DOI:
10.1039/D1NH00254F
(Communication)
Nanoscale Horiz., 2021,
6, 729-743
Gene-editing by CRISPR–Cas9 in combination with anthracycline therapy via tumor microenvironment-switchable, EGFR-targeted, and nucleus-directed nanoparticles for head and neck cancer suppression†
Received
6th May 2021
, Accepted 9th July 2021
First published on 29th July 2021
Abstract
Head and neck cancer (HNC) has a high incidence and a poor prognosis. Epirubicin, a topoisomerase inhibitor, is a potential anthracycline chemotherapeutic for HNC treatment. HuR (ELAVL1), an RNA-binding protein, plays a critical role in promoting tumor survival, invasion, and resistance. HuR knockout via CRISPR/Cas9 (HuR CRISPR) is a possible strategy for the simultaneous modulation of the various pathways of tumor progression. Multifunctional nanoparticles modified with pH-sensitive epidermal growth factor receptor (EGFR)-targeting and nucleus-directed peptides were designed for the efficient delivery of HuR CRISPR and epirubicin to human tongue squamous carcinoma SAS cells and SAS tumor-bearing mice. The pH-sensitive nanoparticles responded to the acidic pH value as a switch to expose the targeting peptides. The cellular uptake and transfection efficiency of these nanoparticles in SAS cells increased via EGFR targeting, ligand-mediated endocytosis, and endosomal escape. These nanoparticles showed low cytotoxicity towards normal oral keratinocyte NOK cells. CRISPR/Cas9 was transported into the nucleus via the nuclear directing peptide and successfully knocked out HuR to suppress proliferation, metastasis, and resistance in SAS cells. The multiple inhibition of EGFR/β-catenin/epithelial–mesenchymal transition pathways was mediated through modulating the EGFR/PI3K/mTOR/AKT axis. The co-treatment of epirubicin and HuR CRISPR in SAS cells further facilitated apoptosis/necroptosis/autophagy and caused cancer cell death. In combination with HuR CRISPR nanoparticles, the efficacy and safety of epirubicin nanoparticles against cancer in SAS tumor-bearing mice improved significantly. Collectively, these nanoparticles showed a tumor pH response, active EGFR targeting, and nuclear localization and thus offered a combinatorial spatiotemporal platform for chemotherapy and the CRISPR/Cas gene-editing system.
New concepts
The accompanying problems associated with treatment failure of head and neck cancer (HNC) include early relapse, distant metastasis, and poor prognosis. Human antigen R (HuR), an RNA binding protein encoded by the ELAVL1 gene, plays an important role in regulating survival, metastasis, invasion, and multidrug resistance of the tumor. To specifically transport the HuR gene-editing plasmid of CRISPR–Cas9 (HuR CRISPR) into the nucleus of cancer cells is a big challenge. Tumor pH-tunable, EGFR-targeted, and nucleus-directed nanoparticles were, for the first time, utilized to deliver HuR CRISPR. In combination with the anthracycline chemotherapeutic, the co-treatment enhanced chemotherapy-induced programmed cell death through suppression of the EGFR/PI3K/mTOR/AKT axis. This combination triggered multiple inhibition of proliferation, metastasis, invasion, and resistance in HNC. The smart nanoparticles may provide a combinatorial spatiotemporal platform against tumors for chemotherapy and the CRISPR/Cas gene-editing system with the advantages of a tumor pH response, active EGFR targeting, and nuclear localization.
|
1. Introduction
Oral squamous cell carcinoma (OSCC) is a common head and neck cancer (HNC) with a poor prognosis, high mortality rate, and an increased likelihood of recurrence.1 The patients of OSCC are often unresponsive to traditional chemotherapeutic agents such as 5-fluorouracil and cisplatin after several treatment regimens.2 The accompanying problems may include early relapse, distant metastasis, and poor prognosis.1,2 Previous studies showed that increased neoplastic transformation in oral tissues is significantly associated with the unusual activation of the Wnt/β-catenin signaling pathway,3,4 which can affect tumor proliferation and migration and trigger cancer progression.5 Among various regulatory proteins, human antigen R (HuR), an RNA binding protein encoded by the ELAVL1 gene, post-transcriptionally modulates mRNA stability and protein translation associated with survival, metastasis, invasion, and multidrug resistance (MDR) of the tumor.6,7 New evidence suggests that overexpression of HuR positively regulates various cancer-related transcripts of survival and resistance in various cancers such as OSCC.2,8 β-catenin, cyclin-D, and c-Myc, which are downstream proteins of the Wnt-activated pathway, are confirmed targets of HuR.7,9 The Wnt/β-catenin-mediated pathway also correlates with the upregulation of P-glycoprotein (P-gp) and multidrug resistance-associated proteins (MRPs),7,10 leading to poor anticancer drug efficacy in the MDR spectrum.11 The dysregulatory epithelial–mesenchymal transition (EMT) pathway is also linked to HuR overexpression12via zinc finger E-box binding 1 (ZEB1)- and transforming growth factor-beta 1 (TGF-β1)-induced Smad signaling to upregulate vimentin and N-cadherin.13–15 HuR modulates the RNA operon of anti-apoptotic genes such as Bcl-2 and Mcl-1 and thus contributes to the survival of cancer cells.16,17 The post-transcriptional silencing of HuR by siRNA (siHuR) in the repression of survival signaling, MDR reversal, and apoptosis induction in colorectal cancer cells (CRC) was partially verified in our previous study.7 However, siHuR only provided a knockdown of HuR expression. We have further developed an effective CRISPR/Cas9 gene-editing system to knock out HuR (HuR CRISPR). CRISPR/Cas9 enables sequence-specific genome editing for transcriptional control via the Cas9 endonuclease directed by single-guide RNA (sgRNA) to identify the protospacer-adjacent motif (PAM) sequence and a complementary 20-nucleotide genomic sequence, which triggers double-strand breaks in the target DNA.18 The following deletions knock out the targeted HuR gene.19 For better knockout efficiency, Cas9 and sgRNA must occur in the same cells.18 Therefore, we designed suitable solid lipid nanoparticles (SLN), namely HuR CRISPR/SLN, for the co-delivery of CRISPR/Cas9 and programmable sgRNA to achieve effective HuR suppression in SAS cells in vitro and in vivo.
The combined treatment of chemotherapy-loaded nanoparticles and HuR CRISPR/SLN can be an effective anticancer strategy. Epirubicin (Epi) is not the drug of choice for the therapy of HNC but was selected in this study as an anthracycline chemotherapeutic, which may be a potential antineoplastic drug for OSCC. Epi acts as a topoisomerase II inhibitor by intercalating with DNA and disrupting DNA and RNA production.10 It is a stereoisomer of doxorubicin but has the advantage of lower cardiotoxicity than doxorubicin.20 However, epirubicin is a substrate of P-gp, MRP1, and MRP2, which may cause resistance in many tumors after epirubicin treatment.7 In our previous studies, Epi showed a strong apoptosis-inducing effect against various types of cancer by triggering intrinsic mitochondrial signaling10,20via the production of intracellular reactive oxygen species (ROS).20 Our recent study demonstrated the lethal role of autophagy in Epi-treated cervical cancer cells, suggesting that promoting autophagy may improve the therapeutic efficacy in resistant tumors.21 In this study, we reported on the design of Epi-loaded liposomes (Epi/Lip) for the sustained release of Epi and to reduce the toxicity of Epi to normal cells. The enhanced cellular uptake and tumor-specific recognition were further modified by peptide conjugation, as described below.
The tumor microenvironment typically exhibits a low pH (pH 6.0–6.5) because of aggressive glycolysis at the tumor site,22 indicating that a pH-responsive biomaterial may be a prospective avenue for tumor-specific drug delivery. In this study, we modified SLN and Lip with three cell-penetrating peptides (CPPs), including the pH-sensitive H-peptide as a switch for the tumor microenvironment, P-peptide for targeting the epithelial growth factor receptor (EGFR), and R-peptide for nucleus directing to form SLN-HPR and Lip-HPR. Also, the PEG chains on the surface of the nanoparticles can protect SLN and Lip from uptake by the reticuloendothelial system (RES) during systemic circulation.23 The H-peptide has a characteristic branched arginine-rich sequence for the CPP function and a pH-responsive histidine-rich sequence. At physiological pH, the H-peptide hides its hydrophilic arginine- and histidine-rich residues to expose lipophilic amino acid residues that interact with the hydrophobic moiety of lipid nanoparticles and form a stable conformation.24,25 In an acidic environment, however, ionization of polyhistidine residues switches the H-peptide from a hydrophobic to a hydrophilic conformation to expose peptide P for targeting the EGFR of tumor cells. The pH-responsive alteration of the H peptide in an acidic environment ensures the appropriate display of the P and R peptides at tumor sites to achieve the following EGFR and nuclear targeting purposes. The P-peptide is a ligand that can target EGFR that is overexpressed in various cancer cells, including OSCC SAS cells.26 Nanoparticles modified with the P-peptide can bind to EGFR-upregulated tumor cells to increase the transfection efficiency of lipopolyplex formulations.27 The R-peptide is derived from the nuclear localization sequence (NLS) of a human phosphatidate phosphatase and shows high deliverability, no immunogenicity, and/or low toxicity.28 The cationic R-peptide can increase the nuclear delivery of drugs, as its specific NLS can affect the subcellular distribution of co-delivered cargos.29 Therefore, we expect that the cellular uptake of these nanoparticles in SAS cells may be increased via EGFR targeting and ligand-mediated endocytosis. After escaping from endosomes and lysosomes, HuR CRISPR and Epi, which were released from SLN-HPR or Lip-HPR, are transported into the nucleus with the help of the R-peptide. The design of Epi/Lip-HPR or HuR CRISPR/SLN-HPR is shown in Fig. 1A.
