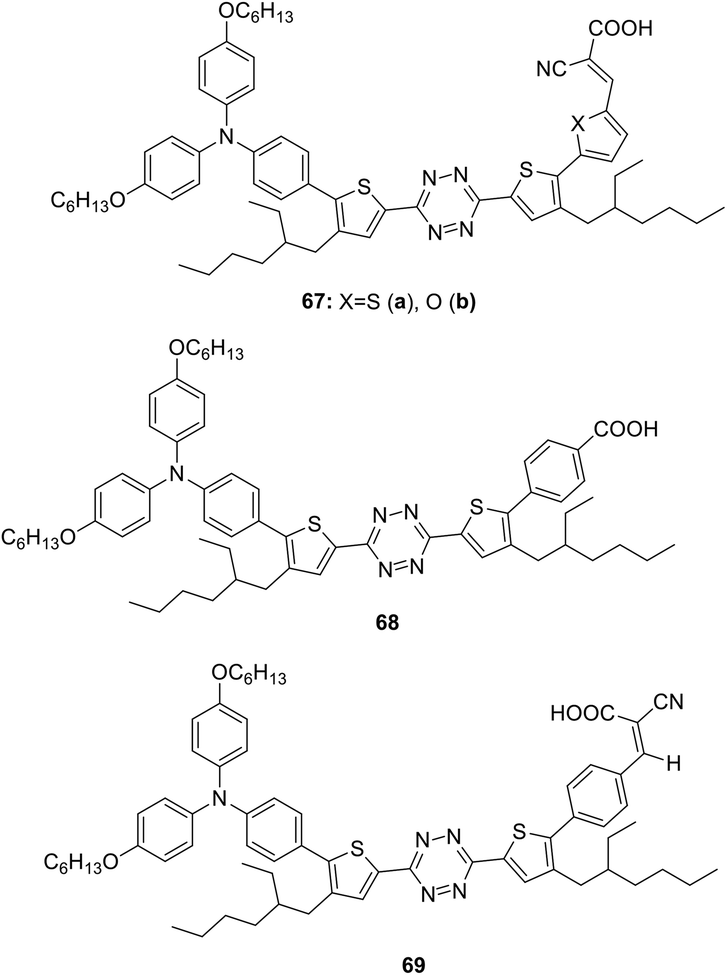 | ||
| Fig. 10 Chemical structure of 3,6-bis(thienyl)tetrazines 67–69. | ||
The synthesis was carried out by means of the bromination of the thienyl rings in 3,6-bis(4-(2-ethylhexyl)-2-thienyl)-1,2,4,5-tetrazine by the action of N-bromosuccinimide, the following cross-coupling reaction with diarylaminophenyl-boronic acid, and then with 5-formyl-2-thienylboronic acid or 5-formyl-2-furylboronic acid and further condensation with cyanoacetic acid. The route to derivative 68 included the cross-coupling process with 4-methoxycarbonylphenylboronic acid at the second step with the subsequent hydrolysis. In the synthesis of compound 69 4-formylboronic acid was used for the second cross-coupling step, and the condensation with cyanoacetic acid was carried out in the last step.51 Dyes 67–69 were characterised by absorption spectroscopy and voltammetry. These compounds exhibited an absorption in the range of 343–426 nm and fluorescence with emission maxima in a range of 538–575 nm. According to CV data, oxidation potentials of the donor triphenylamino groups were 0.81–0.86 V, and the reduction potentials of the tetrazine moiety were in a range from −0.79 V to −0.90 V.
HER activity was studied with triethanolamine as an electron donor component under sunlight irradiation in the absence or in the presence of a co-catalyst (Pt and Cu2WS4). It was concluded that furyl-containing dye 67b was more photoactive than compound 67a due to the higher electronegativity of the oxygen atom.50 Compound 69 was expected to exceed derivative 68 in sensitising activity due to the presence of more electron withdrawing groups and to exhibit a higher molar absorption coefficient. However, the aggregation on the TiO2 surface was observed for dye 69, so, for this reason the compound 68 possessed better properties. Both dyes 68 and 69 were found to be more effective than xanthane eosin-y (EY) dye during hydrogen release due to effective separation of electron–hole pairs.50 Aslan et al. proposed the HER mechanism with participation of dye energy levels, TiO2 and Cu2WS4 co-catalyst.
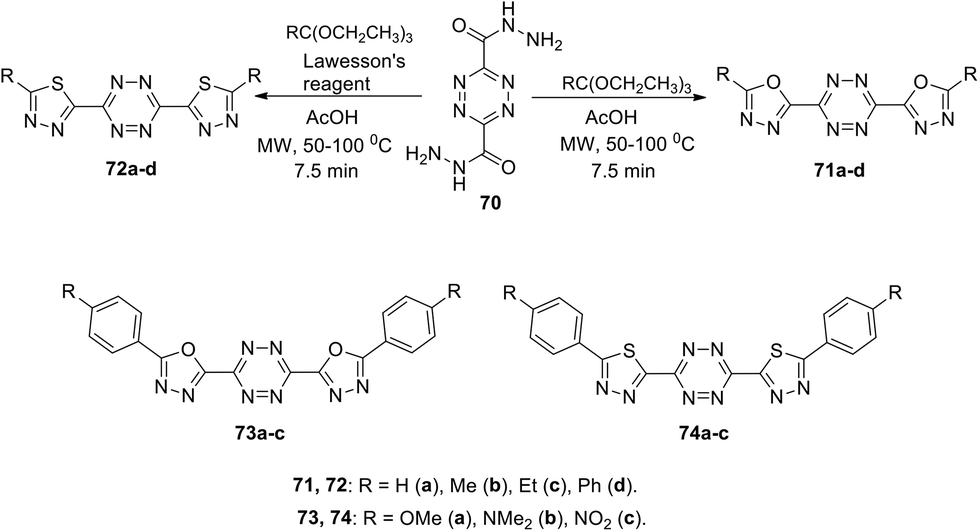
Kedzia et al. reported novel conjugated systems based on tetrazines bound directly to such heterocycles as 1,3,4-oxadiazole (71 and 73) or 1,3,4-thiadiazole (72 and 74) (Scheme 19).52 The oxadiazole framework attracted the attention of the authors due to its ability to transfer electrons and to block holes. Compounds 71 were synthesized by the condensation reaction between tetrazine-3,6-dicarboxydrazide 70 and triethyl orthoesters in acetic acid, while the derivatives 72 were obtained in 51–77% yields under the same conditions using Lawesson's reagent. Compounds 73 and 74 were prepared in 23–77% yields from tetrazine-3,6-dicarbohydrazide 70 and aroyl chlorides, followed by the condensation reaction. In all the cases, the cyclocondensation reaction proceeded quickly and efficiently under microwave irradiation conditions.
 | ||
| Scheme 19 Synthesis of 1,3,4-oxadiazole- and 1,3,4-thiadiazole-containing tetrazines 71–74. | ||
In UV-spectra of chromophores 71–74, intense absorption bands at 265–347 nm were observed, which preceded immediately the emission maximum. Compounds 71–74 exhibited blue fluorescence in solutions with one regular fluorescence maximum in 3D spectra at 327–418 nm, and significant quantum yields from 0.33 to >0.98 were observed in the most cases, except the compounds 71d and 73c (Φf 0.10 and 0.08, respectively). 1,3,4-Oxadiazole and thiadiazole-based derivatives 71a,b, 73a,b, and 74a,b demonstrated the highest luminescence intensity (Φf > 0.98). According to Kedzia et al.,52 these molecules represent perspective building blocks for optoelectronics.
In other publication, a series of symmetrically and unsymmetrically substituted 1,2,4,5-tetrazines 77a–j were presented in which the 1,3,4-oxadiazole cycles were conjugated through the 1,4-phenylene linkers.53 These compounds were prepared via the Pinner reaction of 4-(5-aryl-1,3,4-oxadiazol-2-yl)benzonitrile 76, and the subsequent oxidation of the dihydro derivative with hydrogen peroxide at room temperature (Scheme 20) with final yields of 43–72%.
 | ||
| Scheme 20 Synthesis of 3,6-bis-(1,3,4-oxadiazolylphenyl)-1,2,4,5-tetrazines 77. | ||
Photophysical properties of derivatives 77 were studied, and only one intense absorption band at 284–338 nm was observed in absorption spectra. All the compounds exhibited strong fluorescence emission with maxima located at 348–438 nm. It was shown that the n–π* electron transition during light absorption represents the main source of excited states of tetrazines 77, and the luminescence intensity increases in the following order of substituents in aryl fragments: NO2/NO2 < NO2/t-Bu < NO2/H < OMe/H < NO2/OMe < t-Bu/H < H/H < OMe/t-Bu < t-Bu/t-Bu < OMe/OMe. Three-dimensional fluorescence spectra for all derivatives 77 were recorded (Fig. 11), and it was shown that compounds 77a, 77c, 77f and 77g possess one maximum at 348–374 nm, whereas derivative 77e exhibits two maxima at 359 nm and 432 nm. For compounds 77b, 77d, 77h, 77i and 77j only one fluorescence maximum was detected at 438–384 nm, but there was a second, weaker local maximum that was partially covered by the main maximum. The fluorescence quantum yield of compounds 77 ranged from 0.264 to 0.607. Derivatives with two donor substituents exceeded the unsubstituted ones in quantum yield (Φf = 0.466, 0.607, 0.582, and 0.532 for compounds 77a, 77b, 77c, and 77h, respectively). Based on all mentioned above and large Stokes shift values, these molecules may be referred as promising building blocks for optoelectronics.
 | ||
| Fig. 11 Three-dimensional fluorescence spectra of compounds 77b,c,e,h. Reproduced with permission of Elsevier.53 | ||
Many aryl derivatives of 1,2,4,5-tetrazines, including 3-phenyl-1,2,4,5-tetrazine conjugates with fluorophores, were used in chemical biology to monitor cell proliferation and generate new enzyme inhibitors and for other processes. The reason for this popularity of tetrazine derivatives lies in their ability to participate in non-catalysed transition metal (TM-free) click reactions, accompanied by the transformation of the 1,2,4,5-tetrazine ring into pyridazine one, and this transformation caused a multiple increase in fluorescence. The ability of tetrazine to quench the luminescence of certain dyes was reported for the first time.54,55 Since then, a wide range of dyes were conjugated with tetrazines for further participation in TM-free click reactions; notably, some known fluorophores such as coumarin, xanthene, boron dipyrromethene (BODIPY) and others were applied. Audebert et al. designed turn-on luminescent probes through the Buchwald–Hartwig cross-coupling reaction of 3-(4-bromophenyl)-1,2,4,5-tetrazine with carbazole, phenoxazine, phenothiazine and 9,9-dimethylacridane.56
Among all conjugates, the most accessible were those in which the tetrazine fragment is bound to the fluorescent dye by the amide spacer, since the reaction of 3-(4-benzylamino)-1,2,4,5-tetrazine with the corresponding succinimidyl esters was especially convenient for the synthesis.54,55 As a next step, the synthesized conjugates 78–82 (Fig. 12) were studied in the inverse-electron-demand Diels–Alder (DAinv) reaction with trans-cyclooctenol (TCO). Absorption and emission spectra were recorded, and the quantum yields before and after reaction were determined (Table 1).
 | ||
| Fig. 12 Chemical structure of conjugates 78–82. | ||
| Conjugate | λ abs (nm) | λ em (nm) | Φ f without TCO | Φ f with TCO | Fluorescence enhancement |
|---|---|---|---|---|---|
| 78 | 430 | 480 | 0.01 | 0.03 | 3.3-fold |
| 79 | 505 | 512 | 0.02 | 0.24 | 15.0-fold |
| 80 | 495 | 523 | 0.04 | 0.82 | 18.5-fold |
| 81 | 543 | 573 | 0.02 | 0.40 | 20.6-fold |
| 82 | 669 | 687 | 0.16 | 0.16 | 1.0-fold |
It was shown that conjugation of all dyes, possessing emission at 400–600 nm, with tetrazine led to emission quenching; however, pyridazine-type products exhibiting intense fluorescence were formed through the reaction of conjugates with dienophile. Under conversion the conjugates, exhibiting green and red emission, into cycloaddition products, 15–20-fold increase in emission intensity was recorded in sodium phosphate buffer. Conjugates with dyes exhibiting shorter emission wavelengths were transformed during the click reaction into products with only a 3-fold increase in emission. For the emitting in the near IR region dyes no emission enhancement was observed for the tetrazine conjugate (for example, conjugate 82).
