
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Double annulation of ortho- and peri-C–H bonds of fused (hetero)arenes to unusual oxepino-pyridines†
Majji
Shankar
a,
Raja K.
Rit
 a,
Somratan
Sau
a,
Kallol
Mukherjee
a,
Vincent
Gandon
*bc and
Akhila K.
Sahoo
*a
a,
Somratan
Sau
a,
Kallol
Mukherjee
a,
Vincent
Gandon
*bc and
Akhila K.
Sahoo
*a
aSchool of Chemistry, University of Hyderabad, Hyderabad-500046, India. E-mail: akhilchemistry12@gmail.com; akssc@uohyd.ac.in
bInstitut de Chimie Moléculaire et des Matériaux d'Orsay, CNRS UMR 8182, Université; Paris-Sud, Université; Paris-Saclay, Bâtiment 420, 91405 Orsay Cedex, France. E-mail: vincent.gandon@universite-paris-saclay.fr
cLaboratoire de Chimie Moléculaire (LCM), CNRS UMR 9168, Ecole Polytechnique, Institut Polytechnique de Paris, route de Saclay, 91128 Palaiseau Cedex, France
First published on 27th May 2020
Abstract
Direct difunctionalization of chemically distinct ortho- and peri-C–H bonds of fused hetero(arenes) is illustrated through an unusual one-pot domino {[4 + 2] & [5 + 2]} double annulation with alkynes for the first time. This process is viable under Ru(II)-catalysis using a sulfoximine directing group and builds four bonds [(C–C)–(C–N) and (C–C)–(C–O)] in a single operation. Such synthetic manifestation offers access to uncommon [6,7]-fused oxepino-pyridine skeletons. DFT calculations provide mechanistic insight into this double annulation of naphthoic acid derivatives with alkynes and corroborate the participation of a ruthena-oxabicyclooctene intermediate, which is responsible for the rare 7-membered ring formation.
Introduction
Diversity oriented synthesis provides efficient access to complex molecular architectures that are present in natural products, pharmaceuticals, agrochemicals, and advanced-materials.1 This approach has sustained the development of novel therapeutic agents or probes for molecular biology, based on the resilient interaction of heterocycles with biological systems.2,3 Continuous efforts have therefore been directed towards the conception of straightforward synthetic methods for the construction of complex heteroarenes.3 In this regard, transition-metal (TM) catalyzed annulations of C–H bonds of (hetero)arenes with alkynes have proven invaluable.4,5 In particular, the TM-catalyzed direct functionalization or annulation of the ortho-C(2)–H bond of