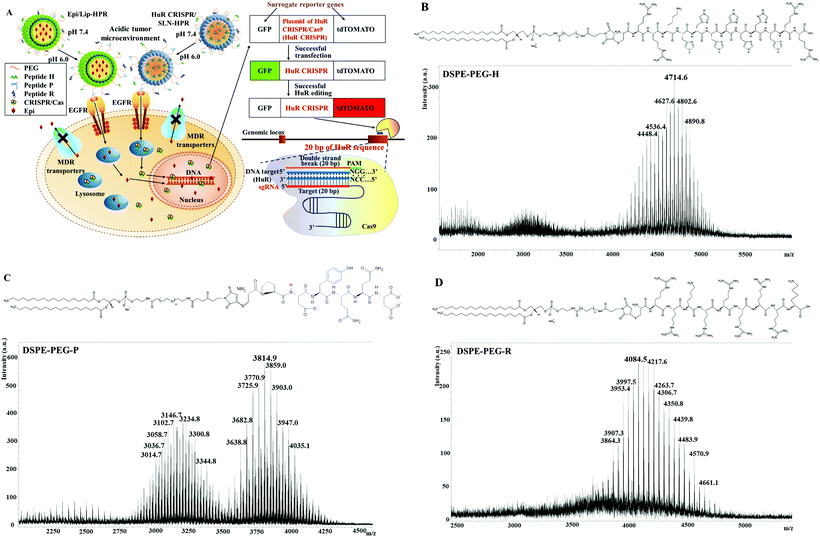 |
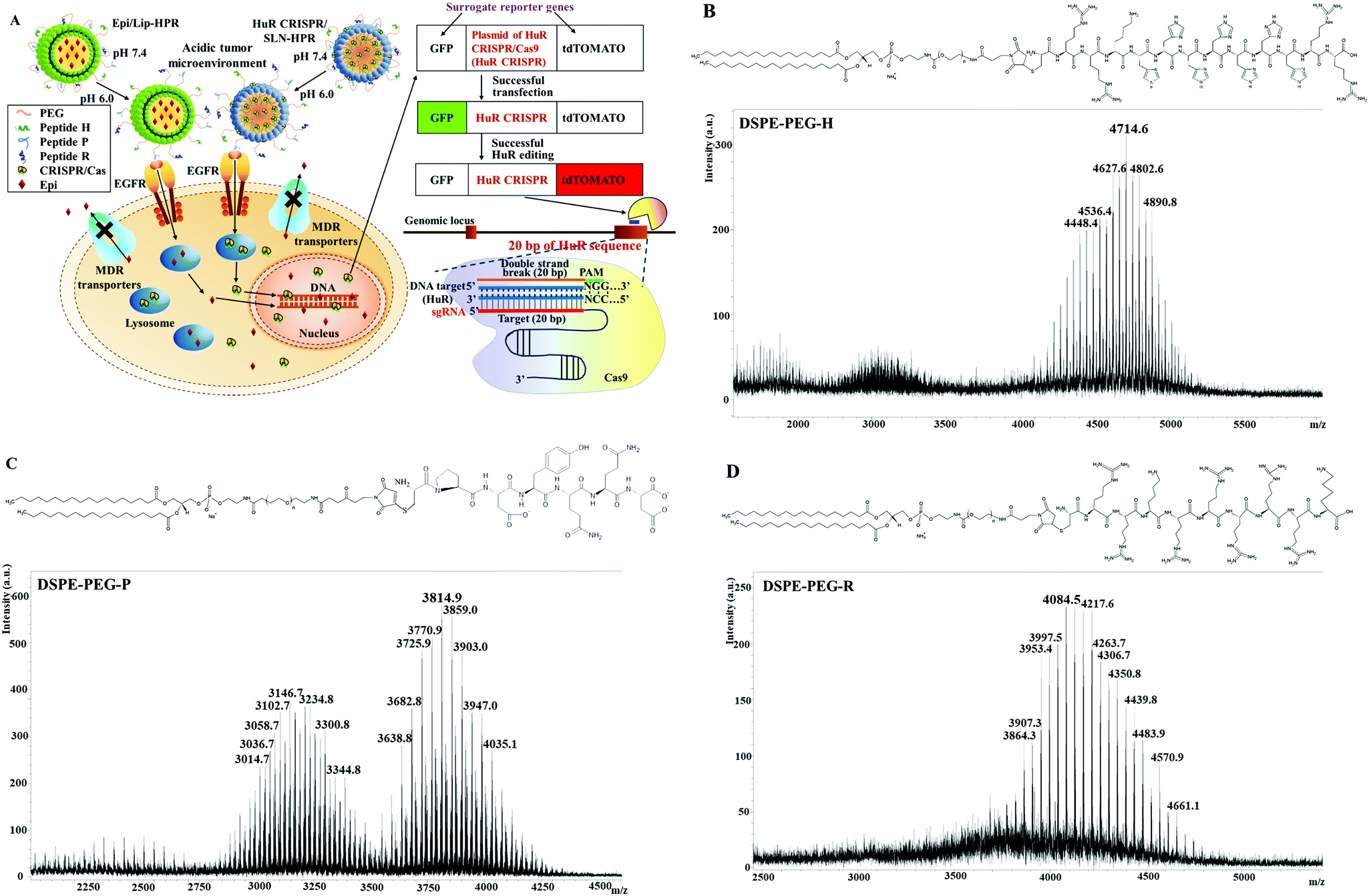
| | Fig. 1 (A) Scheme of the design of pH-responsive and targeted solid lipid nanoparticles (SLN) and liposomes (Lip) encapsulating Epi (Epi) and the HuR CRISPR plasmid, respectively. If HuR CRISPR and the reporter gene system were to enter the cells, the green fluorescence of the green fluorescent protein (GFP) would be illuminated. When the CRISPR/Cas9 system cut off the designed HuR sequence of 20 bp in the target cells, the red fluorescence of the fluorescent tdTomato protein (tdTomato) would be illuminated. (B–D) Conjugation of DSPE-PEG-maleimide to H, P, and R. The mass spectra of the corresponding lipid–peptide conjugates of (B) DSPE-PEG-H, (C) DSPE-PEG-P, and (D) DSPE-PEG-R were detected using a MALDI-TOF mass spectrometer. | |
2. Results and discussion
2.1. Characterization of HuR CRISPR/SLN-HPR and Epi/Lip-HPR
Epi, an anthracycline, still shows the drawbacks of hematologic or cardiac toxicity, even though it displays a more promising therapeutic index than that of doxorubicin at comparable doses.30 Moreover, the common occurrence of MDR further hinders the anticancer efficacy of Epi for various tumor types.7 The CRISPR/Cas system also shows the disadvantages of rapid degradation, off-target effects, and limited cellular uptake.18 Hence, it is necessary to develop pH-sensitive, tumor-targeted, and nucleus-localized delivery systems with excellent uptake of Epi and HuR CRISPR to enhance the treatment of HNC, as illustrated in Fig. 1A.
DSPE-PEG peptides were synthesized by conjugating DSPE-PEG maleimide with the respective H-, P- and R-peptides. The DSPE-PEG-H/P/R structure is shown in Fig. 1B–D, and the mass spectrometric data confirmed that the H-, P-, or R-peptide was successfully conjugated to DSPE-PEG. The size, PDI, zeta potential, encapsulation efficiency (EE%), and drug loading efficiency (DL%) of HuR CRISPR/SLN-HPR and Epi/Lip-HPR are summarized in Table 1. The data in Fig. 1B–D and Table 1 demonstrate that these specially-designed peptides have been successfully linked into lipids to prepare the indicated nanoparticle formulations with high EE% and DL% and homogeneous size distributions.
Table 1 Characterization of the Lip and SLN formulations
| Formulations |
HuR CRISPR/SLN |
HuR CRISPR/SLN-HPR |
Epi/Lip |
Epi/Lip-HPR |
| Results are shown as the mean ± standard deviation (SD). |
| Average size (nm) |
147.30 ± 3.39 |
159.80 ± 3.87 |
152.50 ± 3.46 |
164.50 ± 1.34 |
| PDI |
0.16 ± 0.08 |
0.14 ± 0.01 |
0.08 ± 0.01 |
0.09 ± 0.02 |
| Zeta potential (mV) |
8.55 ± 0.30 |
9.39 ± 0.33 |
−15.70 ± 0.47 |
8.24 ± 0.48 |
| Encapsulation efficiency (%) |
91.28 ± 2.54 |
95.47 ± 4.38 |
92.32 ± 1.23 |
93.45 ± 1.36 |
| Drug-loading capacity (%) |
19.38 ± 4.37 |
7.21 ± 6.59 |
20.58 ± 2.81 |
22.66 ± 2.48 |
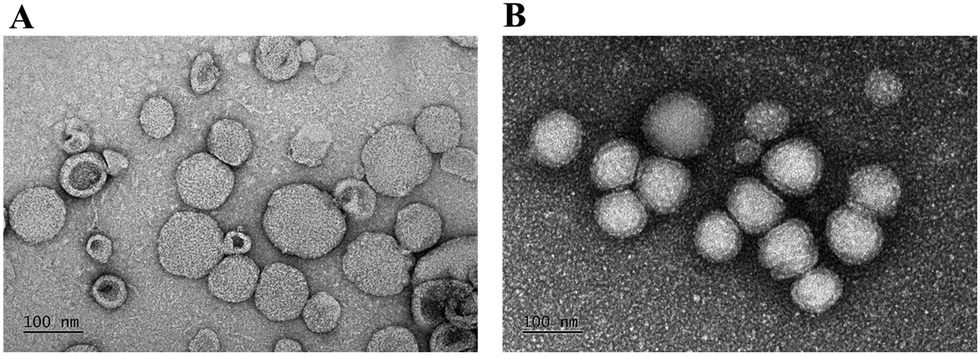
Also, the morphologies of HuR CRISPR/SLN-HPR and Epi/Lip-HPR as detected by transmission electron microscopy (TEM) are shown in Fig. 2A and B. Nanoparticles of a suitable size (20–200 nm) can have the advantage of passing through tumor vessels without uptake by RES.31 Cationic nanoparticles could easily enter the cells by electrostatic interactions due to negative charges on the surface of the plasma membrane.32
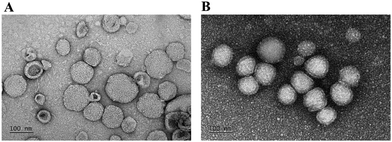 |
| | Fig. 2 TEM images of the nanoparticles. TEM images of (A) HuR CRISPR/SLN-HPR and (B) Epi/Lip-HPR were obtained using a JEM-2000EXIITEM. Bar = 100 nm. For each group, n = 3. A representative image is displayed. | |
2.2. The pH-sensitive cellular uptake, transfection, and drug release
The dominant lactate production by anaerobic glycolysis usually causes an acidic extracellular tumor microenvironment, resulting in the release of therapeutic agents from nanomedicines that respond to tumor acidity.33 Therefore, pH-sensitive nanoparticle-encapsulated drugs can be rapidly released under acidic conditions and can improve the anticancer efficacy in a tumor cell-specific manner.34 The peptide H in this study contains polyhistidine with imidazole groups that can change into cationic and hydrophilic states under acidic conditions.35,36 At physiological pH, the H-peptide is neutral and hydrophobic; it is thus stably covered on the surface of the nanoparticles.37,38
The results of pH-sensitive changes in particle size, PDI, and zeta potential were analyzed with a Zetasizer and are shown in Table 2. The size of HuR CRISPR/SLN-HPR and Epi/Lip-HPR increased as the pH decreased from 7.4 to 6.0. Accordingly, the zeta potential was increased to more positive values for HuR CRISPR/SLN-HPR and Epi/Lip-HPR due to the protonation of polyhistidine residues in the H-peptide under acidic pH conditions (Table 2). Our findings reveal that, in an acidic environment, the conformational alteration after protonation of the imidazole groups in the H-peptide leads to an open and polar structure, thereby exposing the hidden P- and R-peptides of SLN-HPR and Lip-HPR for EGFR binding and nuclear targeting.
Table 2 The pH-sensitive changes in particle size, PDI, and zeta potential of HuR CRISPR/SLN-HPR and Epi/Lip-HPR
| Formulations |
pH |
Size (nm) |
PDI |
Zeta potential (mV) |
| Results are shown as the mean ± SD. |
| HuR CRISPR/SLN-HPR |
7.4 |
159.80 ± 3.87 |
0.14 ± 0.01 |
9.39 ± 0.33 |
| 6.0 |
253.78 ± 16.94 |
3.29 ± 0.41 |
24.55 ± 0.57 |
|
|
| Epi/Lip-HPR |
7.4 |
164.50 ± 1.34 |
0.09 ± 0.02 |
8.24 ± 0.48 |
| 6.0 |
263.31 ± 15.88 |
4.11 ± 0.35 |
29.47 ± 0.61 |
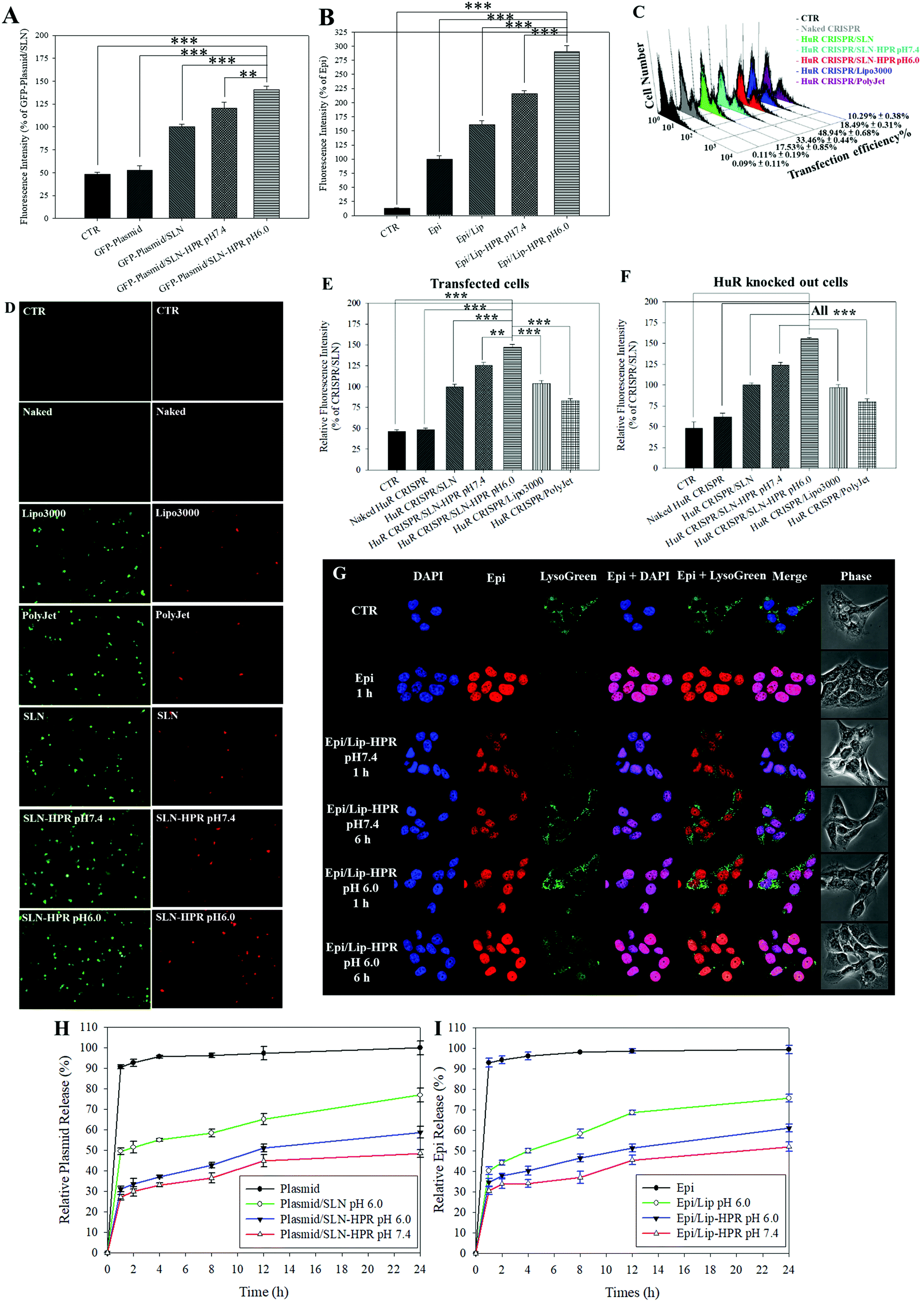
Fig. 3A and B show that the relative cellular uptake percentages of the GFP-plasmid and Epi by SLN-HPR and Lip-HPR in SAS cells were higher at pH 6.0 than at pH 7.4. To compare the relative percentage of the transfection efficiency of HuR CRISPR by different transfection reagents, the fluorescence intensity of the surrogate reporter system of GFP in HuR CRISPR was evaluated (Fig. 3C–E). The green fluorescence of GFP in HuR CRISPR indicated the successful transfection of the CRISPR system and the GFP reporter in SAS cells (Fig. 3C and E, left panels of Fig. 3D). SLN-HPR at pH 6.0 displayed a higher transfection efficiency than commercially available transfection reagents (Fig. 3C–E). SLN-HPR showed the highest transfection efficiency (48.94 ± 0.68%) and the highest percentage of relative fluorescence intensity of transfected cells compared to commercial transfection reagents at pH 6.0 in SAS cells (Fig. 3C and E). When the CRISPR/Cas9 system successfully cuts off the target 20 bp HuR gene in SAS cells, the red fluorescence of tdTomato is illuminated, as shown in Fig. 3F and the right panels of Fig. 3D. SLN-HPR showed more cells with green (transfected; left panels of Fig. 3D) and red (HuR knocked out; right panels of Fig. 3D) fluorescence than other transfection reagents such as Lipofectamine 3000 and PolyJet using a confocal laser scanning microscope (CLSM) (Fig. 3D). SLN-HPR also showed better HuR knockout efficiency than other formulations at pH 6.0 (Fig. 3F). These findings demonstrated that HPR peptides modified on SLN significantly increased the pH-responsive cellular uptake, transfection efficiency, and HuR knockout efficiency compared to other formulations (Fig. 3A and C–F). The results of CLSM also indicated that Epi released from Lip-HPR (pH 6.0) displayed incremental red fluorescence that was co-localized with the nuclear blue fluorescence (stained by DAPI), suggesting the successful endosomal escape of Epi from Lip-HPR at acidic pH to reach its target site in the nucleus (Fig. 3G).