The same team described later new biorthogonal reactions of BODIPY-tetrazine derivatives 83–85 (Fig. 13).33 The synthesis of conjugates 83, 32, and 84 was achieved by the Zn-catalysed reaction of BODIPY nitriles with hydrazine in acetonitrile (for 83, 32) or in water (for 84). Notably the compound 32 was obtained later in higher yield using the cross-coupling reaction.32 Conjugates 85 were prepared from the iodo-derivative of BODIPY. In structures 83–85 the spatial proximity of an acceptor and a donor is enhanced compared to conjugates 79 and 81, providing the orientation of transition dipoles and the possibility for stronger quenching of the fluorescence of the conjugate.
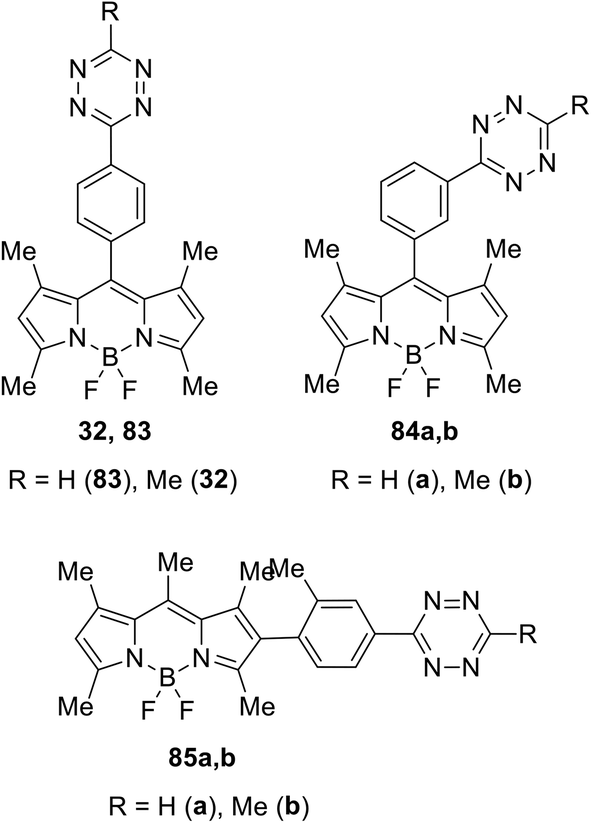 | ||
| Fig. 13 Chemical structure of conjugates 32, 83–85. | ||
The DAinv reactions of conjugates 83–85 with TCO were carried out to confirm the possibility of their use for bio-visualization. The process proceeded rapidly, and the resulting pyridazine derivatives exhibited intense fluorescence. Fluorogenic activity data (Table 2) indicated high efficiency of synthesized conjugates as bioimaging probes. Fluorogenic imaging of epidermal growth factor receptor (EGFR) expression in both fixed and live A-431 cells showed that TCO-labelled targets were readily visualised, with excellent signal intensity and very low background; moreover, the washing step after adding the dye solution was not required.
Coumarin–tetrazine conjugates 86–89 (Fig. 14) for real-time bio-visualisation were synthesized and investigated.57 Compounds were prepared from 3-cyanophenyl-coumarine derivatives and hydrazine through Zn-catalysed tetrazine cycle formation.
 | ||
| Fig. 14 Chemical structure of coumarine–tetrazine conjugates 86–89. | ||
Conjugates 86–89 possess fluorescence in the blue or green region and demonstrated high quantum yields in the click reaction with TCO in phosphate-borate buffer (PBS) (Table 3). These series of compounds were characterised by the highest radiation switching coefficients compared to other probes available at that time.
| Conjugate | λ ex/λem (nm) | ε (M−1 cm−1) | Φ f with TCO | Fluorescence increase |
|---|---|---|---|---|
| 86a | 400/502 | 16000 |
0.41 | 4000-fold |
| 87a | 388/482 | 20000 |
0.38 | 11000-fold |
| 88a | 347/455 | 18500 |
0.29 | 2500-fold |
| 89a | 370/453 | 19000 |
0.49 | 2900-fold |
To demonstrate the applicability of probes as native bio-orthogonal fluorogenic probes, a simple model system was chosen to evaluate the kinetics of probe 89 in extracellular version, namely, EGFR at the cancer cell surface. Cells A-431 were incubated with TCO-labelled anti-EGFR antibody. The addition of conjugate 89 revealed bright, membrane-specific colouring, which coincided with the known receptor distribution. Images were obtained within seconds after the addition of dye. Meimetis et al. noted that such conjugates open great opportunities for in vivo probing.
A series of new conjugates of tetrazines and widely known fluorophores (Fluorescein, Oregon Green, Tetramethylrhodamine, Si-Rhodamine) 90–98 (Fig. 15), which cover the entire emission region, from green to far red, was reported by Wieczorek et al.58 Synthesis of compounds 90, 91 and 95, 96 was carried out by the Stille cross-coupling reaction starting from 3-bromo-6-methyl-1,2,4,5-tetrazine and organotin derivatives of these fluorophores. To prepare tetrazine-rhodamine derivatives 92–94 and 97, and 98, tetrazine benzaldehyde derivatives were reacted with diarylethers or diarylsilanes by means of the Friedel–Crafts alkylation reaction.
 | ||
| Fig. 15 Chemical structure of conjugates 90–98. | ||
Conjugates 90–98 were characterized by means of absorption and emission spectroscopy and changes in their fluorogenic properties in the DAinv cycloaddition reaction with TCO were studied (Table 4, Fig. 16a).
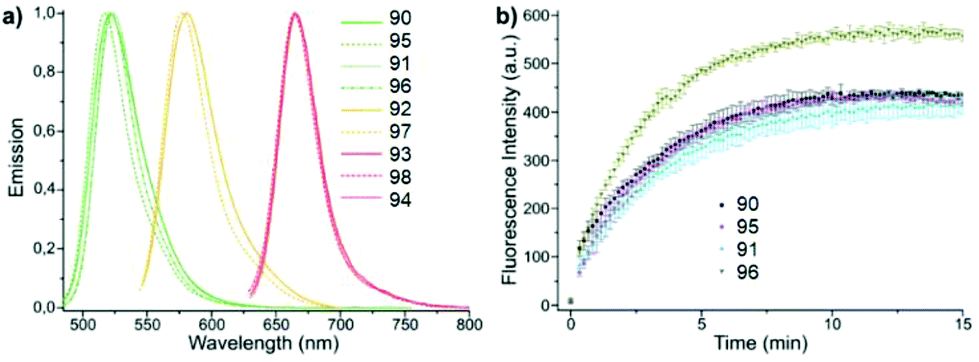 | ||
| Fig. 16 (a) Normalized emission spectra of compounds 90–98. (b) Time course of fluorogenic DAinv between green emitting tetrazine dyes and TCO (500 nM in PBS each). Reproduced with permission of ref. 58. Published by The Royal Society of Chemistry. | ||
| Conjugate | λ abs (nm) | λ em (nm) | ε (M−1 cm−1) | Φ f without TCO | Turn-on (x-fold)a |
|---|---|---|---|---|---|
| a Values in brackets: reaction with TCO-labeled protein. | |||||
| 90 | 495 | 521 | 57000 |
0.0037 | 72 |
| 95 | 495 | 517 | 55000 |
0.0033 | 109 |
| 91 | 495 | 524 | 87000 |
0.0048 | 60 |
| 96 | 495 | 522 | 70000 |
0.0041 | 103 |
| 92 | 556 | 580 | 46000 |
0.037 | 22 |
| 97 | 554 | 577 | 54000 |
0.031 | 12 |
| 93 | 650 | 665 | 58000 |
0.020 | 1.8 (5.6) |
| 98 | 649 | 664 | 62000 |
0.017 | 3.7 |
| 94 | 651 | 656 | 53000 |
0.026 | 2.0 (3.3) |
The increase in fluorescence intensity of green emission of conjugates (90, 95, 91, and 96) in DAinv reactions was monitored over time, and the cycloaddition process was found to complete within 10–15 minutes at equimolar concentrations of reagents (Fig. 16b).
Next, the authors58 successfully applied new fluorogenic probes in experiments on protein selective labelling. Fluorogenic properties of fluorophores 90–98 were successfully applied for the fluorescence microscopy-assisted imaging of the actin cytoskeleton in fixed cells as well as mitochondrial and nuclear proteins in living cells.
Systematic research of a wide series of coumarin–tetrazine conjugates 99 (Fig. 17) for bio-orthogonal fluorescent imaging was reported by Liu et al.59 The influence of the nature of substituents and spacers on photophysical properties of dyes, formed in the reaction of conjugates with two types of dienophiles (TCO and bicyclononyne (BCN)), was studied. The target conjugates were obtained in 12–49% yields after a two step sequence. Thus, the nitrile derivative of the coumarin-spacer was obtained at the first step, and the subsequent interaction of R2-CN with hydrazine hydrate in the presence of Zn(OTf)2 led to target compounds.
 | ||
| Fig. 17 Chemical structure of conjugates 99. | ||
Obtained conjugates 99 were introduced into the DAinv cycloaddition reaction with the axial isomer of (E)-cyclooct-4-enol (axial TCO-OH) and ((1R,8S,9S)-bicyclo[6.1.0]non-4-yn-9-yl)methanol (endo BCN). The photophysical properties of the resulting click-products showed that in most cases the dyes formed from the endo BCN dienophile demonstrated a higher increase in fluorescence compared to the dyes formed from axial TCO. The properties of some click-products are shown in Table 5.
| R1a |
Spacerb | R2c |
Dienophiled | λ abs/λem (nm) | Φ f | Turn-on (x-fold) |
|---|---|---|---|---|---|---|
| a Az – azetydinyl; MP – 1-Me-piperazin-4-yl; M2P – 1-Me2-piperazinium-4. b 3,5-Py – 3,5-pyridinyl; 3,5-Ph – 3,5-phenylene. c 2-Py – pymidin-2-yl; HE – 2-hydroxyethyl. d AT – axial TCO-OH; EB – endo BCN. | ||||||
| Az | 3,5-Py | 2-Py | AT | 375/485 | — | 850 |
| Az | 3,5-Py | 2-Py | EB | 375/470 | 0.032 | 200 |
| Az | 3,5-Py | H | AT | 371/485 | — | 1050 |
| Az | 3,5-Py | H | EB | 371/480 | 0.15 | 500 |
| Az | 3,5-Ph | Me | AT | 374/483 | — | 2900 |
| Az | 3,5-Ph | Me | EB | 376/472 | 0.67 | 3100 |
| Az | 3,5-Ph | HE | AT | 363/483 | — | 2300 |
| Az | 3,5-Ph | HE | EB | 365/479 | 0.57 | 4200 |
| NEt2 | 3,5-Ph | Me | AT | 398/481 | — | 2700 |
| NEt2 | 3,5-Ph | Me | EB | 397/483 | 0.22 | 5000 |
| MP | 3,5-Ph | Ph | AT | 340/460 | — | 70 |
| MP | 3,5-Ph | Ph | EB | 352/451 | 0.38 | 550 |
| M2P | 3,5-Ph | Ph | AT | 352/457 | — | 125 |
| M2P | 3,5-Ph | Ph | EB | 350/452 | 0.58 | 950 |
The formation of pyridazine dyes during the reaction of conjugates with endo BCN proved to be excellent in terms of fluorogenicity. It was also shown that fluorescence of the obtained fluorophores can be tuned by both on steric and electronic effects of substituents at the 1,2,4,5-tetrazine ring and/or coumarin moieties of the tetrazine conjugates. For instance, the presence of the azetidine or N,N-dimethylpiperazine moiety in the coumarin scaffold leads to an increase of fluorescence quantum yield of the obtained pyridazines due to suppressing the formation of twisted intramolecular charge transfer (TICT) upon photoexcitation.59,60 However, cell imaging experiments demonstrated that other factors, such as reaction kinetics and good cell permeability, predominated over fluorescence switching properties.