fused (hetero)arenes with alkynes are successful with acid/amide directing groups (DGs) via 5/7-membered metallacycle (Fig. 1A-I).5 With –OH, –NHR′, and –SR′′ DGs, the reactivity is shifted towards the peri-C(8)–H bond through 5/7-membered metallacycle (Fig. 1A-II).6 On the other hand, the activation of the peri-C(8)–H bond of fused (hetero)arene carboxylic acid derivatives [e.g. 1-naphthoic acid] is much more challenging and underdeveloped, due probably to the involvement of a strained [6,6,6]-fused metallacycle (Fig. 2A).7 Insertion of an alkyne would not even funnel such C–H activation step, as it would lead to an even more strained [6,6,8]-fused metallacycle (Fig. 2A). Thus, the molecular rigidity and conformational strain have hampered the development of such annulations at the peri-C(8)–H bond to form 7-membered fused compounds (Fig. 2A).8,9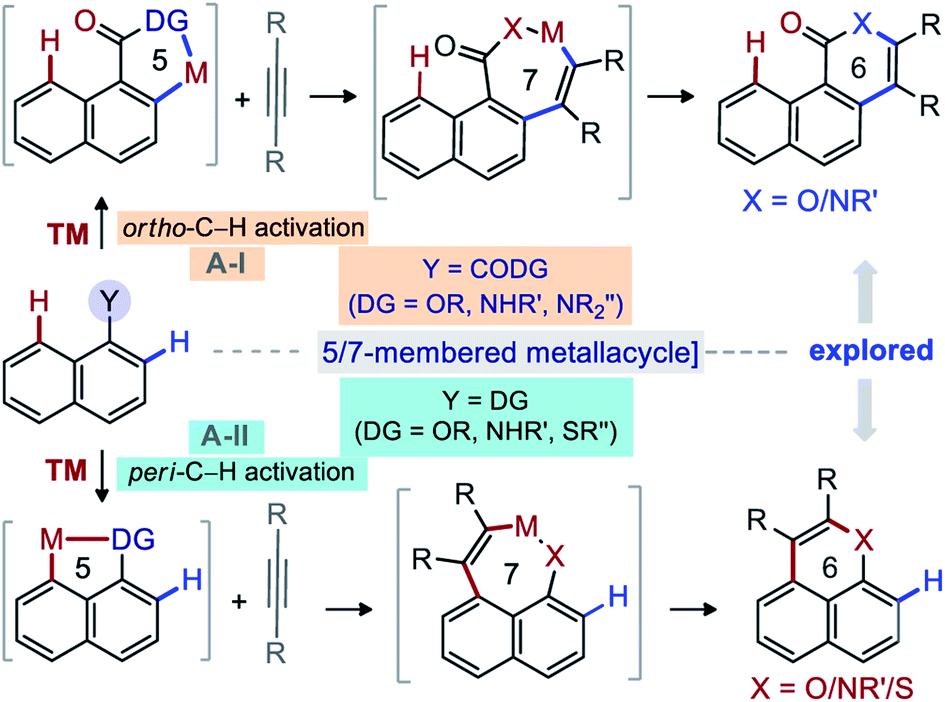 | ||
| Fig. 1 Background: Annulation of ortho-C(2)–H & peri-C(8)–H bond of 1-naphthalene derivatives with alkynes. | ||
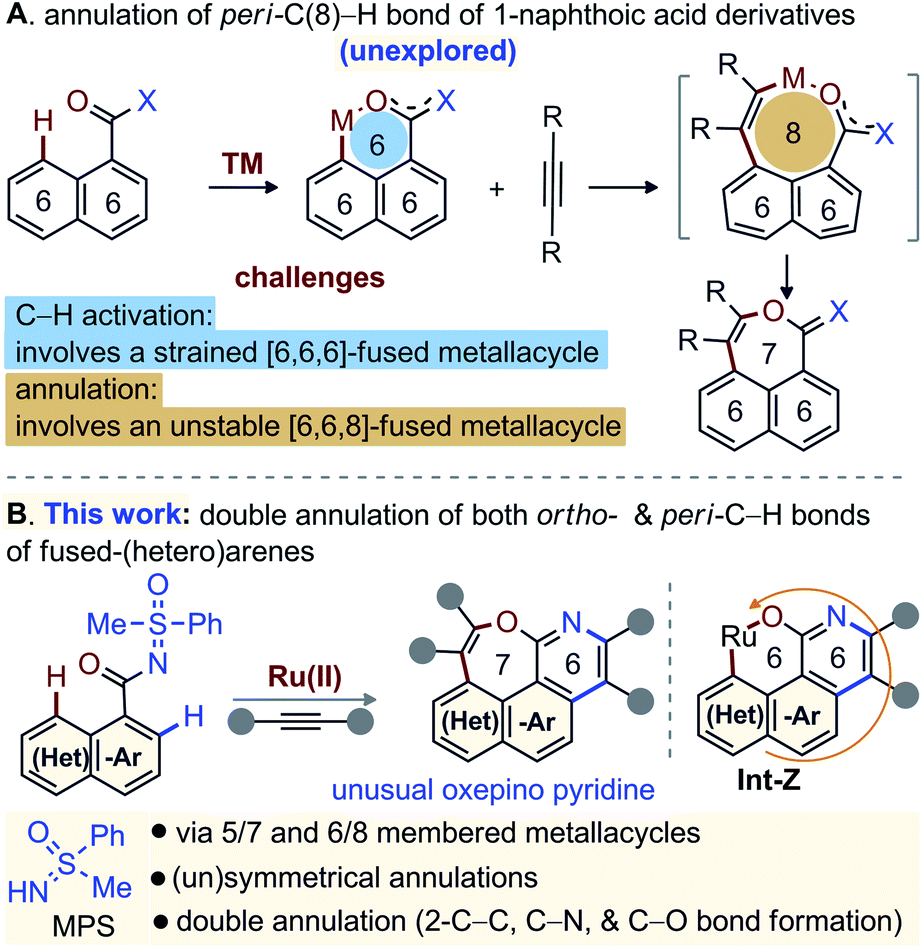 | ||
| Fig. 2 Multiple annulation of (hetero)arenes. | ||
Recent domino one-pot double annulation of o/o′-C–H bonds of (hetero)arenes with alkynes have led to [6,6]-fused heteroaryls.10,11 Although important issues of regio- and chemoselectivity, cumbersome mixtures due to incomplete conversion, catalytic viability, etc., could be addressed,12 such domino double C–H annulations were not extended to the formation of [6,7]-fused heteroarenes. To make such synthetic plan feasible, we hypothesized a Ru-catalyzed double annulation of 1-naphthoic acid derivatives with alkynes.
We believed the reaction would be initiated by N-aided C(2)–H activation and annulation with the alkyne to first form an angularly [6,6,6]-fused benzo[h]isoquinolinol. As peri-C–H bonds of fused-arenes are susceptible to electrophilic substitution, we anticipated an O-directed ruthenation of the proximal peri-C(8)–H bond to provide Int-Z (Fig. 2B). Finally, second alkyne incorporation to Int-Z and reductive elimination would build the unusual [6,7]-fused oxepino-pyridine motif (Fig. 2B). This one-pot domino double annulation uses the methylphenyl sulfoximine (MPS)-DG.12b Thus, the sequential activation of ortho- and peri-C–H bonds and annulation results in the formation of N- and O-enabled 6- and 7-membered rings on fused (hetero)arenes by generating four bonds (C–C & C–N and C–C & C–O) in a single operation (Fig. 2B).