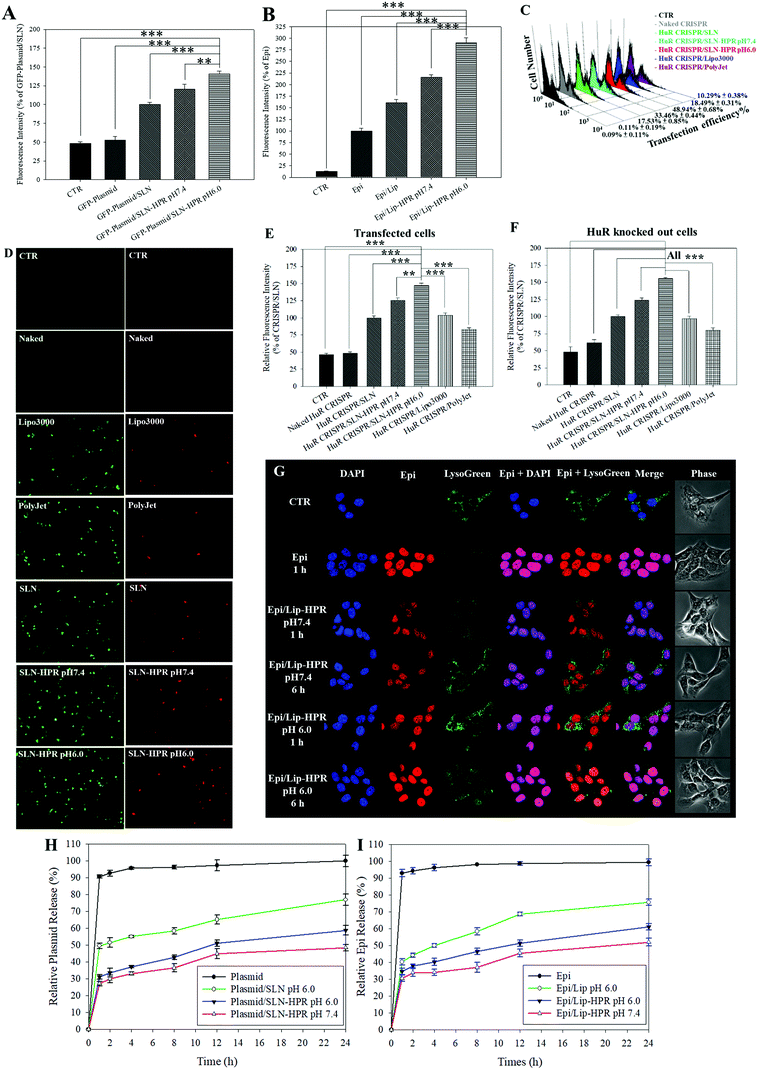 |
| | Fig. 3 pH-sensitive profiles of various formulations. (A and B) Cellular uptake of (A) the GFP-plasmid and (B) Epi in various formulations in SAS cells at pH 7.4 and 6.0, determined by flow cytometry. Results are shown as mean ± standard deviation (SD). *** p < 0.001. (C) The relative percentages of the transfection efficiency of HuR CRISPR by various formulations were measured by flow cytometry. (D) Transfected cells (green fluorescence) and HuR CRISPR-knocked out cells (red fluorescence). SAS cells were detected by fluorescence microscopy. The relative fluorescence intensity percentages of (E) green (transfected cells) and (F) red (HuR CRISPR-knocked out cells) were monitored by flow cytometry. *** p < 0.001. (G) pH-responsive intracellular localization of Epi/Lip-HPR in SAS cells at pH 7.4 and 6.0 by CLSM. DAPI: a nuclear dye; LysoTracker Green (LysoGreen): a lysosomal dye. (H and I) In vitro release profiles of (H) the plasmid and (I) Epi in different formulations at pH 6.0 and 7.4. | |
We also found that more than 90% of the plasmid or Epi was released from the HuR CRISPR plasmid or Epi solution during the first 1 h and the release reached 100% within 24 h (Fig. 3H and I). Nevertheless, the percentage of Epi released from Epi/Lip-HPR up to 24 h was 52.01 ± 2.23% at pH 7.4, which was increased to 61.08 ± 1.92% at pH 6.0. But, Epi released from Epi/Lip was escalated to 75.64 ± 1.89% at pH 6.0 (Fig. 3I). Notably, the release profiles of the plasmid from SLN-HPR also exhibited a comparable pH-dependent tendency to those of Epi from Lip-HPR (Fig. 3H). These findings supported the sustained release patterns of Epi and the plasmid at pH 7.4 and showed dramatic increases in the release percentages from Lip-HPR and SLN-HPR at an acidic pH level, at least partially due to the pH-sensitive H peptide-mediated alteration of these nano-formulations at an acidic pH value.
2.3. Toxicity to noncancerous and cancer cells and intracellular trafficking of SLN-HPR and Lip-HPR
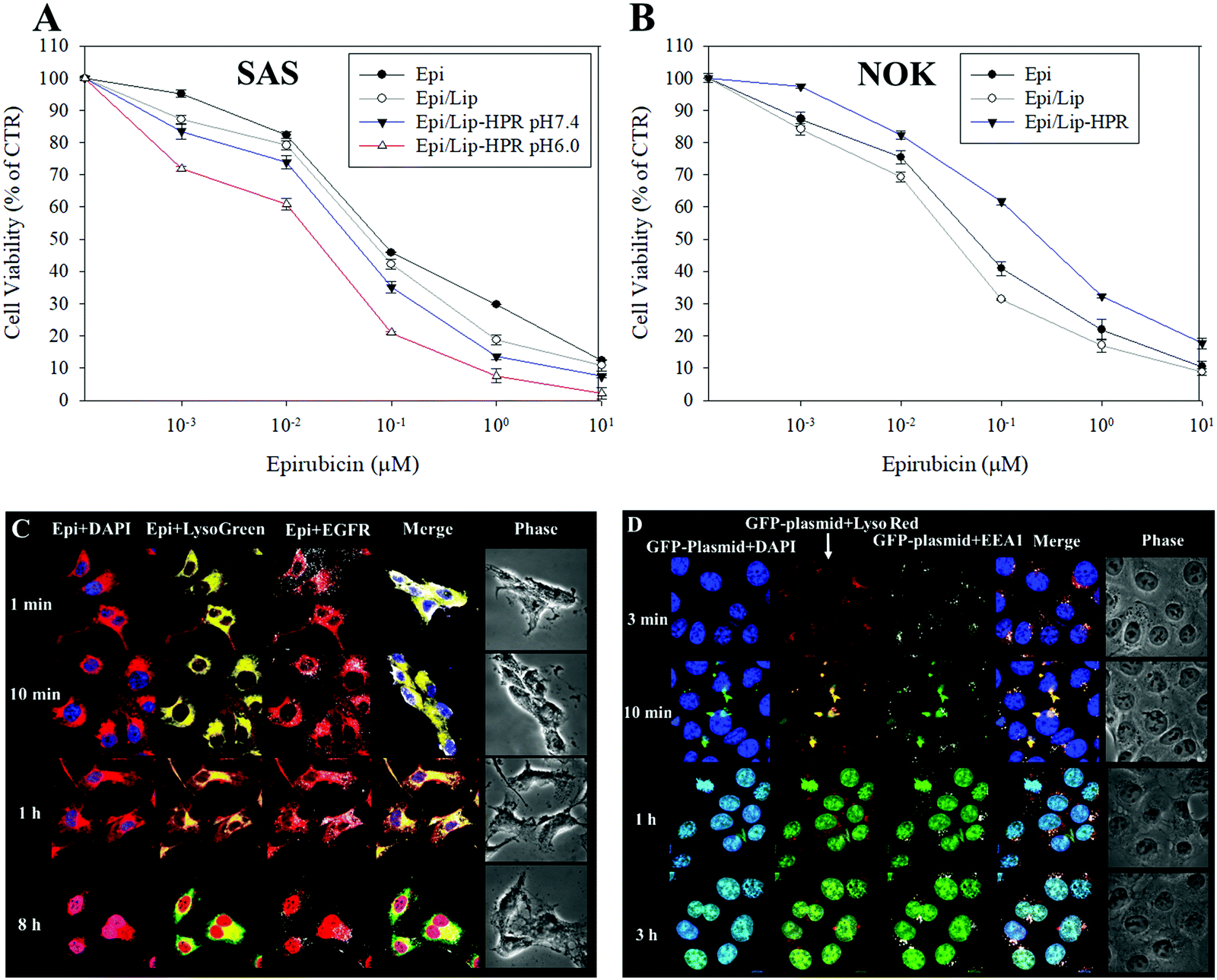
We tested the cytotoxicity of the Epi-loaded formulations against cancerous SAS and non-cancerous normal oral keratinocyte (NOK) cells by SRB assay. Among the Epi, Epi/Lip, and Epi/Lip-HPR formulations, Epi/Lip-HPR showed the highest cytotoxicity for SAS cells at acidic pH (Fig. 4A).