Galeta et al.61 concluded that the best fluorophore was the compound 99, which contained azetidinyl-coumarin bound to 6-(pyrimidin-2-yl)tetrazine through the pyridine linker, since this fluorophore showed excellent results in living cell labelling experiments and allowed realising the fluorogenic imaging without washing for several seconds.
Synthesis and photophysical properties of alkenyltetrazines
While the Knoevenagel reaction (condensation of methyl-substituted hetarenes with aromatic aldehydes) represents the main approach to alkenyl derivatives of different heterocycles, this method was never used for the synthesis of alkenyl-substituted 1,2,4,5-tetrazines due to the inaccessibility and instability of starting methyl-substituted tetrazine precursors. Some other approaches were also developed for alkenyl-tetrazine preparation. Thus, in situ synthesis of (E)-3-substituted 6-alkenyl-1,2,4,5-tetrazine derivatives 101 through the elimination-Heck cascade reaction was reported in 58–99% yields.62 Stable and easily available mesyloxyethyl-substituted derivatives 100 were used as starting reagents. The reaction was carried out in DMF under microwave irradiation in the presence of the palladium catalyst and the ferrocene ligand (Scheme 21). The possibility of preparing bis(styryl) and bis(butadienyl) substituted tetrazines by this method was demonstrated. | ||
| Scheme 21 Synthesis of alkenyltetrazines 101. | ||
This method was also used for the synthesis of tetrazine conjugates with xanthene dyes (102, 103) and BODIPY (104) (Fig. 18).62 Tetrazine derivatives 102–104 were characterized by fluorescence spectroscopy to exhibit low-intensity fluorescence with emission maximum at 520, 575 and 530 nm, respectively. A significant increase in emission intensity was observed for the cyclocondensation reaction products. Thus, reaction of tetrazines 102–104 with various dienophiles led to highly fluorescent pyridazines, which are promising probes for bio-visualisation. The greatest fluorescence enhancement was achieved after for the products of reaction between conjugate 102 and cyclopropene and trans-cyclooctenol: a 135-fold and 400-fold increase in intensity was observed, respectively.
 | ||
| Fig. 18 Chemical structure of conjugates 102–104. | ||
Conjugates similar to 104 were proposed as fluorescent probes for the bio-visualization of nucleic acids, and the tetrazine cycle transformation was carried out under the action of 2-((1R,8S,E)-9-azabicyclo[6.1.0]non-4-en-9-yl)ethan-1-ol. According to Devaraj et al., 20–70 times increase of the emission at 475–540 nm was achieved after carrying out this TM-free click reaction.63
A series of siliconrhodamine conjugates 106 and 107 emitting in the NIR region were reported.64 These compounds were prepared in 11–46% yields by means of Pd-catalysed cross-coupling of bromo-derivatives 105 with the corresponding mesyloxyethyl substituted 1,2,4,5-tetrazines (Scheme 22). The obtained conjugates exhibited fluorescence with an emission maximum at 665–673 nm with quantum yields of 0.015–0.037. The DAinv reaction of these conjugates with axial TCO-OH was studied. In the case of compound 106, a fluorescence increase by 22- or 49-fold, as well as quantum yield increase from 0.016 to 0.34 (conjugate 106a) and from 0.015 to 0.80 (conjugate 106b), was observed. Trifluoromethylphenyl derivatives 107 reacted with axial TCO-OH much faster than compounds 106, but weaker fluorescence increase (6–8 times) was detected.
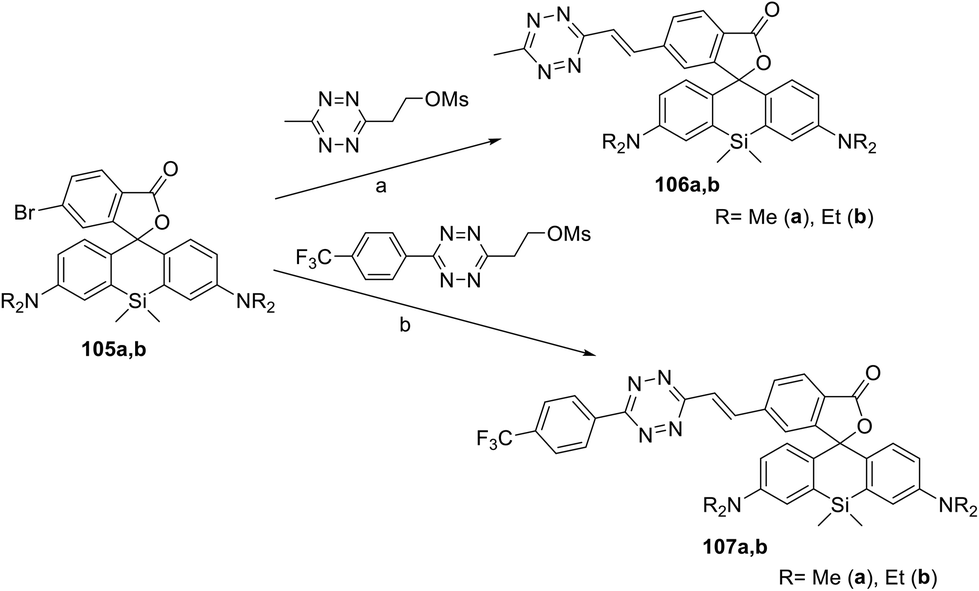 | ||
| Scheme 22 Synthesis of conjugates 106, 107. Reaction conditions: (a) Pd2(dba)3, QPhos, (Cy)2NMe, DMF, 50 °C, 40 min, MW; (b) Pd2(dba)3, QPhos, DIPEA, 1,4-dioxane, 90–110 °C, 16 h. | ||
Notably, siliconrhodamine conjugates represent examples of membrane-permeable labels and allow realising site-specific bio-orthogonal labelling of genetically modified intracellular proteins and subsequent imaging in a high inclusion rate by means of super-resolution microscopy (SRM).
Other examples of alkenyl-tetrazine preparation through cross-coupling reactions were described. Styryl derivative 109 was synthesized by the Stille reaction, and substitution of the methylthio group in tetrazine 108 was carried out in 34% yield in the presence of such monovalent salt as copper thiophene-2-carboxylate (CuTC) in DMF under microwave irradiation and heating (Scheme 23).30
 | ||
| Scheme 23 Synthesis of styryltetrazines 109, 110. | ||
In another work32 3-methyl-6-phenylstyryltetrazine 110 was prepared in 39% yield by the Ag-mediated cross-coupling reaction under mild Pd-catalyzed conditions (Scheme 23).
The previously developed soft organocatalytic approach to unsymmetrically substituted tetrazines, based on application of the thiol-containing catalyst, was also used for the synthesis of simple tetrazines, and the latter were subjected to further functionalization.29
Thus, 3-methyltetrazine phosphonate 112, obtained in 45% yield, proved to be a convenient building block for a highly efficient Horner–Wadsworth–Emmons reaction (HWE) with aldehydes under mild reaction conditions (Scheme 24). Optimization of the reaction conditions was carried out, and lithium 1,1,1,3,3,3-hexafluoroisopropoxide (LiHFI) was proposed as an ideal soft base. A wide series of alkenyltetrazines 113 and conjugates 114 and 115 were obtained in 87–94% yields using various aldehydes 111.
 | ||
| Scheme 24 Synthesis of styryltetrazines 113–115. | ||
The mechanism of the DAinv cycloaddition reaction of trans-cyclooctene to 1,2,4,5-tetrazine and the effect of the dienophile structure were studied for the series of alkenyltetrazines 116 (Scheme 25),65 which are capable of rapidly forming fluorescent products in the abovementioned reaction without introducing extra fluorophore. Two spatial isomers of TCO-OH (axial and equatorial) were synthesized and used. In model experiments with simple 3,6-di-(2-pyridyl)-1,2,4,5-tetrazine as an example, it was shown that axial TCO-OH reacts with the formation of fluorescent 1,4-dihydropyridazine, whereas equa-TCO-OH does not react. Vrabel et al. monitored the reaction by using 1H NMR to analyze the composition of the reaction mixtures. It was found that the reaction proceeds through the formation of an intermediate 4,5-dihydropyridazine derivative, which, in the case of axial TCO-OH, was rapidly isomerized into fluorescent 1,4-dihydropyridazine, but in the case of equa-TCO-OH such isomerization proceeded very slowly. The click-reaction of other tetrazines 116 with axial TCO-OH rapidly afforded the corresponding pyridazines possessing fluorescence with maxima lying at 480–539 nm and up to 195 nm red-shift compared to parent tetrazines. In addition, depending on the nature of the R1 substituent 11–91-fold fluorescence increase was observed for pyridazines compared to parent tetrazines. In all the cases the formation of a second regioisomer was observed as a minor product.
 | ||
| Scheme 25 Reaction of styryltetrazines 116 with two TCO-OH isomers and aza-TCO. | ||
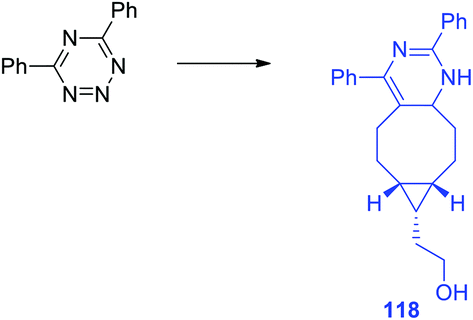
In another work of the same team64 a conformational-restricted aziridine-fused trans-cyclooctene (aza-TCO) dienophile was synthesized, for which the “half-chair” conformation was confirmed by NMR and X-ray methods. This dienophile was introduced in the DAinv process with several tetrazines 116. It was shown that the click-reaction proceeds with the formation of dihydropyridazines 118 (Scheme 25) exhibiting an emission maximum at 475–540 nm, the fluorescence increased by 40–70 times. Kinetics measurement for the click-reaction of simple 3,6-diphenyltetrazine with aza-TCO and axial TCO-OH dienophiles demonstrated that aza-TCO reacts 20 times faster than axial TCO-OH. In the reaction of tetrazines 116 with both aza-TCO and axial TCO-OH the second regioisomer was always observed as the minor product. The potential of aza-TCO dienophile for application in bioimaging was demonstrated by experiments of protein labeling, cell glycoconjugate imaging and living bacteria peptidoglycan imaging.