Results and discussion
This one-pot [4 + 2] & [5 + 2] annulation was developed under Ru-catalysis using N-[1-naphthoyl]methylphenyl sulfoximine (1a) and 4-octyne (2a). The optimization studies are detailed in Table 1.13 The oxepino-pyridine 3aa was detected in 8% yield using {[RuCl2(p-cymene)]2 (5.0 mol%), AgSbF6 (20 mol%), NaOAc (1.0 equiv.)} as catalytic system, in ClCH2CH2Cl (DCE) at 120 °C for 24 h (entry 1). The cleavage of the sulfoximine motif presumably helps the formation of 3aa.11d In general, metal acetates facilitate Ru-mediated C–H activation through CMD (concerted metalation deprotonation), and also act as oxidant in the regeneration of the active catalyst.4 Accordingly, the double annulation was slightly improved when the reaction was conducted in the presence of the redox active bases Mn(OAc)2, AgOAc, and Zn(OAc)2·2H2O (entries 2–4), while Cu(OAc)2·H2O was found more promising as it delivered 3aa in 35% yield (entry 5). Additives such as KPF6, NaPF6, or AgBF4 instead of AgSbF6 were not beneficial (entries 6–8). The reaction efficiency was low when conducted in MeCN, toluene or TCE (entries 9–11). The domino diannulation in 1,4-dioxane provided 3aa in 41% yield (entry 12). The yield of 3aa was significantly improved to 68% when 10 mol% of Ru-catalyst and 40 mol% of AgSbF6 were used (entry 13). Finally, the catalytic conditions comprising [Ru(p-cymene)Cl2]2 (10 mol%), AgSbF6 (40 mol%), and Cu(OAc)2·H2O (1.5 equiv.) in 1,4-dioxane at 120 °C for 24 h were found optimum (entry 14), producing 3aa in 77% yield. Control experiments revealed that the silver salt and the acetate base were crucial (entries 15 and 16).4d|
|
||||
|---|---|---|---|---|
| Entry | Additive 1 (20 mol%) | Additive 2 (1.0 equiv.) | Solvent | Yield 3aab (%) |
| a Conditions: 1a (0.3 mmol), 2a (0.9 mmol), [RuCl2(p-cymene)]2 (5.0 mol%), additive-1 (20 mol%), additive-2 (0.3 mmol), solvent (2.0 mL) at 120 °C. b Isolated yield. c 1H NMR conversion. d [RuCl2(p-cymene)]2 (10 mol%), AgSbF6 (40 mol%) was used. e 2a (1.2 mmol), [RuCl2(p-cymene)]2 (10 mol%), AgSbF6 (40 mol%), Cu(OAc)2·H2O (1.5 equiv.) was used. DCE = ClCH2CH2Cl, TCE = 1,1,2,2-tetrachloroethane. | ||||
| 1 | AgSbF6 | NaOAc | DCE | 8 |
| 2 | 〃 | Mn(OAc)2 | DCE | 12 |
| 3 | 〃 | AgOAc | DCE | 15 |
| 4 | 〃 | Zn(OAc)2·2H2O | DCE | 11 |