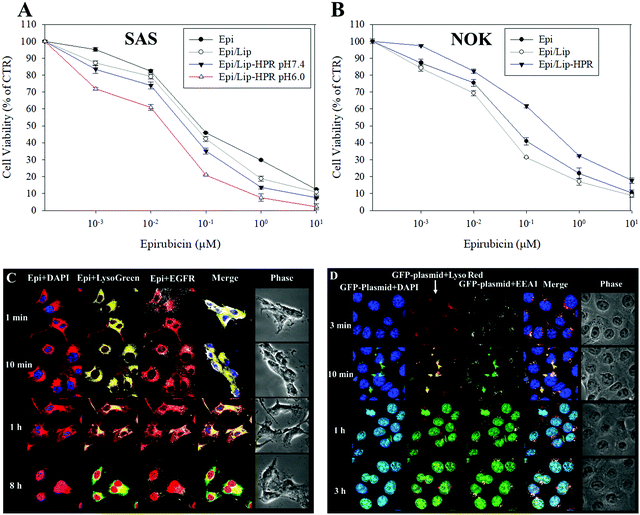 |
| | Fig. 4 Cytotoxicity of various Epi formulations on SAS and NOK cells and intracellular localization of GFP-plasmid/SLN-HPR and Epi/Lip-HPR in SAS cells. (A and B) Cytotoxicity of various Epi formulations on (A) SAS and (B) NOK cells after 48 h, measured by sulforhodamine B (SRB) assay. (C) Epi/Lip-HPR or (D) GFP-plasmid/SLN-HPR were added to SAS cells for the indicated time. Surface EGFR targeting and intracellular trafficking of nanoparticles in SAS cells were observed by CLSM. For (C): blue: DAPI (a nuclear dye); green: LysoGreen (a lysosomal dye); red: Epi; white: EGFR. For (D): blue: DAPI; green: the GFP-plasmid; red: LysoRed (LysoTracker Red; a lysosomal dye); white: EEA1 (early endosome antigen 1; an endosome marker). | |
In comparison, Epi/Lip-HPR caused less cytotoxicity to NOK cells (Fig. 4B), demonstrating the prevention of the release of Epi in normal cells by Lip-HPR at physiological pH. Although both cancerous and non-cancerous cells may have been damaged by Epi, EGFR targeting ligands such as P-peptide modified on the surface of Lip could also play a major role in targeting Epi/Lip-HPR to SAS, an EGFR-overexpressing cancer cell line.39 We observed the EGFR and nucleus-targeted effects of Epi/Lip-HPR in SAS cells by CLSM. CLSM images showed that Lip-HPR was targeted to EGFR after 1 min and then co-localized with lysosomes in SAS cells after 1 h. After escaping from the lysosome, Epi released from Lip-HPR accumulated in the nucleus after 8 h (Fig. 4C). The nucleus is the target site of Epi, which induces topoisomerase II inhibition by intercalation of DNA.21 These results confirmed that peptides P and R on Lip-HPR could help target Lip on EGFR, transport Epi to the nucleus, and increase cancer cell death (Fig. 4A and C). The toxicity to normal cells was reduced due to the dual protective effects of the H- (pH sensitivity) and P-peptides (tumor targeting), as shown in Fig. 4B.
Also, P-peptide-modified nanoparticles can target tumor cells with upregulated EGFR to increase the transfection efficiency of lipopolyplex formulations.27 P-peptide-conjugated nanocarriers suppressed EGFR activation to improve cellular uptake and cargo transport efficiency in tumor cells, thereby inhibiting the progression of cancer cells.27,40 We detected the uptake mechanisms of SLN-HPR and Lip-HPR using various endocytosis or fusion inhibitors. SLN-HPR and Lip-HPR entered SAS cells via multiple ways, including macropinocytosis, and adsorptive and clathrin- and caveolae-mediated endocytosis (Fig. S1, ESI†). Interestingly, the lone P-peptide, an EGFR ligand, also slightly inhibited cellular uptake of SLN-HPR and Lip-HPR in SAS cells, in part because the P-peptide alone could compete with SLN-HPR and Lip-HPR for binding to EGFR (Fig. S1, ESI†). Also, the results of the localization of GFP-plasmid/SLN-HPR in SAS cells indicated that a weak signal from the GFP-plasmid was detected after 3 min. However, the GFP-plasmid was co-localized with early endosomes and lysosomes in SAS cells after 10 min as shown by staining the endosomes with early endosomal antigen 1 (EEA1; an endosomal marker) (Fig. 4D). After 1 h and 3 h, the GFP-plasmid escaped from the endosomes/lysosomes and accumulated in the nucleus (Fig. 4D).
2.4. Knockout of SAS cells with HuR CRISPR/SLN-HPR intensified the cell death triggered by the Epi-formulations
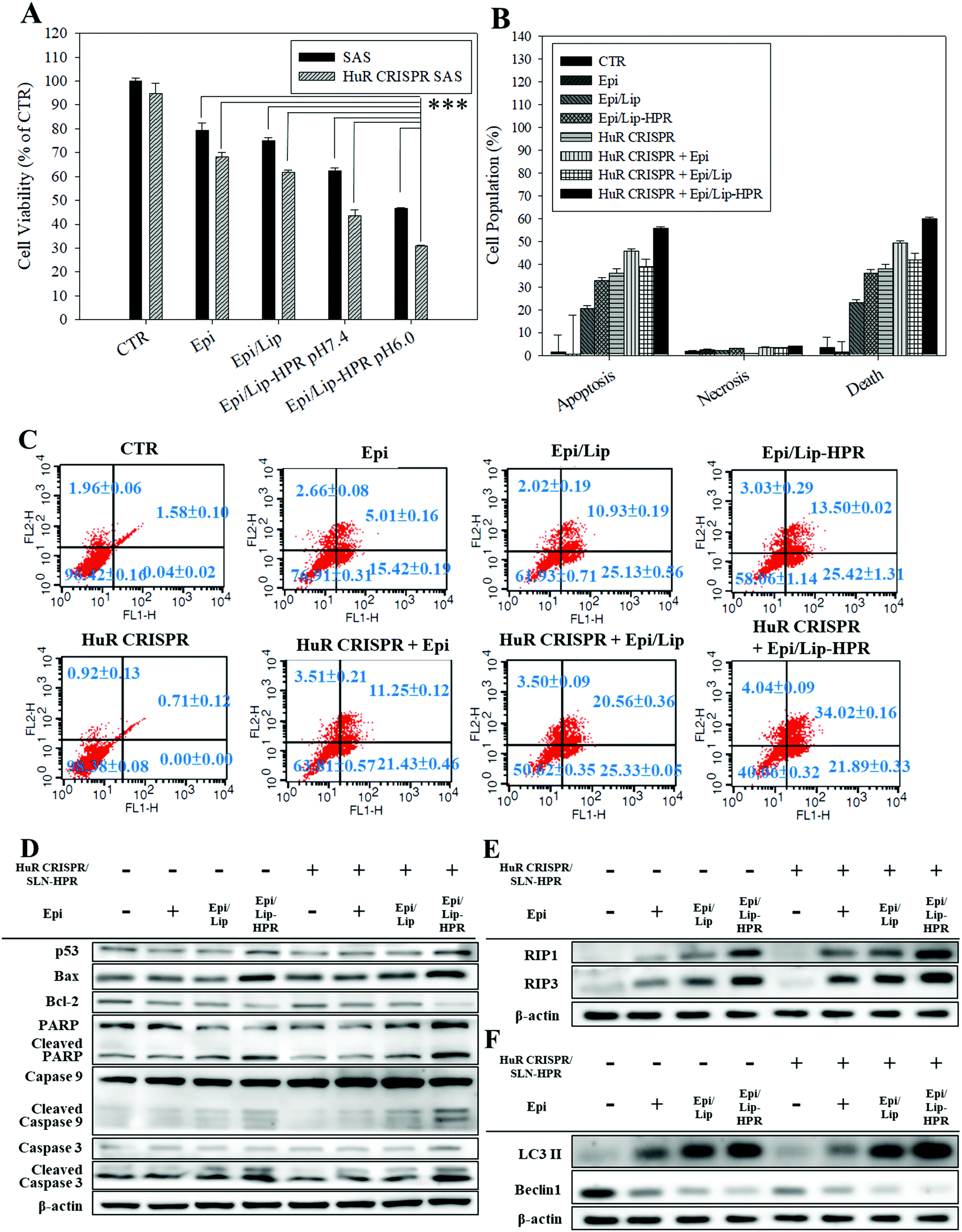
Epi, the model chemotherapeutic agent used in this study, induced apoptosis in various cancer cells, including CRC and cervical cancer, via the mitochondria-mediated pathway associated with suppressing the Wnt/β-catenin pathway.10,20,21 In this study, pretreatment of SAS cells with HuR CRISPR/SLN-HPR followed by Epi in various formulations resulted in significant cytotoxicity for SAS cells as determined by the SRB assay (Fig. 5A). The greatest inhibition of the growth of SAS cells (approximately 69% reduction) was found when co-treatment with HuR CRISPR/SLN-HPR and the Epi/Lip-HPR formulation was carried out (Fig. 5A). These findings suggested that HuR knockout in SAS cells by CRISPR/SLN-HPR was essential to initiate key anti-proliferation signaling pathways (as confirmed below) and thus enhanced the Epi/Lip-HPR cytotoxicity against SAS cells (Fig. 5A). The result of the Annexin V/PI staining assay showed that HuR knockout with CRISPR/SLN-HPR and the combined treatment with Epi/Lip-HPR potentiated apoptosis and necrosis and further led to cell death (Fig. 5B and C). Cell cycle analysis showed that the sub-G1 and G2/M phases of SAS cells were triggered, especially in the treatment groups of HuR CRISPR/SLN-HPR and Epi/Lip-HPR (Fig. S3A, ESI†). Our investigation of the molecular mechanism by RT-PCR and western blot analysis revealed that the mRNA and protein levels in apoptotic, necroptotic, and/or autophagic pathways increased after treatment with HuR CRISPR/SLN-HPR and/or Epi/Lip-HPR, especially their combined treatment (Fig. 5D–F and Fig. S3B and C, ESI†).