Thus, Vrabel et al.65,66 elucidated the role of the dienophile spatial structure in click-reactions with tetrazine derivatives, and it was found that only axially substituted hydroxyl derivatives of trans-cyclooctenes and aza-bicyclononenes act as dienophiles with the formation of fluorescent 1,4-dihydropyridazine products.
In the next work of the same team67 it was shown that in the case of 1,2,4,5-tetrazines bearing specific electron donor substituents the fluorogenic tetrazine cycloaddition can be extended to other dienophiles. The reaction of alkenyltetrazine 116a, containing a strong electron donor, with two TSO-OH isomers was studied (Scheme 25). This tetrazine was found to react with axial TCO-OH giving 1,4-dihydropyridazine 117a (λem = 478 nm) and with equa-TCO-OH to form 4,5-dihydropyridazine 117b (λem = 628 nm). To study the effect of substituents on alkenyltetrazines, the reaction of a wide range of compounds 116 with equa TCO-OH was carried out, and the properties of the resulting products are demonstrated in Table 6. According to the data presented in Table 6, a strong electron donor with a methoxy group is not enough for this reaction. Fluorescence enhancement (15- and 18-fold) was observed in reaction of tetrazines bearing phenyl and thien-3-yl moieties, and pyridine substituents reduced the fluorescence quantum yield.
| Tetrazine | R | R1 | λ abs/λem (nm) | Φ f | Turn-on (x-fold) |
|---|---|---|---|---|---|
| 116a | Ph | NMe2 | 545/626 | 0.55 | 15 |
| 116b | Ph | OMe | 285/— | — | — |
| 116c | Thien-3-yl | NMe2 | 546/626 | 0.52 | 18 |
| 116d | Ph | Azetidinyl | 546/626 | 0.45 | 10 |
| 116e | Pyridin-4-yl | Azetidinyl | 566/643 | 0.21 | 3 |
| 116f | Thien-3-yl | Azetidinyl | 548/626 | 0.47 | 12 |
Thus, tetrazines, containing donor dimethylamino or azetidine groups, are capable of forming fluorescent products in DAinv reactions with different TCOs. Model bio-visualisation experiments have demonstrated the application potential of this fluorogenic DAinv reaction.
It is worth mentioning that recently Boger's group reported the synthesis of novel isomeric tetrazine, namely 2,6-diphenyl-1,3,4,5-tetrazines,68 and its photophysical and electrochemical properties were studied. In the UV–vis spectrum, a strong absorption (ε = 2.52 × 104 L mol−1 cm−1) was observed at 275 nm, corresponding to the π–π* transition of the phenyl rings and tetrazine core along with a weaker absorption peak (ε = 4.90 × 102 L mol−1 cm−1) at 396 nm, corresponding to the n–π* transition of the tetrazine. Compared to 3,6-diphenyl-1,2,4,5-tetrazine, which exhibits weak fluorescence (ϕ = 1.5 × 10−4),40 2,6-diphenyl-1,3,4,5-tetrazine demonstrated no fluorescence. By cyclic voltammetry (CV), 1,3,4,5-tetrazine was found to undergo a quasi-reversible single-electron reduction at −1.37 V (vs. Fc+/Fc, −0.78 V vs. NHE), to reveal its electron-deficient nature. In contrast to reduction potential, no oxidation was observed when a positive scan was performed. According to Li et al., the reduction potential of 2,6-diphenyl-1,3,4,5-tetrazine is almost comparable with the one for 3,6-diphenyl-1,2,4,5-tetrazine 19 (−1.21 V vs. Fc+/Fc),69 and this indicates a similar electron deficiency of both tetrazine isomers.
Next, the reactivity of 2,6-diphenyl-1,3,4,5-tetrazine in inverse electron demand Diels–Alder cycloaddition reactions with various dienophiles, such as heterodienophiles (amidines), electron-rich dienophiles (ynamines, enamines, ketene acetal), and strained alkynes (cyclooctyne), was studied (Scheme 26).70 Isomeric 3,6-diphenyl-1,2,4,5-tetrazine 19 was used as a reference.
 | ||
| Scheme 26 Reaction of 2,6-diphenyl-1,3,4,5-tetrazine with cyclooctyne. | ||
According to Fischer et al.,70 compared to 1,2,4,5-tetrazine 19, 1,2,3,5-tetrazine exhibited an outstanding reactivity toward heterodienophiles (amidines), a comparable reactivity toward electron-rich dienophiles (ynamines, enamines, ketene acetal), and a lower reactivity toward strained olefins and alkynes (cyclooctyne). In the last case the cycloaddition reaction at room temperature under effectively and modestly concentrated reaction conditions (400 mM tetrazine, 4 eq. cyclooctyne) afforded fused pyrimidine 23 in 97% yield, while under more diluted conditions (10–50 times) the reaction yields went down to 0%. In all the cases only a single mode of cycloaddition was observed (C4/N1 vs. N2/N5).
Synthesis and photovoltaic properties of 3,6-dithienyl-1,2,4,5-tetrazine oligomers and polymers
The simplest way to use solar energy is to convert it into electricity by photovoltaic (PV) technology. Organic polymer solar cells (PSCs) have attracted great attention of researchers due to their low cost and ease of industrial production. A key part of polymer solar cells is the active layer, which usually represents a mixture of donor and electron acceptor. In numerous materials with photovoltaic properties the tetrazine cycle acts as the electron acceptor, and thiophene derivatives play a role of donors. Thus, novel copolymer 119, in which the thiophene bridge was introduced between tetrazine and cyclopenta[2,1-b:3,4-b′′]dithiophene (CPDT), was prepared (Fig. 19).69 The synthesis was realised through the Stille reaction from the dibromo derivative of 3,6-dithienyltetrazine, obtained by the Pinner method from 4-hexylthiophene nitrile. The polymer demonstrated good thermal stability and a wide absorption band, covering the area of 450–700 nm. The energy levels HOMO and LUMO were estimated as −5.34 and −3.48 eV, and the energy gap was 1.86 eV according to CV data. Solar cells fabricated from the mixture of polymer 119 and fullerene-based donor PC71BM ([6.6]-phenyl-C71-butyric acid methyl ester) demonstrated the calibrated energy conversion efficiency of 5.4%.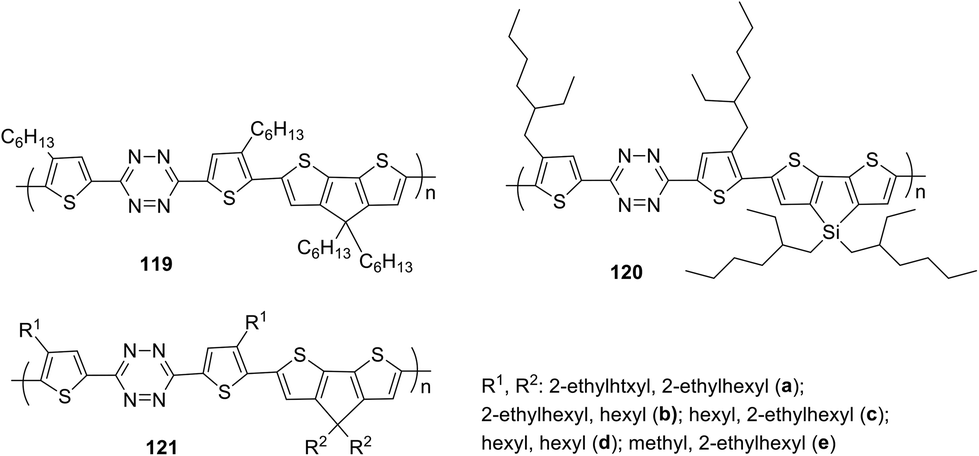 | ||
| Fig. 19 Chemical structure of polymers 119–121. | ||
In another work of the same team,71 the modified polymer 120 (Fig. 19), in which tetrazine is bound to dithieno[3,2-b:2′,3′-d]silole (DTS) as the electron donor component, was described. This polymer is thermally stable and exhibits strong absorption in the visible region (molar coefficient 4.67 × 104 M−1 cm−1 in toluene), and low energy gap (1.65 eV). Solar cells from a mixture of this polymer and PC71BM demonstrated energy conversion efficiency (PCE) up to 4.2%.
In the next work of the same team,72 a series of copolymers 121a–e with various substituents in the thiophene cycle and CPDT fragment (Fig. 19) were studied to elucidate the effect on solubility, optical, electrochemical characteristics and interchain attraction of polymers. According to the authors these polymers have very promising properties for application in functional materials, such as thermostability at 220 °C, a long wavelength absorption band at 546–588 nm (chlorobenzene) with a large molar coefficient (from 4.4 × 104 to 5.6 × 104 M−1 cm−1), a low energy gap of 1.60–1.72 eV and HOMO/LUMO values around −5.35 eV/−3.52 eV accordingly.
To prove the applicability of these polymers bulk heterojunction solar cells were fabricated by mixing the polymers with PC71BM, and the observed power conversion efficiency was up to 5.53% (in case of polymer 121d bearing linear hexyl substituents) under simulated AM 1.5 G irradiation of 100 mW cm−2. The morphological structures of the active layers from different polymers or under different processing conditions were then analyzed by atomic force microscopy (AFM) and correlated with their device performance.
In another report, copolymers 122 and 123 (Fig. 20), in which tetrazine cycle and carbazole or fluorene units as electron donor components are bound through the thiophene spacer, were synthesized and studied.73
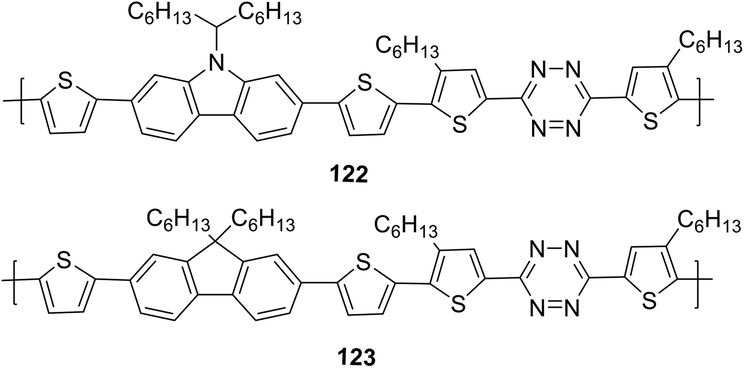 | ||
| Fig. 20 Chemical structure of copolymers 122, 123. | ||
Polymers 122 and 123 are thermally stable to 260 °C and 245 °C, respectively, and both polymers have wide UV-absorption bands to ∼700 nm with maxima at 495 nm (122) and 498 nm (123) in chloroform. It was noted that carbazole-containing copolymer 122 exhibited a higher absorption coefficient than polymer 121d, and a long-wavelength shift of about 30 nm was observed in the films of both polymers. According to CV data, energy levels HOMO and LUMO and band gaps for polymers 122 and 123 were: −5.32 eV and −5.60 eV, −3.09 eV and −3.12 eV, 2.23 eV and 2.49 eV, respectively. Bulk heterojunction photovoltaic devices were made using copolymer 122 and PC71BM at a weight ratio of 1:1, 1:2, 1:3, or 1:4. The best of them with a ratio of 1:1 had a high open-circuit voltage (1.0 V) under AM 1.5 G illumination at 100 mW cm−2 with a power conversion efficiency of 2.13%.