| 5 | 〃 | Cu(OAc)2·H2O | DCE | 35 |
| 6 | KPF6 | 〃 | DCE | <5c |
| 7 | NaPF6 | 〃 | DCE | 6 |
| 8 | AgBF4 | 〃 | DCE | 30 |
| 9 | AgSbF6 | 〃 | MeCN | <5c |
| 10 | 〃 | 〃 | Toluene | 7 |
| 11 | 〃 | 〃 | TCE | 22 |
| 12 | 〃 | 〃 | 1,4-Dioxane | 41 |
| 13d | AgSbF6 | Cu(OAc)2·H2O | 1,4-Dioxane | 68 |
| 14 | AgSbF 6 | Cu(OAc) 2 ·H 2 O | 1,4-Dioxane | 77 |
| 15 | AgSbF6 | — | 1,4-Dioxane | <5c |
| 16 | — | Cu(OAc)2·H2O | 1,4-Dioxane | <5c |
|
||||

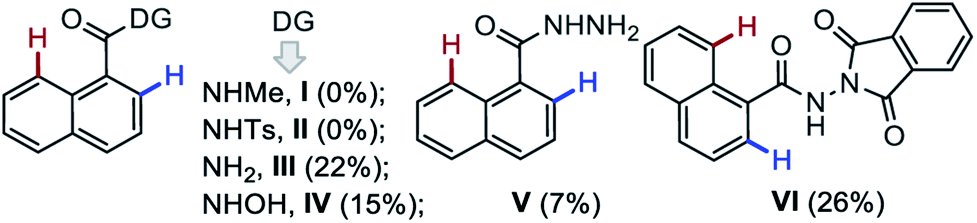
To validate the role of DGs in this one-pot domino {[4 + 2] & [5 + 2]} double annulation strategy, various DG-enabled 1-naphthyl bearing amides (I–VI) were subjected to the annulation with 2a under the optimized conditions (bottom of Table 1). The substrates having NH-Me (I) and NH-tosyl (II) DGs proved unreactive, whereas, simple 1-naphthylamide (III) underwent this domino annulations with 2a producing 3aa in poor yield.6 The N-oxidizable group protected amides [IV (with N–O bond), V, and VI (with N–N bond)] provided 3aa in 15%, 7%, and 26% yield, respectively. Thus, the MPS-DG was found most effective for the construction of the [6,7]-fused oxepino-pyridine skeleton.13
The generality of this annulation among fused (hetero)arenes exhibiting peri-C–H bonds and unactivated alkynes was explored under the optimized catalytic conditions (Scheme 1). The annulation of naphthalene derivatives 1a–l, bearing either electron-donating (Me, OMe, OEt), labile halo (F, Cl, Br), electron-withdrawing (CO2Me, COMPS), arene (Ph, pyrene), and OBn substituents at position 4, 5, or 6, with 2a, was successful in producing the respective 6,7-fused oxepino-pyridine 3aa–la in 45–87% yield. The tolerance of modifiable functionalities (i.e. F, Cl, Br, CO2Me, COMPS) offers the possibility of further functionalization. The core structure of 3ha and 3ja were elucidated by X-ray crystallographic analysis.14,15 Likewise, this double-annulation of 1a with the other internal alkynes 5-decyne (2b) and 3-hexyne (2c) delivered 3ab (79%) and 3ac (80%), respectively. Moreover, the gram scale synthesis of3ac(1.15 g) with recovery of PhSOMe (0.44 g) showed the robustness of the catalytic system and the transformable nature of the MPS group.5g Polyarene bearing scaffolds, for example: phenanthrene (1m), pyrene (1n), and perylene (1o), delivered 3ma, 3nb and 3oa, albeit in moderate yield.