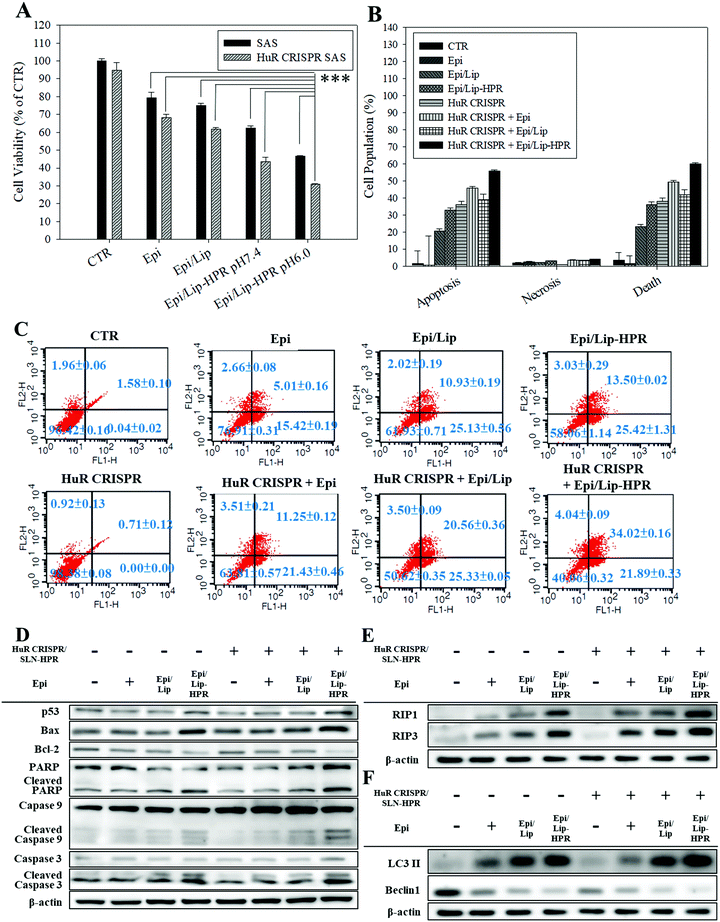 |
| | Fig. 5 Effect of different formulations for 48 h on the percentage of death and protein expression of apoptosis, necroptosis, and autophagy-associated pathways in SAS cells (*** statistical significance at p < 0.001). (A) Measurement of cell viability by SRB assay. (B) The relative percentages of apoptosis, necrosis, and death of cell populations. (C) Cell population distribution using the Annexin V/PI assay. (D–F) The protein expressions of (D) apoptosis, (E) necroptosis, and (F) autophagy pathways, as determined by western blot analysis. | |
2.5. Various pathways regulated by the formulations of Epi and/or HuR CRISPR inhibit proliferation, resistance, and migration of SAS cells
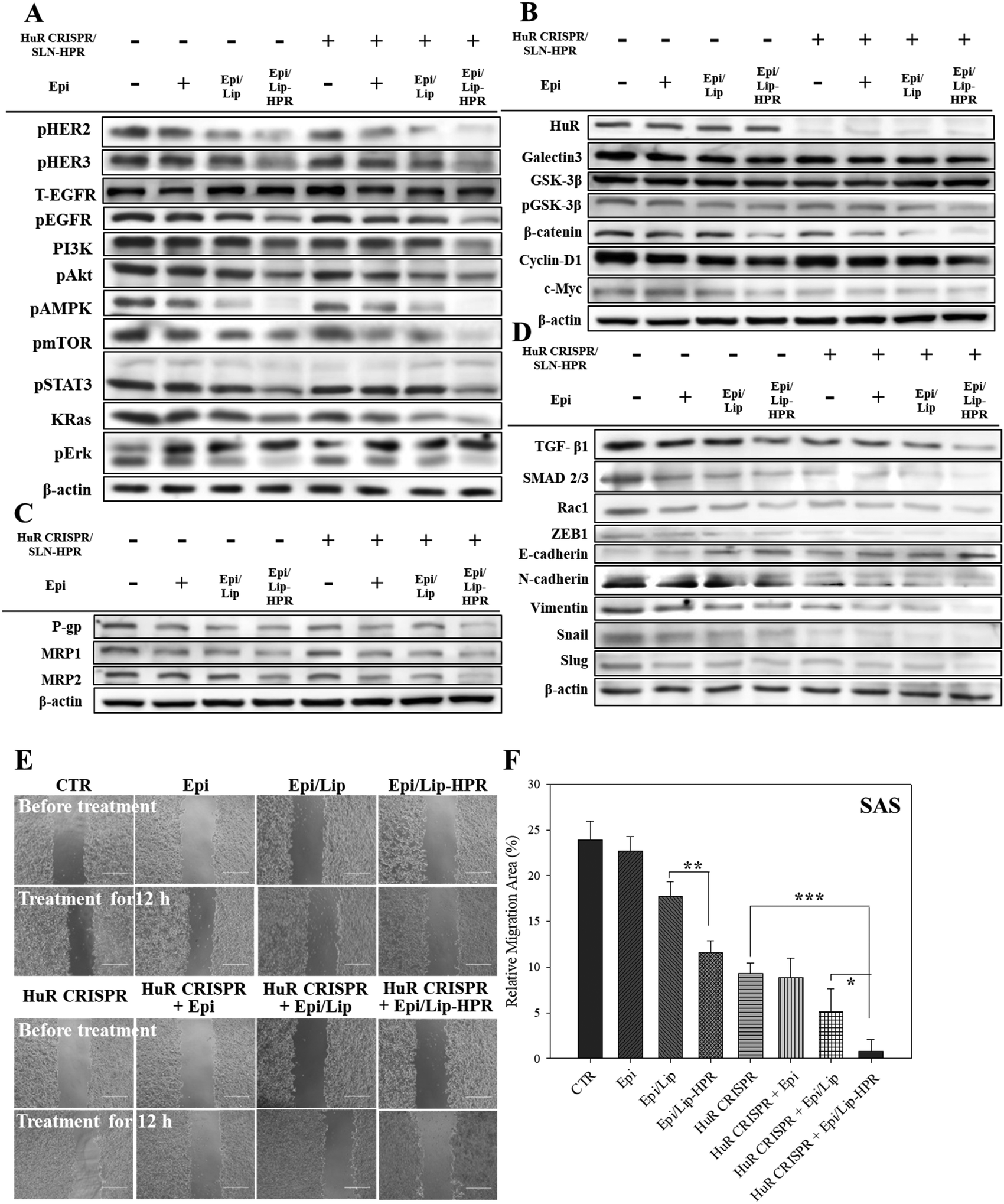
The results of western blot analysis showed that pretreatment of SAS cells with HuR CRISPR/SLN-HPR followed by Epi/Lip-HPR substantially decreased the protein expression of the phosphorylated forms of EGFR, HER2, HER3, PI3K, and KRas, and phospho forms of Akt, AMPK, mTOR, STAT3, and Erk (Fig. 6A). The HuR knockout by CRISPR/SLN-HPR and co-treatment with EGFR-targeted Epi/Lip-HPR significantly reduced EGFR and HER2 phosphorylation in SAS cells and activated the suppression of the PI3K/AKT/Ras/STAT3 axis significantly, whereby the expression was further downregulated by AMPK, mTOR, and Erk (Fig. 6A). Consistently, a previous study also shows that the co-treatment of human oral squamous cell carcinoma SCC25 cells with doxorubicin and LY294002 (a PI3K/AKT inhibitor) improves the efficacy of doxorubicin by suppressing the phosphorylation of ERK1/2 and p38 MAPK in OSCC cells.41
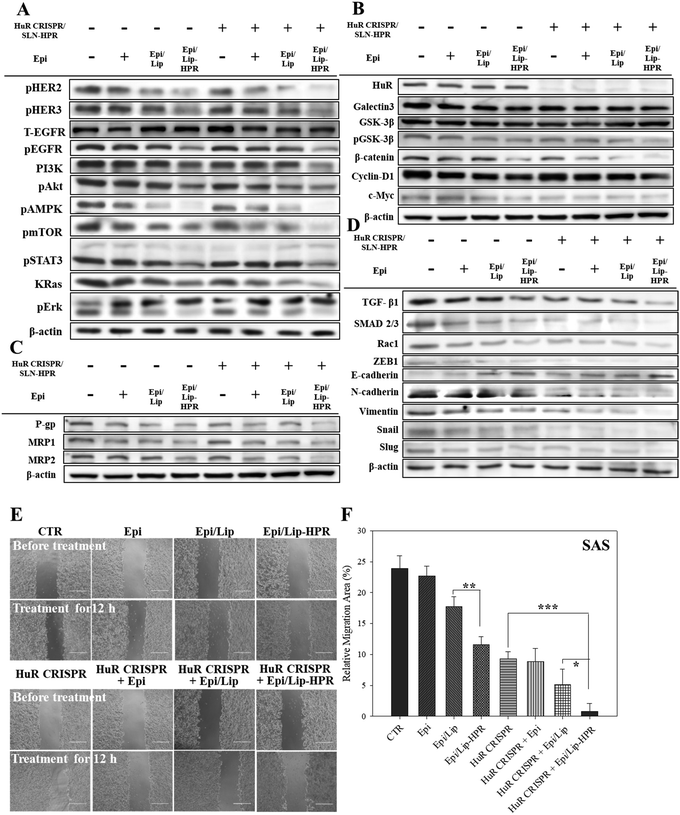 |
| | Fig. 6 The effects of various formulations on the protein expression of the signaling pathways of EGFR/HuR/Wnt/MDR/EMT and the relative migration percentages in SAS cells. Protein expressions of the (A) EGFR, (B) HuR and Wnt/β-catenin, (C) MDR, and (D) EMT pathways as determined by western blot analysis. (E) Migration assay of SAS cells after different treatments for 12 h. (F) Relative percentages of the cell migration area (* statistical significance at p < 0.05; ** p < 0.01; *** p < 0.001). | |
The results of DNA electrophoresis and western blot analysis confirmed that HuR was successfully knocked out by the CRISPR/Cas9 system (Fig. 6B and Fig. S2 and S4A, ESI†). The protein and mRNA levels of β-catenin, cyclin-D1, and c-Myc, which were the targeted transcripts of HuR in the Wnt/β-catenin pathway,9 declined after HuR silencing in SAS cells (Fig. 6B and Fig. S4A and B, ESI†). The decrease in the protein and mRNA levels in the Wnt/β-catenin pathway was markedly intensified by the pretreatment of SAS cells with HuR CRISPR and follow-up treatment with the Epi formulations (Fig. 6B and Fig. S4A and B, ESI†).
Also, the suppression of the Wnt/β-catenin pathway is positively associated with MDR inhibition.7,10 Previously, HuR knockdown by siRNA further repressed the expression of P-gp and MRPs in CRC cells and thereby increased the apoptosis triggered by Epi by hindering galectin-3/β-catenin signaling.7 In this study, pretreatment of SAS cells with HuR CRISPR decreased the expression of P-gp, MRP1, and MRP2 at the mRNA and protein levels (Fig. 6C and Fig. S4C and D, ESI†), suggesting the superior anticancer efficacy of Epi/Lip-HPR and HuR CRISPR/SLN-HPR as supported by findings from SRB and Annexin/PI assays (Fig. 5A–C). Also, TGF-β1, SMAD 2/3, Rac1, and ZEB1, which are associated with promoting EMT, migration, and/or invasion of cancer cells, positively correlated with HuR modulation.13,14 We performed a wound healing assay and western blot analysis to confirm that the Epi-loaded formulations effectively inhibited the expression of TGF-β1, SMAD 2/3, Rac1, and ZEB1 and that HuR CRISPR strengthened the inhibitory effect of Epi on SAS cells (Fig. 6D–F and Fig. S4E, ESI†). Also, HuR knockout decreased the protein levels of EMT-associated proteins such as N-cadherin, vimentin, snail, and slug in SAS cells (Fig. 6D and Fig. S4E, ESI†). The following treatment with Epi potentiated the inhibitory effect of HuR CRISPR/SLN-HPR on EMT-related proteins and the associated migration in SAS cells (Fig. 6D–F and Fig. S4E, ESI†).
2.6. Antitumor efficacy of HuR CRISPR/SLN-HPR and Epi/Lip-HPR in SAS/luc-bearing mice
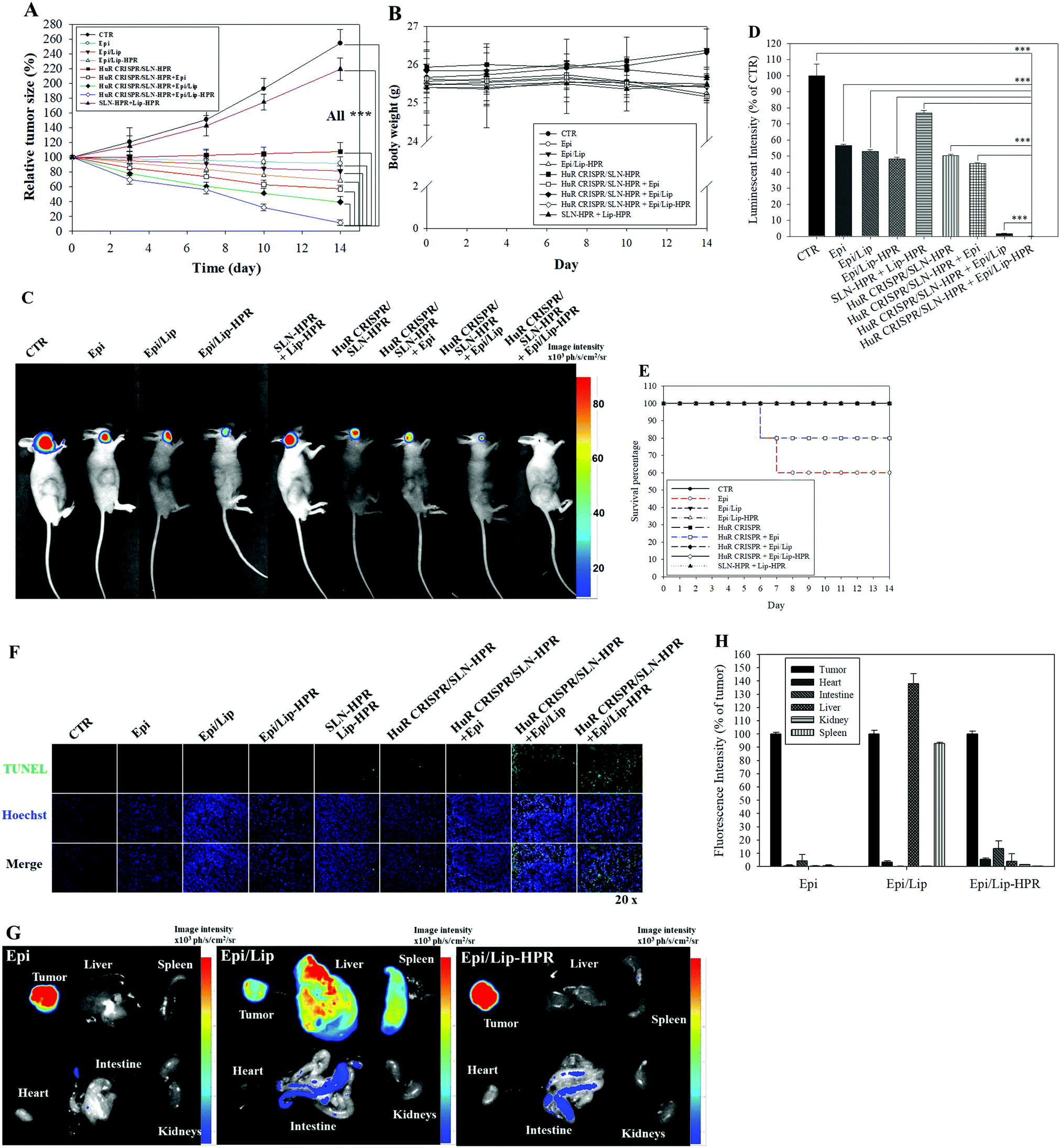
We established a SAS/luc-bearing mouse model to examine the antitumor efficacy of various HuR CRISPR and/or Epi formulations in vivo. These formulations were administered individually to the tail veins of SAS/luc-bearing mice twice weekly. The tumor size was measured twice weekly with a digital caliper. The pre-knockout of HuR in SAS cells by HuR CRISPR/SLN-HPR and co-treatment with Epi/Lip-HPR resulted in the most significant antitumor therapeutic efficacy in SAS/luc-bearing mice (Fig. 7A). No significant differences in the body weights of tumor-bearing mice were found between the groups (Fig. 7B). Also, the results of IVIS images showed that the combined treatment of HuR CRISPR/SLN-HPR and Epi/Lip-HPR displayed the best antitumor efficacy (Fig. 7C and D). However, the survival percentage indicated that Epi without nanocarriers resulted in 40% death of the tumor-bearing mice, possibly due to Epi-associated toxicity (Fig. 7E). Co-treatment of HuR CRISPR and Epi without Lip maintained 80% survival of the mice (Fig. 7E). All Epi-nanoformulations with or without knockout by HuR CRISPR maintained 100% survival of the mice (Fig. 7E). The combined treatment of Epi/Lip-HPR and HuR CRISPR/SLN-HPR induced apoptosis in more tumor cells than the other treatments, as shown in Fig. 7F. The biodistribution results indicated that most of the Epi formulations were accumulated in tumors. Nevertheless, Epi/Lip was also found in the liver and spleen (Fig. 7G and H). Expectedly, Epi/Lip-HPR was targeted to tumor sites and avoided accumulation in the liver and spleen (Fig. 7G and H).