In the last few years, application of small molecules in organic solar cells (OSCs) has attracted increasing attention due to easier purification methods to reach higher purity, well-detailed by using common physical methods, molecular structure, workability of solutions due to better solubility (because of lower molecular weight), and, finally, certain molecular weight, which can be determined by common methods.
The design and synthesis of the series of highly coplanar tetrazine-containing oligomers 124–126 with the A–D–A structure were reported.74 These molecules were prepared by the Pd-catalysed Stille cross-coupling reaction of 3-(5-bromo-4-hexyl-2-thienyl)-1,2,4,5-tetrazine with bis(trimethylstannyl)thiophene derivatives. In all the oligomers, non-alkylated oligothiophene acts as the central donor fragment, and tetrazine units on both sides act as acceptors while alkylated thiophene units act as the π-bridge between donor and acceptor parts (Fig. 21).
 | ||
| Fig. 21 Chemical structure of coplanar molecules bearing two tetrazine rings 124–126. | ||
Molecules 124–126 (Fig. 21) proved to be highly soluble in conventional organic solvents and exhibit high thermal stability (Td > 257 °C). These molecules possessed an ICT band in the visible region with a high molar coefficient: λmax 447 nm and εmax 84500 M−1 cm−1 for 124, λmax 463 nm and εmax 96300 M−1 cm−1 for 125, λmax 473 nm and εmax 82600 M−1 cm−1 for 126. According to CV data, the LUMO energy levels were practically independent of the number of thiophene cycles in oligothiophene: −3.54 eV, −3.55 eV, and −3.41 eV for 124, 125 and 126, respectively. HOMO energy values changed more significantly: −5.50 eV, −5.18 eV, and −5.02 eV for 124, 125 and 126, respectively. The band gaps were 1.96 eV, 1.63 eV, and 1.61 eV for 124, 125, and 126, respectively. It was shown that band gaps of these three SMs and energy levels can be tuned through changing the number of oligothiophenes (OTs) based on the A–D–A structure, and also the A–D–A structure SMs can lead to high absorption coefficients.
To study the photovoltaic properties, bulk heterojunction photovoltaic devices were fabricated using the above-mentioned SM as the donor, and PC61BM or PC71BM as the acceptor. Active layers for the devices were obtained from chloroform solutions of SM and PC61BM or PC71BM by spin-coating. The optimized current–voltage curves of oligomers 124–126 are presented in Fig. 22. The cells based on 124/PC61BM showed an open circuit voltage (Voc) of 0.73 V, a short circuit current density (Jsc) of 1.77 mA cm−2, and a fill factor (FF) of 0.38, giving a PCE of 0.49%. Interestingly, with the increasing number of thiophene units in the backbone, the photovoltaic cell based on 125/PC61BM exhibited Voc of 0.74 V, Jsc of 5.77 mA cm−2, FF of 0.52, and significantly increased PCE to 2.22%, which is more than three times higher than that of 124. However, the photovoltaic performances of 126 with increasing thiophene number in the backbone decreased greatly. The results indicated that hierarchical structures controlled by adjusting the conjugate moiety length of small molecules represent an effective way to improve the performance of OSCs. Thus, the photovoltaic device based on 125/PC71BM showed the best performance, with a Jsc of 7.87 mA cm−2 and a PCE of 3.24%.
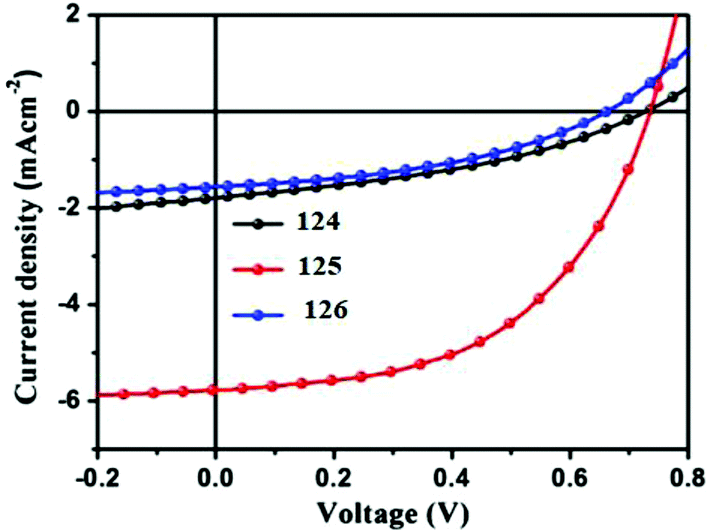 | ||
| Fig. 22 Current–voltage characteristics of optimized photovoltaic cells based on 124: PC61BM, 125: PC61BM and 126: PC61BM under illumination of AM 1.5, 100 mW cm−2 white light. Reproduced with permission of Elsevier.74 | ||
Tetrazine-containing polymers 127 and 128 (Fig. 23), including benzo[1,2-b:4,5-b]dithiophene (BDT) as the electron donor fragment with a rigid planar structure and increased mobility, as well as thiophene spacers between tetrazine and BDT parts to improve the solubility of copolymers, were obtained by the cross-polymerization Stille reaction.74 The determined molecular weight was 7.8 kg mol−1 for copolymer 127 and 30.0 kg mol−1 for copolymer 128. Polymers demonstrated high thermal stability (Td = 295 °C for 127 and 305 °C for 128). The absorption spectrum of polymer 127 in o-dichlorobenzene exhibited two bands in the visible region with maxima at 440 nm and 510 nm, while polymer 128 exhibited one band with a maximum at 494 nm. In the film, the absorption maximum underwent a long-wave shift of 20–25 nm for 127 and 48 nm for 128. According to CV data, the HOMO and LUMO energy levels and the energy gap had the following values for copolymer 127: −5.39 eV, −3.58 eV and 1.81 eV, for copolymer 128: −5.28 eV, −3.60 eV and 1.68 eV. Both polymers possessed a low HOMO level and narrow band gap.
 | ||
| Fig. 23 Chemical structure of polymers 127 and 128. | ||
To study the photovoltaic properties of the obtained polymers, several structural PSCs were created by depositing copolymers 127 or 128 and PC71BM on indium-tin oxide/glass substrates. It was shown that the solvent composition had a significant effect on the device performance (a mixture of 80% o-dichlorobenzene and 20% chlorobenzene was proved to be the best one). Analysis of the characteristics of fabricated devices showed that promising PCE > 5.0% was achieved at high open circuit voltage (Voc) ∼1.0 V.
Novel small molecules (SMs) 129 and 130 with a D2–A–D1–A–D2 structure including benzo[1,2-b:4,5-b]dithiophene as the central building block, tetrazine as the acceptor component, and bi- or terthiophene as terminal donor fragments (Fig. 24) as potential candidates for OSCs were reported.75 Compounds 129 and 130 were prepared by using the Stille cross-coupling reaction between 3-(5-bromo-4-hexyl-2-thienyl)-6-(3,50-dihexyl-2,20-bithiophen-5-yl)-1,2,4,5-tetrazine and 3-(50-bromo-3,40-dihexyl-2,20-bithiophen-5-yl)-6-(3,40,50-trihexyl-2,20-50,20-terthiophene-5-yl)-1,2,4,5-tetrazine and bis (trimethyltin)-4,8-bis(2-ethylhexyloxy)benzo[1,2-b:4.5-b]dithiophene using Pd(PPh3)4 as the catalyst. In UV-spectra, molecules 129 and 130 exhibited wide absorption bands at 300–600 nm in chloroform solution with maxima at 479 nm (129) and 489 nm (130) and high molar coefficients (7.29 × 104 M−1 cm−1 for 129 and 1.27 × 105 M−1 cm−1 for 130). Due to high organic solvent solubility thin films of these SMs were easily fabricated, and in these films the greater widening of the absorption bands along with a 43–49 nm red-shift of the absorption maxima was observed compared to the solutions, thus indicating strong intermolecular π–π interactions in the solid state for the both oligomers.
 | ||
| Fig. 24 Chemical structure of molecules 129 and 130. | ||
The redox behaviour of compounds 129 and 130 was studied by the CV method and it was shown that the energy levels of the HOMO and LUMO and the band gap have the following values for compound 129: −5.51 eV, −3.67 eV and 1.84 eV and for compound 130: −5.30 eV, −3.64 eV and 1.66 eV. Thus, the elongation of the π-bridge and the increase in end groups practically did not affect the LUMO energy level but reduced the HOMO level and the band gap width. For the study of photovoltaic properties, bulk heterojunction organic solar cells were fabricated by blending of molecules 129 and 130 with PC71BM on indium tin oxide/glass substrates. Devices using SMs 129 and 130 showed a promising power conversion efficiency of 5.01% and 5.29% with a large Voc of 0.98 V and 0.88 V, respectively.
The synthesis of tetrazine-quaterthiophene co-polymer 131 (Fig. 25) and data on its optical, structural, and photovoltaic properties were described.76 In this polymer core the 1,2,4,5-tetrazine moiety as an acceptor part combines with two 4-(2-octyl)dodecyl-substituted thiophene units and one non-substituted bithiophene unit as a donor part. Similarly to the above reported cases, the synthesis of copolymer 131 was carried out by the Stille cross-coupling reaction between 3,6-dithiophenetetrazine dibromo derivative and 5,5′-bis (trimethylstannyl)-2,2′′′-bithiophene using Pd(dba)3 as the catalyst.
 | ||
| Fig. 25 Chemical structure of polymers 131–133. | ||
In the thin film, the polymer 131 was characterized by a wide absorption band with a maximum at 583 nm and an absorption coefficient of 1.87 × 105 cm−1, as well as a band gap value of 1.8 eV. The HOMO and LUMO values determined from CV data were −5.58 eV and −3.10 eV respectively, and the value of the band gap was 2.48 eV (greater than optical value). In order to study crystallization behavior and molecular packaging, two-dimensional measurements of wide-angle X-ray scattering of the thin films of copolymer 131 were performed, which demonstrated that the preferred orientation of the packaging of polymer chains in the film was face-to-face, and the d-distance of about 0.40 nm indicates π–π stacking. Next, to fabricate the OSCs this polymer was mixed with a non-fullerene acceptor (1:1) and applied for the absorption layer. ((3,9-Bis(2-methylene-(3-(1,1-dicyanomethylene)-6-fluoroindanone))-5,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:20,30-d0]-s-indaceno[1,2-b:5,6-b0]dithiophene (ITIC-F) was used as an acceptor layer as its energy levels and optical properties match well with those for copolymer 131. Bulk-heterojunction non-fullerene OSCs were manufactured in inverted and normal device configurations. They demonstrated high Voc of 0.9 V, the highest PCE of 2.6% was obtained in the case of inverted solar cell architecture.