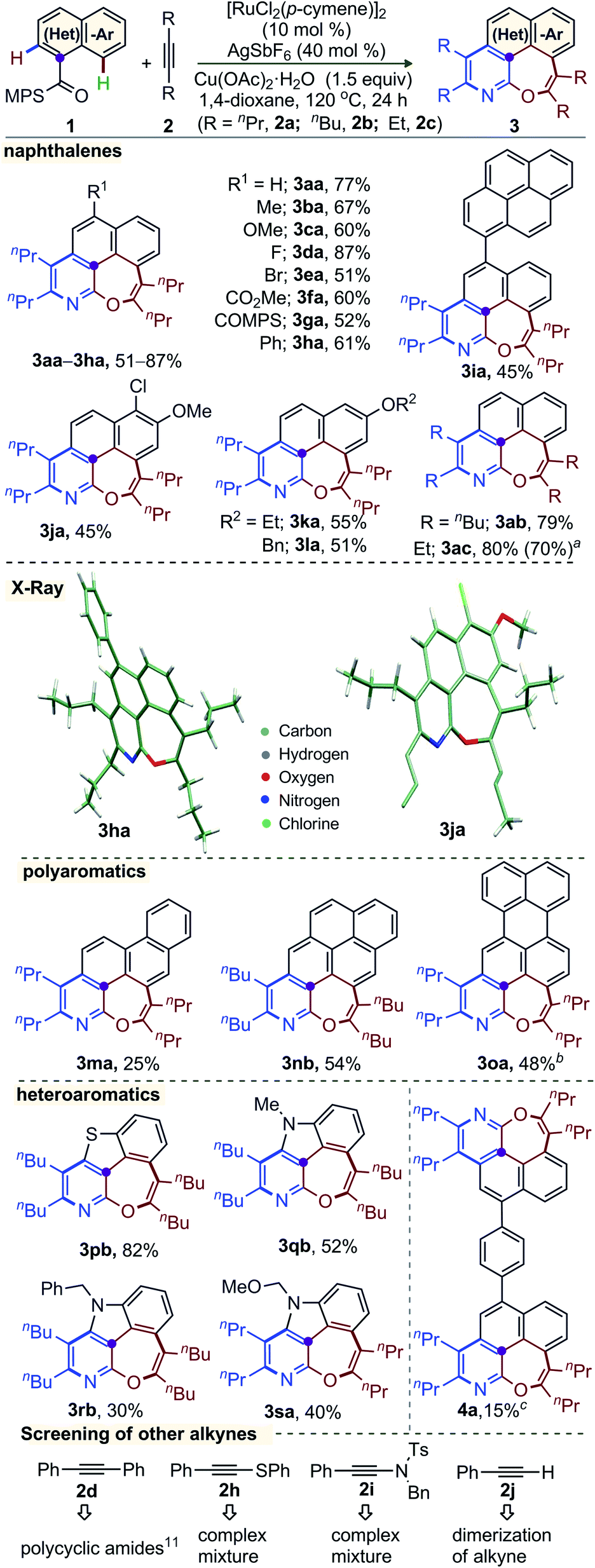 | ||
| Scheme 1 Synthesis of 6,7-oxepino[2,3-b]pyridine. Reactions were carried out with 1 (0.3 mmol) and 2 (1.2 mmol). aGram scale: 1a (1.54 g, 5.0 mmol); PhS(O)Me (63%) was isolated. bReactions were carried out in DCE. c2a (1.8 mmol). | ||
Importantly, benzothiophene derivative 1p smoothly reacted with 2b to afford 3pb in 82% yield. Indole-3-carboxylic acid derivatives 1q–s were used in this double annulation with 2b and 2a. The respective complex heteroarenes 3qb, 3rb, and 3sa were reliably accessed. The common N-protecting groups benzyl and MOM did not prevent the reaction. The yields are moderate in these cases, but the construction of these molecular scaffolds with three heteroatoms (i.e. S–N–O, N–N–O) in a 5,6,7-fused system is remarkable. Notably, the current synthetic plan was successful in making 8 bonds (4 C–C, 2 C–N, and 2 C–O) in a single operation; thus, an extended π-conjugated system 4a with two oxepino-pyridine motifs was made. The reaction of 1a with diphenylacetylene provided polycyclic amides through linear diannulation.11,14 On the other hand, the reaction of a thioalkyne or an ynamide with 1a produced complex mixtures (Scheme 1). Lastly, the terminal alkyne phenylacetylene underwent dimerization under the optimized oxidative condition.