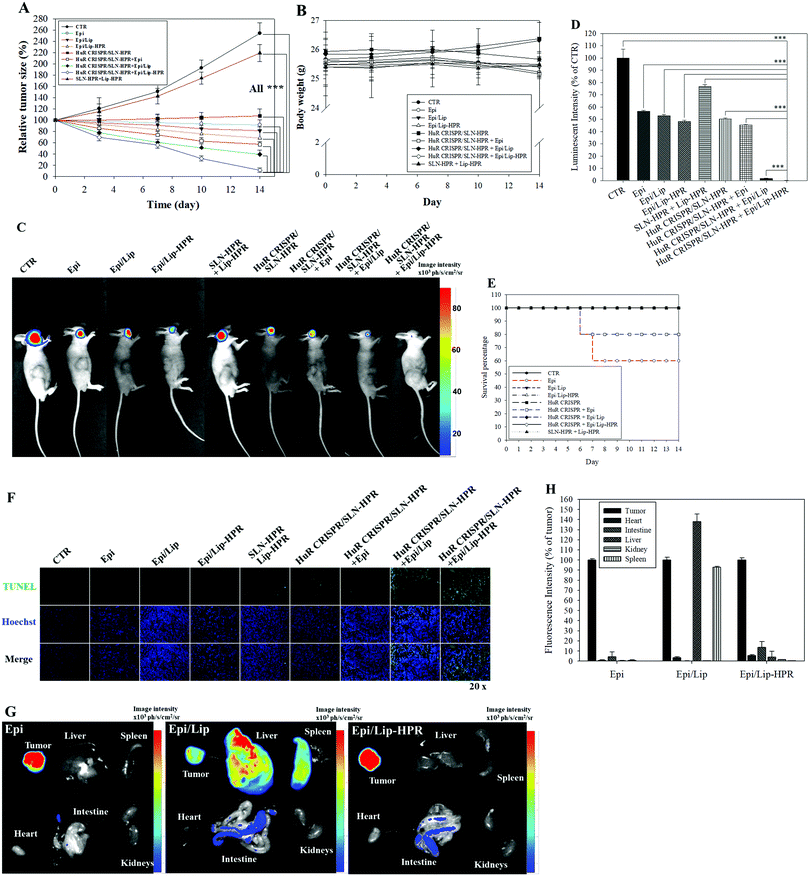 |
| | Fig. 7 Antitumor efficacy and biodistribution of the Epi and/or HuR-CRISPR formulations in SAS/luc-bearing mice. (A) The antitumor efficacy of SAS/luc-bearing mice administered various formulations by intravenous injection. Tumor growth was measured twice weekly using a digital caliper (*** statistical significance at p < 0.001). (B) Body weights of SAS/luc-bearing mice. (C) IVIS images of SAS/luc-bearing mice treated with different formulations for 14 days. (D) The relative percentage of the luminescence intensity of the IVIS images. *** p < 0.001. (E) Overall survival percentages of SAS/luc-bearing mice treated with different formulations. (F) Evaluation of in vivo apoptosis (stained green) by TUNEL assay in SAS tumor cells on the day after the last administration of various formulations. The nuclei were marked blue by Hoechst staining. Scale bar 100 μm. (G) The biodistribution study of Epi-loaded formulations in SAS-bearing mice. (H) The relative percentage of fluorescence intensity of the biodistribution result. | |
2.7. Biosafety assessment and multiple pathways regulated by HuR CRISPR/SLN-HPR and/or Epi/Lip-HPR in SAS/luc-bearing mice
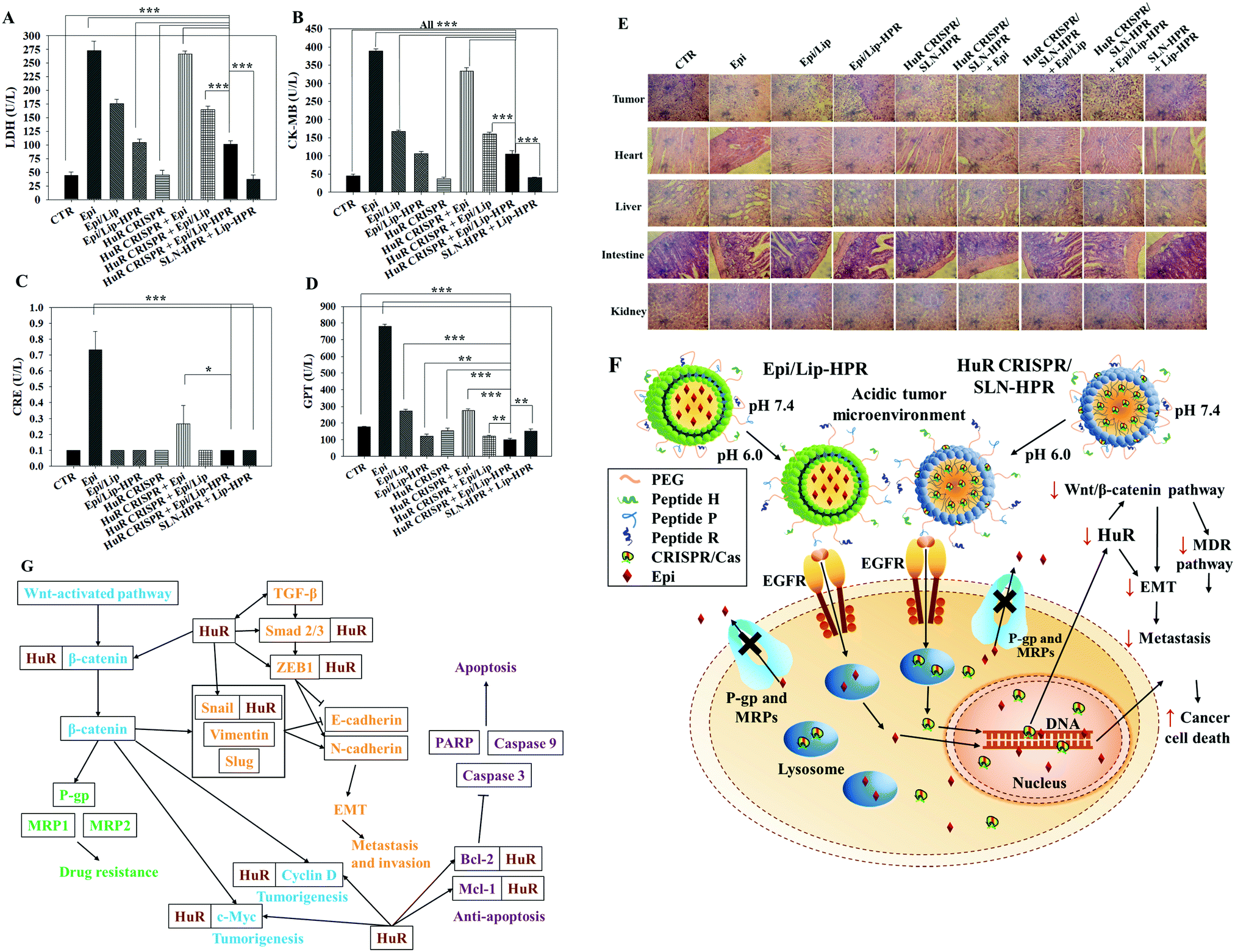
To evaluate the biosafety of various formulations in SAS-bearing mice, we performed biochemical tests and H&E staining studies on major organs. Epi/Lip-HPR in combination with HuR CRISPR/SLN-HPR significantly reduced Epi-induced cardiac toxicity, as shown by the decreases in LDH and CK-MB in Fig. 8A and B. The co-treatment of Epi/Lip-HPR and HuR CRISPR/SLN-HPR also exhibited the least toxicity to the kidney and liver, as indicated by the reductions in CRE and GPT levels, as illustrated in Fig. 8C and D. H&E staining showed similar results. In tumor tissues, Epi/Lip-HPR in combination with HuR CRISPR/SLN-HPR displayed the most remarkable apoptosis/necrosis induction to cause tumor-killing (top panels of Fig. 8E). Although Epi treatment alone resulted in more damaged cells or signs of inflammation in most of the examined organs such as the heart, liver, intestines, and kidneys, the toxicity was reduced by encapsulation with Lip or Lip-HPR and the combined treatment with HuR CRISPR/SLN-HPR did not increase organ toxicity, as shown in Fig. 8E. The finding of pronounced antitumor efficacy of Epi/Lip-HPR alone or in combination with HuR CRISPR/SLN-HPR suggested that the inhibition of EGFR-mediated signaling via the P-peptide (an EGFR ligand) in Lip-HPR and SLN-HPR was enhanced by the effect of the HuR knockout on the EGFR/PI3K/mTOR/AKT axis (Fig. 6A–D), demonstrating the superior therapeutic potential to treat SAS-bearing mice with EGFR overexpression and PI3K/AKT signaling activation (Fig. 8F and G).
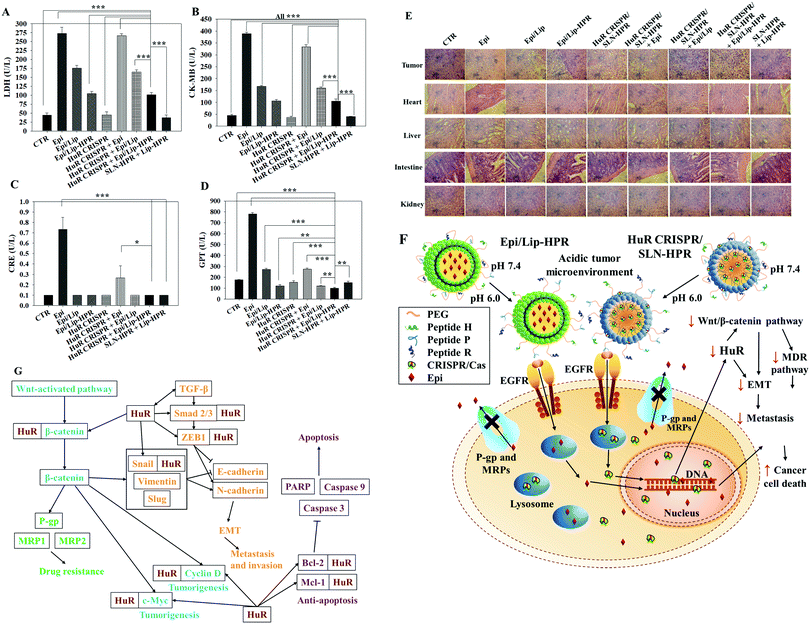 |
| | Fig. 8 Biochemical tests, HE staining, and the overall scheme of the signaling regulation influenced by HuR CRISPR/SLN-HPR and Epi/Lip-HPR on various pathways in SAS cells. (A–D) Blood biochemical indices of the heart, kidney, and liver of the mice treated with different formulations. (E) Histological photomicrographs of the tumor, kidney, liver, and intestine stained with H&E. (F) Scheme showing the alteration of HuR CRISPR/SLN-HPR and Epi/Lip-HPR in an acidic tumor microenvironment via the conformational change of the H-peptide. These nanoparticles entered SAS cells through endocytosis or fusion with the help of exposed P and R peptides. The HuR gene was knocked out by the CRISPR/Cas9 system and the protein expressions in the HuR, EGFR, β-catenin, MDR, EMT, and apoptosis pathways were modulated. Epirubicin accumulated in the cell nucleus and caused cancer cell death. (G) Schematic of the multiple signaling pathways regulated by HuR CRISPR/SLN-HPR and Epi/Lip-HPR in the OSCC model. | |
3. Conclusions
Collectively, we designed two different nano vehicles, namely SLN-HPR and Lip-HPR, to deliver the CRISPR/Cas9 system and Epi, respectively. The functions of these nanocarriers include: (1) sustaining blood circulation through PEG modification of both nanoparticles and passive tumor targeting via the enhanced permeability and retention (EPR) effect; (2) improvement of EGFR targeting and cellular internalization via EGFR ligand P-mediated endocytosis; (3) facilitating endosomal escape and nuclear localization via the pH-responsive and nucleus-targeted sequences of H and R peptides; and (4) nuclear and/or cytoplasmic release of Epi and/or CRISPR/Cas-9 to act as a topoisomerase inhibitor or HuR knockout system to suppress tumor progression and multiple pathways of survival and metastasis and to modulate resistance in HNC cells. The anticancer efficacy and safety of anthracycline therapeutics such as Epi for treating OSCC can be significantly improved in combination with the gene-editing of HuR by the CRISPR–Cas9 system.