Two new polymers 132 and 133 (Fig. 25) were synthesized by Stille coupling polymerization and studied for PSCs.77 In polymer 132, the tetrazine moiety was used as an acceptor part and tetrathiophene derivatives with 2-octyldodecy side chains introduced for better solubility were used as electron-donating moieties. In polymer 133 hexyl- and 2-octyldodecyl-substituted hexathiophene fragments were introduced to suppress excessive molecular aggregation and to tune energy levels as well as morphology of the blend film. The obtained polymers exhibited high thermal stability (Td = 299.8 °C for 132 and 418 °C for 133). Molecular geometry and HOMO and LUMO energy levels of polymers 132 and 133 were calculated by using DFT at the B3LYP/6-31G(d,p) level, and hexyl, octyl and dodecyl residues were replaced with ethyl to simplify the theoretical study. Based on the calculated and X-ray diffraction data, it was concluded that polymer 133 with a hexathiophene fragment possesses relatively lower molecular coplanarity, a larger stacking distance but more orderly molecular stacking compared to tetrathiophene polymer 132. Next, the UV-absorption spectra of these polymers were measured in chlorobenzene at temperatures from 35 to 95 °C (Fig. 26a and b). At 35 °C, two UV-bands were observed for polymer 132 with maxima at 564 and 618 nm, which were attributed to intramolecular charge transfer (ICT) and intermolecular π–π stacking respectively. The latter is higher than the former, which indicates that intermolecular π–π stacking is stronger than ICT in polymer 132. For the polymer 133 these two bands also appeared at longer wave lengths (lmax = 581 and 634 nm). Films 132 and 133 exhibit strong absorption in the visible light band between 400 nm and 700 nm, but they possess little absorption in the short wavelength band (<380 nm) and long wavelength band (>700 nm), while PC71BM and ITIC possess strong absorption in the short wavelength band (300–600 nm) and long wavelength band (550–900 nm) respectively (Fig. 26c). Peng et al. demonstrated that two polymers possess complementary absorption spectra with PC71BM and ITIC. This fact implies that the polymers could construct efficient fullerene- and nonfullerene-based PSCs as well as the corresponding ternary PSCs. According to voltammetry data, the HOMO, LUMO and band gap values were respectively −5.63 eV, −3.75 eV, and 1.88 eV for polymer 132, and −5.34 eV, −3.64 eV, and 1.70 eV for polymer 133.
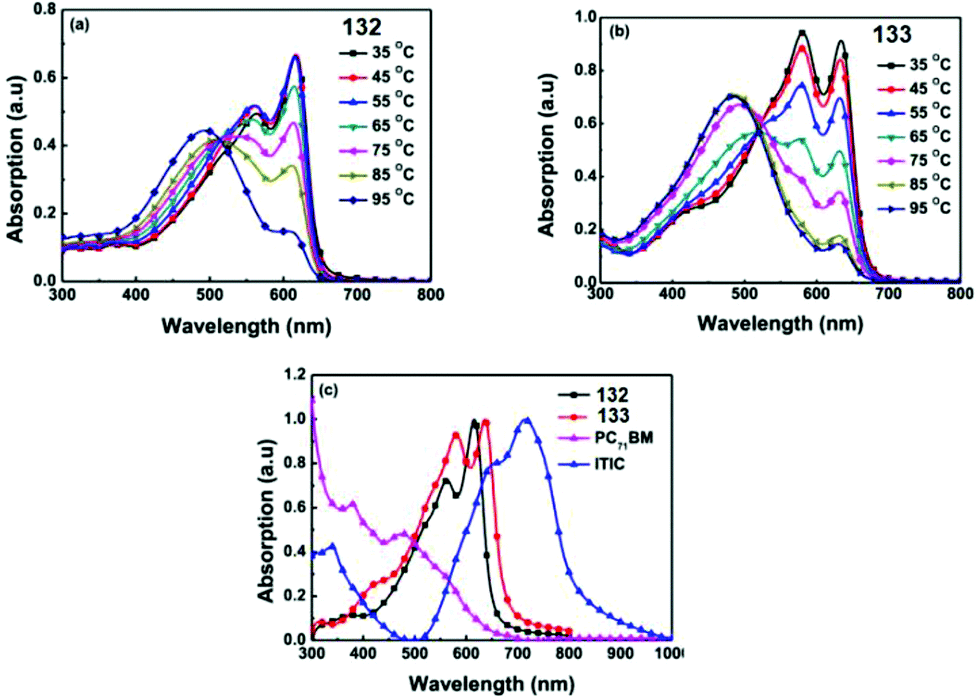 | ||
| Fig. 26 Temperature-dependent absorption spectra of (a) 132 and (b) 133 in chlorobenzene solution, and (c) absorption spectra of 132, 133, PC71BM and ITIC in film. Reproduced with permission of Elsevier.77 | ||
To analyze exciton dissociation and electron transfer behavior of the blend films, the steady state photoluminescence measurements were carried out for the films of polymers 132 and 133, as well as their mixed samples (132/PC71BM), (133/PC71BM) and (133/PC71BM/ITIC) were measured. The film prepared from polymer 132 demonstrated an emission maximum at 652 nm and the intensity in the mixed film at this wavelength maintained at 33.9%. The film of polymer 133 had an emission maximum at 667 nm, the mixed film (133/PC71BM) retained intensity at this wavelength by only 6.1%, and the film of the triple mixture (133/PC71BM/ITIC) had only 1.5% of retained intensity. The mixed films of polymer 133 exhibit higher fluorescence quenching than polymer 132, and the triple blend film 133 possessed the most effective exciton dissociation and charge transfer to electron acceptors. The hole mobility of mixed films was studied, and it was shown that the triple mixture film demonstrates the highest value.
Photovoltaic properties of polymers 132 and 133 were investigated by using inverted PSC devices with an architecture of ITO/ZnO (30 nm)/polymer: acceptor/MoO3 (5 nm)/Al (100 nm), in which very active layers were spin-coated from chlorobenzene solution. Optimal polymer/acceptor ratios were found as follows: 132/PC71BM (1/2), 133/PC71BM (1/2), 133/PC71BM/ITIC (1/1.8/0.2). For devices with such active layers, the Voc values of 1.02, 0.84, 0.88 V and PCEmax 5.16, 6.69 and 7.88%, respectively, were obtained, i.e., the triple blend layer showed better results.
Conclusion and future perspectives
In this review we have demonstrated that tetrazine-based systems display a wide set of promising chemical, photophysical and redox properties, which makes them ideal candidates for various practical applications.First of all, 3,6-disubstituted 1,2,4,5-tetrazine-based systems possess a strong electron deficient s-tetrazine core and a strong election acceptor, and for this reason many research articles in the last decade were focused on the synthetic design of tetrazine-based push–pull systems/NLO-devices. Original and convenient approaches for the synthesis of new s-tetrazine derivatives have been developed, such as various types of metal-catalysed cross-coupling reactions, as well as metal-catalysed or metal free C–H functionalisation or their combinations. Some general important tools for tuning photophysical characteristics and solubility of molecules were applied: incorporation of amino-derived electron donors possessing highly positive mesomeric effects, extension and planarization of the π-system, and introduction of long alkyl chains to provide good solubility of the chromophore.78 Tetrazine-based push–pull systems with an unprecedently high fluorescence quantum yield (>0.98) and considerable Stokes shifts were recently reported52 as potentially useful building blocks for optoelectronics.
Some new tetrazine derivatives were used for creating advanced all-organic materials with high electrochemical energy storage capacity and a good electrocatalytic reduction effect,43 as well as for creating advanced two-photon absorbing materials.47,48 Semiconductor organic donor–acceptor dyes bearing tetrazine moieties as acceptor units were designed to sensitize titanium dioxide in the photochemical hydrogen evolution reaction (HER).50,51
Tetrazine-based pre-fluorophore systems are now widely used in chemical biology as synthetic precursors for “turn on” fluorogenic probes for bioimaging applications. Such probes are generated by means of the DAinv reactions between tetrazines and various dienophiles to afford pyridazine, and this is accompanied by a dramatic enhancement in fluorescence. Various tetrazine derivatives, which demonstrated excellent results in living cell labelling experiments and enabled no-wash fluorogenic imaging on a timescale of seconds, were reported.61 Depending on the nature of a dienophile and the reaction conditions the fluorescence of the resulted pyridazine can be enhanced dramatically and this improves the efficiency of tracking/staining biomolecules in cellulo.79 Along with 1,2,4,5-tetrazine derivatives the recently reported 1,2,3,5-tetrazine diens69 were proven to be successfully involved in DAinv reactions with dienophiles to afford pyrimidines, which might be even more advanced in terms of photophysical properties. In addition, the antibody pretargeting approach for radioimmunotherapy (RIT) using the DAinv reaction between tetrazines and trans-cyclooctenes was recently reported as an emerging theranostic approach for solid cancers.80 Another important application of 1,2,4,5-tetrazines in bioimaging can be associated with their ability to form so called through-bond energy transfer (TBET) donor–acceptor systems. Fluorescence resonance energy transfer (FRET)-based ratiometric probes are widely used for bioimaging applications. However, in such systems it is necessary to provide a sufficiently large spectral overlap between the donor emission and the acceptor absorption, which would limit the resolution of double-channel (bio)images. The s-tetrazine-based through-bond energy transfer (TBET) systems do not need to follow such conditions and, thus, they could afford a large wavelength difference between the two emissions with improved imaging resolution and higher energy transfer efficiency than that of the classical FRET system and that is more favorable for designing more advance ratiometric probes for bioimaging applications.33,57,58,62,81,82 Finally, exclusive use of the tetrazine moiety as a photoquencher in the design of advanced fluorogenic probes was reported recently.83 In contrast to common nitro-phenyl-based phototriggers in this study upon light irradiation the tetrazine-based quenchers were photodecomposed to afford novel photoactivatable fluorophores with an enhanced fluorescence.
In numerous advanced small-molecule-based, oligomeric or polymeric photovoltaic materials of D–A types the appropriate combination of the s-tetrazine units (A) and thiophene derivatives (D) results in materials with a relatively broad light harvesting range and appropriate energy levels for both fullerene and non-fullerene-based acceptors,76,84 which results in promising power conversion efficiencies over 5.0%.75 It should be noted that the creation of highly effective organic materials with thermally activated delayed fluorescence (TADF) which represent an alternative to phosphorescent OLEDs was achieved for some tetrazine derivatives.56,85
Based on all the above, in our opinion, the future applications of tetrazine-based scaffolds should be concentrated on creating more advanced materials for bioimaging, photovoltaic and NLO materials as well as on the ones for energy storage.
Author contributions
Galina N. Lipunova: investigation; Emiliya V. Nosova: data curation; Grigory V. Zyryanov: validation; Valery N. Charushin: project administration; Oleg N. Chupakhin: supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Russian Foundation for Basic Research (grant number 19-33-90014) and Grants Council of the President of the Russian Federation (grant number NSh-2700.2020.3).Notes and references
- A. Pinner, Ueber die Einwirkung von Hydrazin auf Imidoäther, Chem. Ber., 1893, 26, 2126 CrossRef.
- S. M. Weinreb and R. R. Staib, Synthetic aspects of diels-alder cycloadditions with heterodienophiles, Tetrahedron, 1982, 38, 3087 CrossRef CAS.
- D. L. Boger, Diels-Alder reactions of azadienes, Tetrahedron, 1983, 39, 2869 CrossRef CAS.
- D. H. Levy, The spectroscopy, photophysics and photochemistry of clusters of s-tetrazine, J. Chem. Soc., Faraday Trans. 2, 1986, 82, 1107 RSC.
- D. L. Boger, Diels-Alder reactions of heterocyclic aza dienes. Scope and applications, Chem. Rev., 1986, 86, 781 CrossRef CAS.
- A. S. Shawali and S. M. Elsheikh, Annelated[1,2,4,5]Tetrazines, J. Heterocycl. Chem., 2001, 38, 541 CrossRef CAS.
- W. Kaim, The coordination chemistry of 1,2,4,5-tetrazines, Coord. Chem. Rev., 2002, 230, 127 CrossRef CAS.