The site-specific introduction of a novel functionality on an unreactive site of a complex motif has tremendous significance to the field of complex molecule synthesis and is often termed as late stage functionalization (LSF).16 In particular, LSF through C–H functionalization is very useful in drug discovery and draws significant attention from the scientific community. Accordingly, a range of biologically relevant motifs moulded with MPS-bearing naphthalene-1-carboxylic acid (5a–g) were synthesized and were independently subjected to the optimized reaction conditions with 2a and 2c (Scheme 2). Thus, the desired oxepino-pyridines 6aa-β-citronellol, 6bc-camphorsultam, 6ca-(−)-boreneol, 6ec-cholesterol, 6fc-estrone, and 6gc-lithocholic acid were constructed without any structural (chemical and stereochemical) changes of the complex architecture.14 The poor-to-moderate synthetic yields are due to low conversions. Isolation of unreacted precursors justifies the mass balance of the transformation.
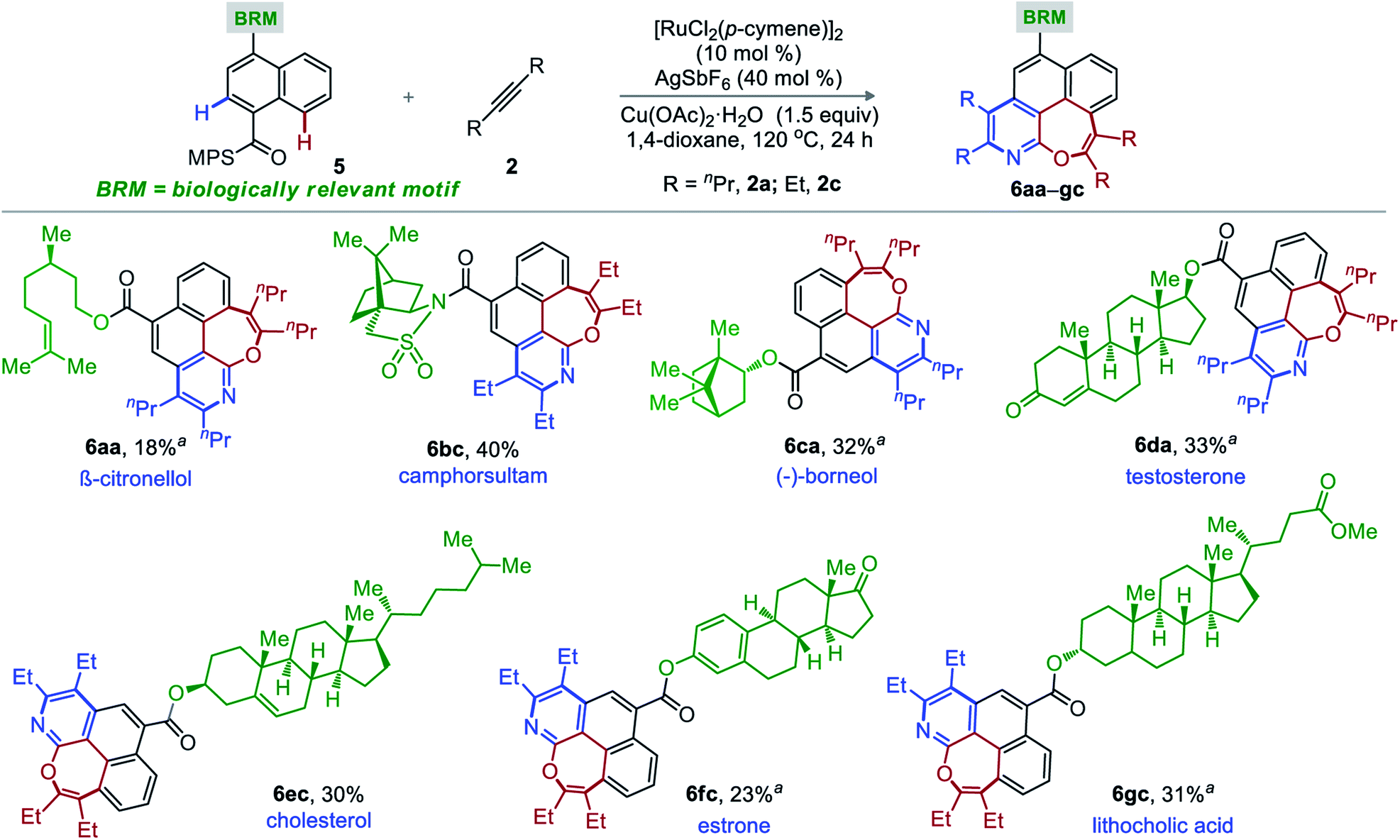 | ||