4. Experimental section
Materials
All lipids were purchased from Avanti (Alabaster, AL, USA). H, P, and R peptides were custom synthesized by Kelowna (Taipei, Taiwan). All cell culture media and reagents were ordered from Gibco (Grand Island, NY, USA) or HyClone (Logan, UT, USA). Most of the other chemicals or reagents were obtained from either Sigma-Aldrich (St. Louis, Missouri, USA) or Merck (Darmstadt, Germany).
Synthesis of peptide-conjugated lipids
DSPE-PEG-maleimide and the respective H-, P- and R-peptide in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (lipid/peptide) were dissolved in chloroform/methanol. After reacting overnight at room temperature, the mixture was evaporated to remove the organic solution and dialyzed against water using a 3.5–5 kDa membrane (Spectrum Laboratories, CA, USA). The final product of the DSPE-PEG peptide was lyophilized, and the structure was confirmed using matrix-assisted mass spectrometry with laser desorption ionization-time of flight (MALDI-TOF MS; Applied Biosystems, MA, USA).
:1 (lipid/peptide) were dissolved in chloroform/methanol. After reacting overnight at room temperature, the mixture was evaporated to remove the organic solution and dialyzed against water using a 3.5–5 kDa membrane (Spectrum Laboratories, CA, USA). The final product of the DSPE-PEG peptide was lyophilized, and the structure was confirmed using matrix-assisted mass spectrometry with laser desorption ionization-time of flight (MALDI-TOF MS; Applied Biosystems, MA, USA).
Preparation of peptide-conjugated and HuR CRISPR-loaded SLN (HuR CRISPR/SLN-HPR)
SLN were prepared by mixing L-α-phosphatidylcholine (PC), cholesterol, DOTAP, and the DSPE-PEG peptide (molar ratio 1:0.1:0.1:0.1) in methanol/dichloromethane. Then 0.1% Tween 80 was added to the mixture with stirring. The pre-designed plasmid of CRISPR/Cas9 was carefully dispersed in SLN-HPR in a volume ratio of 1:2 to form HuR CRISPR/SLN-HPR.
Preparation of peptide-conjugated and Epi-loaded liposomes (Epi/Lip-HPR)
Lip-HPR was prepared via a thin film hydration method by mixing DSPC, cholesterol, the DSPE-PEG-peptide, and DSPE-omPEG (molar ratio of 1:0.1:0.1:0.1) in methanol. After removing methanol using a rotary evaporator, the lipid thin film was resuspended in distilled water. The mixture was extruded through a 200 nm and 100 nm membrane filter and dialyzed against PBS for 24 h. Epi was loaded into Lip-HPR to make Epi/Lip-HPR.
Characterization of the HuR CRISPR-loaded SLN and Epi-loaded Lip formulations
The size distribution and the zeta potential of HuR CRISPR/SLN and Epi/Lip with and without HPR modification were determined using a Zetasizer Nano-ZS particle size analyzer (Malvern Instruments Ltd, Malvern, Worcestershire UK). Also, the nanoparticles were fixed on carbon-coated copper grids and stained with uranyl acetate to observe the morphology via TEM (JEM-2000EXII, Japan).
Encapsulation efficiency (EE%) and drug loading capacity (DL%)
A dispersion of Epi or HuR CRISPR plasmid-incorporated nanoparticles was centrifuged for 30 minutes at 15000 rpm and 4 °C through an ultracentrifuge filter (Amicon®). The filtrate was examined with a microplate reader (TECAN Sunrise, Zurich, Switzerland) or NanoDrop (Thermo Fisher, Massachusetts, USA). Besides, the harvested nanoparticles were broken by Triton X 100 and the remaining nanoparticles were re-dissolved in methanol/chloroform. Epi or the plasmid was then analyzed separately by high performance liquid chromatography (HPLC) and a NanoDrop (Thermo Fisher, Massachusetts, USA). Each dispersion was detected in triplicate. The EE% or DL% of Epi in Lip or Lip-HPR and the plasmid in SLN or SLN-HPR were calculated by the following equations| | | EE% = [(We − Wf)/We] × 100% | (1) |
| | | DL% = [(We − Wf)/Wt] × 100% | (2) |
where We is the weight of added Epi or the plasmid, Wf is the weight of Epi or the plasmid in the filtrate, and Wt is the total nanoparticle weight.
HPLC analysis
Epi was detected by HPLC as described in our previous study.21 The HPLC system is composed of a Primaide 1110 pump (Hitachi, Tokyo, Japan), an autosampler (Primaide 1210), and a Luna® 5 μm C18 100 Å LC column (250 × 4.6 mm; Phenomenex, Torrance, CA, USA), accompanied by a L2420 UV-VIS detector (Hitachi). The mobile phase comprises methanol and water at a volume ratio of 75:25. The flow rate was established at 1.2 mL min−1 and the detection wavelength was set at 254 nm. All experiments were repeated three times.
pH-induced changes in size, zeta potential, and drug release
The nanoparticles were incubated with PBS at pH 7.4 and pH 6.0 at 37 °C for 24 hours. The size and the zeta potential of the pH-sensitive nanoparticles were measured with a Zetasizer Nano-ZS. Furthermore, Epi- or plasmid-loaded formulations were placed in a dialysis bag (1000–3500 MWCO) and dialyzed, respectively, against PBS at pH 6.0 and 7.4 for the indicated time intervals. The samples were withdrawn as scheduled and the concentrations of Epi and the plasmid were measured by HPLC and a NanoDrop, correspondingly, to calculate the relative drug release%.
Cell lines and cell culture
Human tongue squamous carcinoma SAS cells and normal oral keratinocyte NOK cells were cultured in Dulbecco's modified Eagle medium with 10% FBS and 1% penicillin/streptomycin.
Design of HuR sgRNA
The human HuR sequence (ELAVL1) was obtained from the NCBI gene database (National Center for Biotechnology Information). The exon 2 sequence of HuR was analyzed with the CRISPR sgRNA design tool. We selected two sgRNA sequences with higher targeting specificity and lower off-target efficiency and used these sequences to perform the following experiments.
Identification of HuR CRISPR cells
The designed surrogate reporter system comprises genes that encode two different fluorescent proteins GFP and tdTomato, which are linked to the targeting HuR sequence. This design can help identify the cells where the target HuR gene is knocked out by the CRISPR system. The green fluorescence of GFP on the reporter is emitted when the surrogate reporter has been successfully transfected into the cells, and the red fluorescence from tdTomato is expressed after knocking out the targeting HuR sequence by the CRISPR/Cas9 system. SLN-HPR-encapsulated HuR CRISPR plasmids and surrogate reporter genes at 37 °C for 24 h. After confirmation of the fluorescence signal using a fluorescence microscope, the cells with the signals of green and red fluorescence were sorted by a cell sorter (BD FACSAria, San Jose, CA, USA).
DNA from wild-type and HuR knock-out cells was extracted and DNA electrophoresis was performed to identify the knock-out effect of HuR CRISPR. Briefly, DNA (1 μg) was amplified by PCR with dNTP, primers, and DNA polymerase. The amplified DNA samples were loaded into the gel with 12% polyacrylamide. After running the gel at 60 V, the gel was stained with ethidium bromide at room temperature in the dark. Finally, the gel was visualized and scanned using the DigiGel Gel Documentation System.
Analysis of transfection efficiency and knock-out efficiency
The quantification of the transfection efficiency and the knock-out efficiency of SAS cells was performed by flow cytometry. The cells were seeded in 24-well plates. After incubation with the HuR CRISPR plasmid (1 μg mL−1) and surrogate reporters in various transfection reagents including SLN and SLN-HPR, and commercial transfection agents such as Lipofectamine 3000 and PolyJet, the cell pellets were collected. The transfection efficiency (green fluorescence) and the knockout efficiency (red fluorescence) of the HuR CRISPR/Cas9 system in SAS cells were measured with a FACSCalibur™ flow cytometer (BD Biosciences, San Jose, CA, USA). The results were analyzed by CellQuest (BD Biosciences, San Jose, CA, USA).
Quantification of cellular uptake
The cellular uptake by SAS cells was quantized by flow cytometry. The cells were seeded in 24-well plates and incubated for 8 hours with GFP-plasmid/SLN-HPR (1 μg mL−1) or Epi/Lip-HPR (1 μM). After centrifugation, the collected cell pellets were washed and suspended in PBS. The efficiency of nanoparticle uptake by the cells was measured with a FACSCalibur™ flow cytometer, based on the fluorescence intensity of the loaded GFP plasmid (Ex/Em = 488/507 nm) and Epi (Ex/Em = 480/560 nm). The results were analyzed by CellQuest.
Identification of cellular uptake mechanisms
SAS cells were seeded in 24-well plates and pre-incubated with different specific endocytosis or fusion inhibitors at 37 °C for 1 h. After co-incubation with Epi/Lip-HPR (1 μM) or GFP plasmid/SLN-HPR (1 μg mL−1) for an additional 3 h, the collected cell pellets were washed with PBS after centrifugation. The fluorescence intensity was then determined using a FACSCalibur™ flow cytometer.
Identification of the EGFR targeting effect of the nanoparticles
After different incubation times with Epi/Lip-HPR (1 μM), the cells were fixed in 4% paraformaldehyde before they were stained with LysoGreen, a lysosomal marker. EGFR was identified by anti-EGFR antibodies (Cell Signaling Technology, Ipswich, MA, USA) using immunofluorescent staining. The cells were then stained with DAPI at 37 °C to reveal the nuclei. The images were taken by CLSM (FV10i; OLYMPUS, Tokyo, Japan).
Cell viability assay
SAS and NOK cells were seeded in 96-well plates overnight and treated with various concentrations of Epi, Epi/Lip, and Epi/Lip-HPR with or without HuR CRISPR/SLN-HPR for 48 hours at 37 °C. 1% trichloroacetic acid was added and incubated at 4 °C. Then 0.04% SRB was added, and each well was washed three times with 1% acetic acid. The absorbance was measured with an ELISA reader at a wavelength of 540 nm.
Identification of intracellular localization
SAS cells were incubated for the specified time with GFP plasmid/SLN-HPR (1 μg mL−1) or Epi/Lip-HPR (1 μM); the cells were incubated before staining with LysoGreen fixed in 4% paraformaldehyde. Early endosomes were then identified by EEA1 antibodies (Cell Signaling Technology) using immunofluorescent staining. The cells were also stained with DAPI at 37 °C to localize the nuclei. The photos were taken by CLSM.
Apoptosis Annexin V–PI assay
The relative percentages of cells in the live, apoptotic, and necrotic stages were detected with the Annexin V/PI Dual Stain Detection Kit (Strong Biotech Corporation, Taiwan). After seeding SAS cells overnight, various formulations were added to the cells at 37 °C for 48 h. The cells were then stained in the dark with Annexin V-PI labeling solution. The collected cells were washed with PBS and detected with a FACSCalibur™ flow cytometer.