- N. Saracoglu, Recent advances and applications in 1,2,4,5-tetrazine chemistry, Tetrahedron, 2007, 63, 4199 CrossRef CAS.
- G. Clavier and P. Audebert, s-Tetrazines as Building Blocks for New Functional Molecules and Molecular Materials, Chem. Rev., 2010, 110, 3299 CrossRef CAS PubMed.
- K. Rajen and D. Suranjan, Push-Pull Unit Decorated Oligomer for Organic Electronics, Adv. Chem. Res., 2019, 53, 21 Search PubMed.
- J. Yang, M. R. Karver, W. Li, S. Sahu and N. K. Devaraj, Metal–Catalyzed One–Pot Synthesis of Tetrazines Directly from Aliphatic Nitriles and Hydrazine, Angew. Chem., Int. Ed., 2012, 51, 5222 CrossRef CAS PubMed.
- F. Gückel, A. H. Maki, F. A. Neugebauer, D. Schweitzer and H. Vogler, Chem. Phys., 1992, 164, 217 CrossRef.
- F. A. Neugebauer and H. Fischer, Tetrahedron Lett., 1986, 27, 5367 CrossRef CAS.
- M. L. Blackman, M. Royzen and J. M. Fox, Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels–Alder Reactivity, J. Am. Chem. Soc., 2008, 130, 13518 CrossRef CAS PubMed.
- H. Neunhoeffer, Tetrazines and Pentazines, in Comprehensive Heterocyclic Chemistry I, ed. A. R. Katritzky, Pergamon, Frankfurt, Germany, 1984, vol. 3, p. 531 Search PubMed.
- J. Sauer, 1,2,4,5-Tetrazines, in Comprehensive Heterocyclic Chemistry II, ed. A. J. Boulton, Elsevier, Oxford, U.K., 1996, vol. 6, p. 901 Search PubMed.
- O. Stetsiuk, A. Abhervé and N. Avarvari, Dalton Trans., 2020, 49, 5759 RSC.
- M. O. Abdel, M. A. Kira and M. N. Tolba, A direct synthesis of dihydrotetrazines, Tetrahedron Lett., 1968, 9, 3871 CrossRef.
- P. Audebert, S. Sadki, F. Miomandre, G. Clavier, M. C. Vernieres, M. Saoud and P. Hapiot, Synthesis of new substituted tetrazines: electrochemical and spectroscopic properties, New J. Chem., 2014, 38, 387 Search PubMed.
- D. L. Boger and S. M. Sakya, Inverse electron demand Diels-Alder reactions of 3,6-bis(methylthio)-1,2,4,5-tetrazine. 1,2-Diazine introduction and direct implementation of a divergent 1,2,4,5-tetrazine, J. Org. Chem., 1988, 53, 1415 CrossRef CAS.
- M. D. Coburn, G. A. Buntain, B. W. Harris, M. A. Hiskey, K. Y. Lee and D. G. Ott, An improved synthesis of 3,6-diamino–1,2,4,5-tetrazine. II. From triaminoguanidine and 2,4-pentanedione, J. Heterocycl. Chem., 1991, 28, 2049 CrossRef CAS.
- D. E. Chavez and M. A. Hiskey, Synthesis of the bi–heterocyclic parent ring system 1,2,4-triazolo[4,3-b][1,2,4,5]tetrazine and some 3,6-disubstituted derivatives, J. Heterocycl. Chem., 1998, 35, 1329 CrossRef CAS.
- D. E. Chavez and M. A. Hiskey, 1,2,4,5-Tetrazine based energetic materials, J. Energy Mater., 1999, 17, 357 CrossRef CAS.
- Z. Novak, B. Bostai, M. Csekei, K. Lorincz and A. Kotschy, Selective Nucleophilic Substitutions on Tetrazines, Heterocycles, 2003, 60, 2653 CrossRef CAS.
- Z. Novak and A. Kotschy, First cross-coupling reactions on tetrazines, Org. Lett., 2003, 5, 3495 CrossRef CAS PubMed.
- Y. Xie, Y. Fang, Z. Huang, A. M. Tallon, C. W. Am Ende and J. M. Fox, Divergent Synthesis of Monosubstituted and Unsymmetrical 3,6- Disubstituted Tetrazines from Carboxylic Ester Precursors, Angew. Chem., 2020, 59, 16967 CrossRef CAS PubMed.
- H. Liu and Y. Wei, Novel synthesis of 3,6-disubstituted-1,2,4,5-tetrazine derivatives from hydrazones by using [hydroxyl(tosyloxy)iodo]benzene, Tetrahedron Lett., 2013, 54, 4645 CrossRef CAS.
- Y. Qu, F.-X. Sauvage, G. Clavier, F. Miomandre and P. Audebert, Metal-Free Synthetic Approach to 3-Monosubstituted Unsymmetrical 1,2,4,5-Tetrazines Useful for Bioorthogonal Reactions, Angew. Chem., Int. Ed., 2018, 57, 12057 CrossRef CAS PubMed.
- W. Mao, W. Shi, J. Li, D. Su, X. Wang, L. Zhang, L. Pan, X. Wu and H. Wu, Organocatalytic and Scalable Syntheses of Unsymmetrical 1,2,4,5-Tetrazines by Thiol-Containing Promotors, Angew. Chem., 2019, 131, 1118 CrossRef.
- N. Leconte, A. Keromnes-Wuillaume, F. Suzenet and G. Guillaumet, Efficient Palladium-Catalyzed Synthesis of Unsymmetrical (Het)aryltetrazines, Synlett, 2007,(2), 204 CAS.
- A. M. Bender, T. C. Chopko, T. M. Bridges and C. W. Lindsley, Preparation of Unsymmetrical 1,2,4,5-Tetrazines via a Mild Suzuki Cross-Coupling Reaction, Org. Lett., 2017, 19, 5693 CrossRef CAS.
- W. D. Lambert, Y. Fang, S. Mahapatra, Z. Huang, C. W. Ende and J. M. Fox, Installation of Minimal Tetrazines through Silver-Mediated Liebeskind–Srogl Coupling with Arylboronic Acids, J. Am. Chem. Soc., 2019, 141, 17068 CrossRef CAS PubMed.
- J. C. T. Carlson, L. G. Meimetis, S. A. Hildebrandt and R. Weissleder, BODIPY–Tetrazine Derivatives as Superbright Bioorthogonal Turn–on Probes, Angew. Chem., Int. Ed., 2013, 52, 6917 CrossRef CAS PubMed.
- C. Testa, E. Gigot, S. Genc, R. Decreau, J. Roger and J. C. Hierso, Ortho-Functionalized Aryltetrazines by Direct Palladium-Catalyzed C-H Halogenation: Application to Fast Electrophilic Fluorination Reactions, Angew. Chem., Int. Ed., 2016, 55, 5555 CrossRef CAS PubMed.
- C. D. Mboyi, C. Testa, S. Reeb, S. Genc, H. Gattey, P. F. Ltssard, J. Roger and J. C. Hierso, Building Diversity in ortho-Substituted s-Aryltetrazines By Tuning N-Directed Palladium C–H Halogenation: Unsymmetrical Polyhalogenated and Biphenyl s-Aryltetrazines, ACS Catal., 2017, 7, 8493 CrossRef CAS.
- H. Xiong, Y. Gu, S. Zhang, F. Lu, Q. Ji, L. Liu, P. Ma, G. Yang, W. Hou and H. Xu, Iridium-catalyzed C–H amidation of s-tetrazines, Chem. Commun., 2020, 56, 4692 RSC.
- C. D. Mboyi, A. Daher, N. Khirzada, C. H. Devillers, H. Cattey, P. Fleurat-Lessard, J. Roger and J.-C. Hierso, Synthesis and structural characterisation of bulky heptaaromatic (hetero)aryl o-substituted s-aryltetrazines, New J. Chem., 2020, 44, 15235 RSC.
- C. D. Mboyi, D. Vivier, A. Daher, P. Fleurat-Lessard, H. Cattey, C. H. Devillers, C. Bernhard, F. Denat, J. Roger and J.-C. Hierso, Bridge Clamp Bis-Tetrazines Stacked by [N]8-π-Interactions and Azido-s-Aryl Tetrazines: New Classes of Doubly Clickable Tetrazines, Angew. Chem., 2020, 59, 1149 CrossRef CAS.
- E. Kurach, D. Djurado, J. Rimarcik, A. Kornet, M. Wlostowski, V. Lukes, J. Pecaut, M. Zagorska and A. Pron, Effect of substituents on redox, spectroscopic and structural properties of conjugated diaryltetrazines—a combined experimental and theoretical study, Phys. Chem. Chem. Phys., 2011, 13, 2690 RSC.
- M. Plugge, V. Alain-Rizzo, P. Audebert and A. M. Brouwer, Excited State Dynamics of 3,6-Diaryl-1,2,4,5-tetrazines, J. Photochem. Photobiol., A, 2012, 234, 12 CrossRef CAS.
- D. K. Hwang, R. R. Dasari, M. Fenoll, V. Alain-Rizzo, A. Dindar, J. W. Shim, N. Deb, C. Fuentes-Hernandes, S. Barlow, D. G. Bucknall, P. Audebert, S. R. Marder and B. Kippelen, Stable Solution–Processed Molecular n–Channel Organic Field–Effect Transistors, Adv. Mater., 2012, 24, 4445 CrossRef CAS PubMed.
- P. Zhang, C. Li, Y. Zhao, Y. Li and Y. Tu, Controlling Morphology of Active Layer by Tuning Coplanarity of the Centrality in Acceptor–Donor–Acceptor Small Molecules for Photovoltaic Application, Chin. J. Chem., 2013, 31, 1439 CrossRef CAS.
- C. Li, H. Ge, B. Yin, M. She, P. Liu and X. Li, Novel 3,6-unsymmetrically disubstituted-1,2,4,5-tetrazines: S-induced one-pot synthesis, properties and theoretical study, RSC Adv., 2015, 5, 12277 RSC.
- M. Moral, G. García, A. Garzón, J. M. Granadino-Roldán, M. A. Fox, D. S. Yufit, A. Peňas, M. Melguizo and M. Fernández-Gómez, Electronic Structure and Charge Transport Properties of a Series of 3,6-(Diphenyl)–s–tetrazine Derivatives: Are They Suitable Candidates for Molecular Electronics?, J. Phys. Chem. C, 2014, 118, 26427 CrossRef CAS.
- C. Quinton, V. Alain-Rizzo, C. Dumas-Verdes, F. Miomandre and P. Audebert, Design of New Tetrazine–Triphenylamine Bichromophores – Fluorescent Switching by Chemical Oxidation, Eur. J. Org. Chem., 2012, 1394 CrossRef CAS.
- C. Quinton, V. Alain-Rizzo, C. Dumas-Verdes, F. Miomandre and P. Audebert, Tetrazine–triphenylamine dyads: Influence of the nature of the linker on their properties, Electrochim. Acta, 2013, 110, 693 CrossRef CAS.
- C. Quinton, S.-H. Chi, C. Dumas-Verdes, P. Audebert, G. Clavier, J. W. Perry and V. Alain-Rizzo, Novel s-tetrazine-based dyes with enhanced two-photon absorption cross-section, J. Mater. Chem. C, 2015, 3, 8351 RSC.
- S. Pluczyk, P. Zassowski, C. Quinton, P. Audebert, V. Alain-Rizzo and M. Lapkowski, The influence of the linker on electrochemical and spectroelectrochemical properties of donor-acceptor-donor triphenylamine-s-tetrazine derivatives, Electrochim. Acta, 2016, 216, 160 CrossRef CAS.
- C. Quinton, V. Alain-Rizzo, C. Dumas-Verdes, G. Clavier, L. Vignau and P. Audebert, Triphenylamine/tetrazine based π-conjugated systems as molecular donors for organic solar cells, New J. Chem., 2015, 39, 9700 RSC.
- E. Aslan, M. Karaman, G. Yanalak, M. Can, F. Ozel and I. H. Patir, The investigation of novel D–π-A type dyes (MK-3 and MK-4) for visible light driven photochemical hydrogen evolution, Dyes Pigm., 2019, 171, 107710 CrossRef CAS.
- E. Aslan, M. Karaman, G. Yanalak, H. Bilgili, M. Can, F. Ozel and I. H. Patir, Synthesis of novel tetrazine based D–π-A organic dyes for photoelectrochemical and photocatalytic hydrogen evolution, J. Photochem. Photobiol., A, 2020, 390, 112301 CrossRef CAS.
- A. Kedzia, A. Kudelko, M. Swiatkowski and R. Kruszynski, Microwave-promoted synthesis of highly luminescent s-tetrazine-1,3,4-oxadiazole and s-tetrazine-1,3,4-thiadiazole hybrids, Dyes Pigm., 2020, 172, 107865 CrossRef.
- A. Kedzia, A. Kudelko, M. Swiatkowski and R. Kruszynski, Highly fluorescent 1,2,4,5-tetrazine derivatives containing 1,3,4-oxadiazole ring conjugated via a 1,4-phenylene linker, Dyes Pigm., 2020, 183, 108715 CrossRef CAS.
- N. K. Devaraj, S. A. Hilderbrand, R. Upadhyay, R. Mazitschek and R. Weissleder, Bioorthogonal turn-on probes for imaging small molecules inside living cells, Angew. Chem., Int. Ed., 2010, 49, 2869 CrossRef CAS PubMed.
- N. K. Devaraj, R. Upadhyay, J. B. Haun, S. A. Hilderbrand and R. Weissleder, Fast and sensitive pretargeted labeling of cancer cells through a tetrazine/trans-cyclooctene cycloaddition, Angew. Chem., Int. Ed., 2009, 48, 7013 CrossRef CAS PubMed.
- Y. Qu, P. Pander, O. Vybornyi, M. Vasylieva, R. Guillot, F. Miomandre, F. B. Dias, P. Skabara, P. Data, G. Clavier and P. Audebert, Donor–Acceptor 1,2,4,5-Tetrazines Prepared by the Buchwald–Hartwig Cross-Coupling Reaction and Their Photoluminescence Turn-On Property by Inverse Electron Demand Diels–Alder Reaction, J. Org. Chem., 2020, 85, 3407 CrossRef CAS PubMed.
- L. G. Meimetis, J. C. T. Carlson, R. J. Giedt, R. H. Kohler and R. Weissleder, Ultrafluorogenic coumarin-tetrazine probes for real-time biological imaging, Angew. Chem., Int. Ed., 2014, 53, 7531 CrossRef CAS.
- A. Wieczorek, P. Werther, J. Euchner and R. Wombacher, Green- to far-red-emitting fluorogenic tetrazine probes – synthetic access and no-wash protein imaging inside living cells, Chem. Sci., 2017, 8, 1506 RSC.
- X. Liu, Q. Qiao, W. Tian, W. Liu, J. Chen, M. J. Lang and Z. Xu, Aziridinyl Fluorophores Demonstrate Bright Fluorescence and Superior Photostability by Effectively Inhibiting Twisted Intramolecular Charge Transfer, J. Am. Chem. Soc., 2016, 138, 6960 CrossRef CAS PubMed.
- X. Lv, C. Gao, T. Han, H. Shi and W. Guo, Improving the quantum yields of fluorophores by inhibiting twisted intramolecular charge transfer using electron-withdrawing group-functionalized piperidine auxochromes, Chem. Commun., 2020, 56, 715 RSC.
- J. Galeta, R. Dzijak, J. Oboril, M. Drazinsky and M. Vrabel, A Systematic Study of Coumarin–Tetrazine Light–Up Probes for Bioorthogonal Fluorescence Imaging, Chem. – Eur. J., 2020, 26, 9945 CrossRef CAS PubMed.
- H. Xu, J. Yang, J. Seckute and N. K. Devaraj, In situ synthesis of alkenyl tetrazines for highly fluorogenic bioorthogonal live-cell imaging probes, Angew. Chem., Int. Ed., 2014, 53, 5805 CrossRef PubMed.
- X. Wu, B. T. Cisneros, C. M. Cole and N. K. Devaraj, Bioorthogonal Tetrazine-Mediated Transfer Reactions Facilitate Reaction Turnover in Nucleic Acid-Templated Detection of MicroRNA, J. Am. Chem. Soc., 2014, 136, 17942 CrossRef PubMed.
- E. Kozma, G. E. Girona, G. Paci, E. A. Lemke and P. Kele, Bioorthogonal double-fluorogenic siliconrhodamine probes for intracellular super-resolution microscopy, Chem. Commun., 2017, 53, 6696 RSC.
- A. Vazquez, R. Dzijak, M. Drazinsky, R. Rampmaier, S. J. Siegl and M. Vrabel, Mechanism–Based Fluorogenic trans–Cyclooctene–Tetrazine Cycloaddition, Angew. Chem., 2017, 129, 1354 CrossRef.
- S. J. Siegl, A. Vazquez, R. Dzijak, M. Drazinsky, J. Galeta, R. Rampmaier, B. Klepetarova and M. Vrabel, Design and Synthesis of Aza–Bicyclononene Dienophiles for Rapid Fluorogenic Ligations, Chem. – Eur. J., 2018, 24, 2426 CrossRef CAS PubMed.
- S. J. Siegl, J. Galeta, R. Dzijak, A. Vazquez, M. D. Rio-Villanueva, M. Drazinsky and M. Vrabel, An Extended Approach for the Development of Fluorogenic trans–Cyclooctene–Tetrazine Cycloadditions, ChemBioChem, 2019, 20, 886 CrossRef CAS PubMed.
- Z.-C. Wu and D. L. Boger, Synthesis, Characterization, and Cycloaddition Reactivity of a Monocyclic Aromatic 1,2,3,5-Tetrazine, J. Am. Chem. Soc., 2019, 141, 16388 CrossRef CAS PubMed.
- Z. Li, J. Ding, N. Song, J. Li and Y. Tao, Development of a New s-Tetrazine-Based Copolymer for Efficient Solar Cells, J. Am. Chem. Soc., 2010, 132, 13160 CrossRef CAS PubMed.
- H. Fischer, T. Muller, I. Umminger, F. A. Neugebauer, H. Chandra and M. C. R. Symons, Radical Cations and Anions of 1,2,4,5-Tetrazines: An Electron Spin Resonance and Cyclic Voltammetric Study, J. Chem. Soc., Perkin Trans. 2, 1988, 413 RSC.
- J. Ding, N. Song and Z. Li, Synthesis, characterization and photovoltaic applications of a low band gap polymer based on s-tetrazine and dithienosilol, Chem. Commun., 2010, 46, 8668 RSC.
- Z. Li, J. Ding, N. Song, X. Du, J. Zhou, J. Lu and Y. Tao, Alternating Copolymers of Dithienyl-s-Tetrazine and Cyclopentadithiophene for Organic Photovoltaic Applications, Chem. Mater., 2011, 23, 1977 CrossRef CAS.
- M.-J. Baek, W. Jang, S.-H. Lee and Y.-S. Lee, Synthesis of new alternating conjugated copolymers consisting of tetrazine/carbazole or tetrazine/fluorene derivatives along with thiophene spacers for photovoltaic applications, Synth. Met., 2012, 161, 2785 CrossRef.
- Y. Chen, C. Li, P. Zhang, Y. Li, X. Yang, L. Chen and Y. Tu, Solution-processable tetrazine and oligothiophene based linear A–D–A small molecules: Synthesis, hierarchical structure and photovoltaic properties, Org. Electron., 2013, 14, 1424 CrossRef CAS.
- P. Ma, C. Wang, S. Wen, L. Wang, L. Shen, W. Guo and S. Ruan, Small molecules based on tetrazine unit for efficient performance solution-processed organic solar cells, Sol. Energy Mater. Sol. Cells, 2016, 155, 30 CrossRef CAS.
- A.-C. Knall, S. F. Hoefier, M. Hollauf, F. Thaler, S. Noesberger, L. Hanzu, H. Ehmann, M. Nobisch, S. Spirk, S. Wen, F. Yang, T. Flath and G. Trimmel, Synthesis of a tetrazine-quaterthiophene copolymer and its optical, structural and photovoltaic properties, J. Mater. Sci., 2019, 54, 10065 CrossRef CAS PubMed.
- L. Peng, Y. Yu, J. Lu, P. He, G. Wang, M. Huang, B. Zhao, Y. Pei and S. Tan, Development of s-tetrazine-based polymers for efficient polymer solar cells by controlling appropriate molecular aggregation, Dyes Pigm., 2019, 171, 107717 CrossRef CAS.
- F. Bureš, Fundamental aspects of property tuning in push–pull molecules, RSC Adv., 2014, 4, 58826 RSC.
- B. Pinto-Pacheco, W. P. Carbery, S. Khan, D. B. Turner and D. Buccella, Fluorescence Quenching Effects of Tetrazines and Their Diels–Alder Products: Mechanistic Insight Toward Fluorogenic Efficiency, Angew. Chem., 2020, 59, 22140 CrossRef CAS PubMed.
- J.-B. Béquignat, N. Ty, A. Rondon, L. Taiariol, F. Degoul, D. Canitrot, M. Quintana, I. Navarro-Teulon, E. Miot-Noirault, C. Boucheix, J.-M. Chezal and E. Moreau, Optimization of IEDDA bioorthogonal system: Efficient process to improve trans-cyclooctene/tetrazine interaction, Eur. J. Med. Chem., 2020, 203, 112574 CrossRef.
- G. Knorr, E. Kozma, A. Herner, E. A. Lemke and P. Kele, New Red-Emitting Tetrazine-Phenoxazine Fluorogenic Labels for Live-Cell Intracellular Bioorthogonal Labeling Schemes, Chem. – Eur. J., 2016, 22, 8972 CrossRef CAS PubMed.
- Y. Lee, W. Cho, J. Sung, E. Kim and S. B. Park, Monochromophoric Design Strategy for Tetrazine-Based Colorful Bioorthogonal Probes with a Single Fluorescent Core Skeleton, J. Am. Chem. Soc., 2018, 140, 974 CrossRef CAS PubMed.
- A. Loredo, J. Tang, L. Wang, K.-L. Wu, Z. Peng and H. Xiao, Tetrazine as a general phototrigger to turn on fluorophores, Chem. Sci., 2020, 11, 4410 RSC.
- C. Wang, C. Li, S. Wen, P. Ma, G. Wang, C. Wang, H. Li, L. Shen, W. Guo and S. Ruan, Enhanced Photovoltaic Performance of Tetrazine-Based Small Molecules with Conjugated Side Chains, ACS Sustainable Chem. Eng., 2017, 5, 8684 CrossRef CAS.
- F. Miomandre and P. Audebert, 1,2,4,5-Tetrazines: An intriguing heterocycles family with outstanding characteristics in the field of luminescence and electrochemistry, J. Photochem. Photobiol., C, 2020, 44, 100372 CrossRef CAS.
| This journal is © the Partner Organisations 2021 |