Wound healing assay
SAS cells with or without HuR knockout were treated with various Epi formulations at 37 °C for 12 hours. The images were taken by optical microscopy and the migration area was measured and quantified by ImageJ. The migration percentage was calculated by the following equation.| | | Migration area (% of area before treatment) = 100% − (blank area(aftertreatment)/blank area(beforetreatment) × 100%) | (3) |
Western blot assay
After overnight seeding of SAS cells, the cells were incubated with various formulations for 24 h. Proteins from the cells were lysed with RIPA (Cell Signaling, Beverly, MA, USA) and the protein concentrations were measured by BCA protein assay (Thermo Fisher Scientific, Waltham, MA, USA). Various protein samples were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Bio-Rad, Hercules, CA, USA). The blots were incubated overnight at 4 °C with primary antibodies against various proteins. After conjugation with horseradish peroxidase-conjugated immunoglobulin G (Jackson ImmunoResearch Inc., PA, USA), the blots were visualized with enhanced chemiluminescence kits (Millipore, Billerica, MA, USA). The images were taken using a luminescence imaging system (Amersham™ Imager 600; GE Healthcare, Chicago, USA).
RNA extraction and real-time RT-PCR
Total RNA was extracted from the cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the protocol. The RT-PCR process was performed with a high-capacity RNA-to-cDNA™ kit (Thermo Fisher; Waltham, MA, USA) and KAPA SYBR® FAST qPCR kits (Kapa Biosystems, Roche; Basel, Switzerland). Real-time PCR was performed using a StepOnePlus™ real-time PCR system (Thermo Fisher; Waltham, MA, USA).
Establishment of the SAS-bearing mouse model and evaluation of antitumor efficacy and body weight
Male BALB/c nude mice weighing approximately 20 g were purchased from the National Laboratory Animal Center (Taipei, Taiwan) and the animal protocol was approved by the Institutional Animal Care and Use Committees (IACUC) of National Yang Ming Chiao Tung University (NYCU). The animals were treated under the appropriate national and university guidelines and all the experiment procedures were performed in compliance with the IACUC policy of NYCU for the use and ethics of laboratory animals in research and teaching. 1 × 105 SAS-luc cells were inoculated subcutaneously on the right cheek of each mouse and the tumors were grown to about 60 mm3. The mice were randomly grouped into 5 mice per group. The tumor-bearing mice with or without HuR knock out by CRISPR/SLN-HPR were administered intravenously with saline (CTR) and various Epi-loaded formulations twice a week. The concentration of Epi in each injection was equivalent to 10 mg per kg per mouse. The treatment was completed on the 14th day. The percentage of survival was defined as the number of surviving mice at the specified time divided by the initial number of mice. The body weight and tumor size of the mice were monitored with a digital caliper twice a week, and the tumor volume was calculated according to the following equation| | | V (mm3) = 0.5 × L × W2 | (4) |
where L is the longest diameter (mm), and W is the shortest diameter (mm) perpendicular to the longest axis.
IVIS imaging detection and biochemical tests of SAS tumor-bearing mice
Mouse bioluminescent images were captured and imaged a day after final treatment using an IVIS Spectrum (PerkinElmer, Waltham, MA, USA). After taking the images, 170 μL of blood was sampled from the orbital sinus of the mice. Serum levels of LDH, CK-MB, CRE, and GPT were detected with the respective activity assay kits (Fujifilm, Tokyo, Japan) using a clinical dry chemistry analyzer (Fuji Dri-Chem 7000 V, Fujifilm Corp.) for functional evaluation of the heart, kidney, and liver.
Biodistribution of the Epi formulations, H&E staining study, and TUNEL assay of SAS tumor-bearing mice
The mice were sacrificed and the tumor and organs of the mice were collected in cold PBS. The tumor tissues were washed with PBS and lysed in lysis buffer to extract the proteins for western blot assay. Tumor tissues were preserved in formaldehyde and embedded in paraffin for H&E staining study and TUNEL assay. Also, the organs and tumor were scanned by Animal Optical and Computed Tomography Core (National Yang-Ming Chiao Tung University) and recorded using dynamic optical imaging systems (PhotonIMAGER™; Biospace Lab, Nesles la Vallée, France).
Statistical analysis
Results are reported as mean ± SD. Statistical analysis was performed using Student's t-test to compare significant differences between the two treatment groups. The differences were statistically significant based on a p-value less than 0.05.
Author contributions
Yu-Li Lo conceived and designed the experiments; Chen-Shen Wang and Chih-Hsien Chang conducted the experiments; Yu-Li Lo, Chen-Shen Wang, Chih-Hsien Chang, and Tsai-Yu Tzeng analyzed the data; Yu-Li Lo and Anya Maan-Yuh Lin contributed reagents/materials/analysis tools; Yu-Li Lo wrote the paper.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This research was funded by grants from the Ministry of Science and Technology of Taiwan (MOST 107-2320-B-010-015-MY3), Veterans General Hospitals and University System of Taiwan (VGHUST109-V7-3-2; VGHUST110-G2-2-2), National Yang Ming Chiao Tung University and Cheng Hsin General Hospital Foundation (CY10935; CY11004), Center for Advanced Pharmaceutics and Drug Delivery Research, National Yang Ming Chiao Tung University. We thank the Taiwan Mouse Clinic, Academia Sinica, and the Taiwan Animal Consortium for technical assistance with animal imaging experiments.
References
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394 CrossRef PubMed.
- J. Weiße, J. Rosemann, V. Krauspe, M. Kappler, A. W. Eckert, M. Haemmerle and T. Gutschner, Int. J. Mol. Sci., 2020, 21, 6835 CrossRef.
- Z. Javed, H. Muhammad Farooq, M. Ullah, M. Zaheer Iqbal, Q. Raza, H. Sadia, R. Pezzani, B. Salehi, J. Sharifi-Rad and W. C. Cho, Asian Pac. J. Cancer Prev., 2019, 20, 995 CrossRef CAS PubMed.
- M. Reyes, T. Flores, D. Betancur, D. Peña-Oyarzún and V. A. Torres, Int. J. Mol. Sci., 2020, 21, 4682 CrossRef CAS PubMed.
- T. Zhan, N. Rindtorff and M. Boutros, Oncogene, 2016, 36, 1461 CrossRef PubMed.
- J. Wang, Y. Guo, H. Chu, Y. Guan, J. Bi and B. Wang, Int. J. Mol. Sci., 2013, 14, 10015 CrossRef PubMed.
- G. L. Lin, H. J. Ting, T. C. Tseng, V. Juang and Y. L. Lo, PLoS One, 2017, 12, e0185625 CrossRef PubMed.
- J. D. Cha, H. K. Kim and I. H. Cha, Head Neck, 2014, 36, 1168 CrossRef PubMed.
- I. Kim, J. Hur and S. Jeong, Biochem. Biophys. Res. Commun., 2015, 457, 65 CrossRef CAS PubMed.
- Y. K. Lee, T. H. Lin, C. F. Chang and Y. L. Lo, PLoS One, 2013, 8, e82478 CrossRef.
- H. Lage, Cell. Mol. Life Sci., 2008, 65, 3145 CrossRef CAS.
- C. Lin, S. Zhang, Y. Wang, Y. Wang, E. Nice, C. Guo, E. Zhang, L. Yu, M. Li, C. Liu, L. Hu, J. Hao, W. Qi and H. Xu, Clin. Cancer Res., 2018, 24, 486 CrossRef CAS PubMed.
- J. Sun, X. Gu, N. Wu, P. Zhang, Y. Liu and S. Jiang, Respir. Res., 2018, 19, 109 CrossRef PubMed.
- N. Wang, D. Yan, Y. Liu, Y. Liu, X. Gu, J. Sun, F. Long and S. Jiang, Respir. Res., 2016, 17, 117 CrossRef PubMed.
- Y. Zhang, L. Xu, A. Li and X. Han, Biomed. Pharmacother., 2019, 110, 400 CrossRef CAS.
-
Y. Kuwano, I.-E. Gallouzi and M. Gorospe, Role of the RNA-binding protein HuR in apoptosis and apoptosome function, in Apoptosome: an up-and-coming therapeutical tool, ed. F. Cecconi and M. D'Amelio, Springer Netherlands, Dordrecht, 2010, p. 203 Search PubMed.
- D. Durie, S. M. Lewis, U. Liwak, M. Kisilewicz, M. Gorospe and M. Holcik, Oncogene, 2011, 30, 1460 CrossRef CAS.
- C. Liu, L. Zhang, H. Liu and K. Cheng, J. Controlled Release, 2017, 266, 17 CrossRef CAS PubMed.
- H. X. Wang, M. Li, C. M. Lee, S. Chakraborty, H. W. Kim, G. Bao and K. W. Leong, Chem. Rev., 2017, 117, 9874 CrossRef CAS.
- Y. L. Lo, W. Wang and C. T. Ho, Toxicology, 2012, 302, 221 CrossRef CAS PubMed.
- V. Juang, H. P. Lee, A. M. Lin and Y. L. Lo, Int. J. Nanomed., 2016, 11, 6047 CrossRef CAS.
- C. Wang, T. Zhao, Y. Li, G. Huang, M. A. White and J. Gao, Adv. Drug Delivery Rev., 2017, 113, 87 CrossRef CAS PubMed.
- L. Kong, F. Campbell and A. Kros, Nanoscale Horiz., 2019, 4, 378 RSC.
- H. Derakhshankhah and S. Jafari, Biomed. Pharmacother., 2018, 108, 1090 CrossRef CAS.
- S. G. Patel, E. J. Sayers, L. He, R. Narayan, T. L. Williams, E. M. Mills, R. K. Allemann, L. Y. P. Luk, A. T. Jones and Y.-H. Tsai, Sci. Rep., 2019, 9, 6298 CrossRef PubMed.
- M. Kriegs, T. S. Clauditz, K. Hoffer, J. Bartels, S. Buhs, H. Gerull, H. B. Zech, L. Bußmann, N. Struve, T. Rieckmann, C. Petersen, C. S. Betz, K. Rothkamm, P. Nollau and A. Münscher, Sci. Rep., 2019, 9, 13564 CrossRef PubMed.
- R. Bofinger, M. Zaw-Thin, N. J. Mitchell, P. S. Patrick, C. Stowe, A. Gomez-Ramirez, H. C. Hailes, T. L. Kalber and A. B. Tabor, J. Pept. Sci., 2018, 24, e3131 CrossRef PubMed.
- S. Lim, W. J. Kim, Y. H. Kim and J. M. Choi, Mol. Cells, 2012, 34, 577 CrossRef CAS PubMed.
- H. An, C. Rabesahala de Meritens, V. L. Buchman and T. A. Shelkovnikova, Mol. Brain, 2020, 13, 77 CrossRef CAS PubMed.
- G. L. Plosker and D. Faulds, Drugs, 1993, 45, 788 CrossRef CAS.
- U. Bazylinska, J. Pietkiewicz, J. Rossowska, G. Chodaczek, A. Gamian and K. A. Wilk, Macromol. Biosci., 2017, 17, 1600356 CrossRef PubMed.
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577 CrossRef.
- P. Zhang, Y. Zhang, X. Ding, C. Xiao and X. Chen, Biomater. Sci., 2020, 8, 3052 RSC.
- A. U. Rehman, Z. Omran, H. Anton, Y. Mely, S. Akram, T. F. Vandamme and N. Anton, Eur. J. Pharm. Biopharm., 2018, 133, 331 CrossRef CAS PubMed.
- Y. Zhao, W. Ren, T. Zhong, S. Zhang, D. Huang, Y. Guo, X. Yao, C. Wang, W. Q. Zhang, X. Zhang and Q. Zhang, J. Controlled Release, 2016, 222, 56 CrossRef CAS PubMed.
- L. Zhang, J. Xu, F. Wang, Y. Ding, T. Wang, G. Jin, M. Martz, Z. Gui, P. Ouyang and P. Chen, Langmuir, 2019, 35, 3513 CrossRef CAS PubMed.
- O. Rötzschke, J. M. Lau, M. Hofstätter, K. Falk and J. L. Strominger, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16946 CrossRef PubMed.
- S. T. Hong, H. Lin, C. S. Wang, C. H. Chang, A. M. Lin, J. C. Yang and Y. L. Lo, J. Nanobiotechnol., 2019, 17, 89 CrossRef PubMed.
- W. Chen, B. Shen and X. Sun, Mol. Imaging, 2019, 18, 1 CrossRef CAS PubMed.
- C. Y. Han, L. L. Yue, L. Y. Tai, L. Zhou, X. Y. Li, G. H. Xing, X. G. Yang, M. S. Sun and W. S. Pan, Int. J. Nanomed., 2013, 8, 1541 Search PubMed.
- D. Smolensky, K. Rathore, J. Bourn and M. Cekanova, J. Cell. Biochem., 2017, 118, 2615 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nh00254f |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *abdf
*abdf