| Scheme 2 Double annulation of MPS-bearing naphthalene-1-carboxylic acid moulded in biologically relevant motifs. Reactions were carried out with 5 (0.3 mmol), 2 (1.2 mmol), [RuCl2(p-cymene)]2 (10 mol%), AgSbF6 (40 mol%), 1,4-dioxane (2.0 mL) at 120 °C for 24 h. aIsolation of unreacted precursors (20–55%). | ||
Encouraged by the broad range of oxepino-pyridines derivatives obtained (Schemes 1 and 2), the title reaction was next envisaged with two different alkynes. However, the difference in reactivity, regio- and chemoselectivity with different alkynes led to unexploitable annulation mixtures.12 To make this challenging unsymmetrical transformation viable, a two-step annulation sequence was tested. Accordingly, benzo[h]isoquinolinone 7a (0.5 mmol, 75%) was accessed from 1a and 2a when the reaction was carried out in presence of AcOH under Ru-catalysis (Scheme 3, Conditions A). Presumably the acid suppresses the second annulation through proto-demetallation.11 Next, the annulation of 7a with 1,2-diaryl alkynes (2d–g) led to the respective [6,7]-fused oxepino-pyridines (8ad–ag) in moderate yields (Scheme 3). The structure of 8ae was unambiguously confirmed by X-ray crystallography.14,15 A deuterium scrambling study and competition experiments were then performed to gain some mechanistic insight into this annulation (Scheme 4).
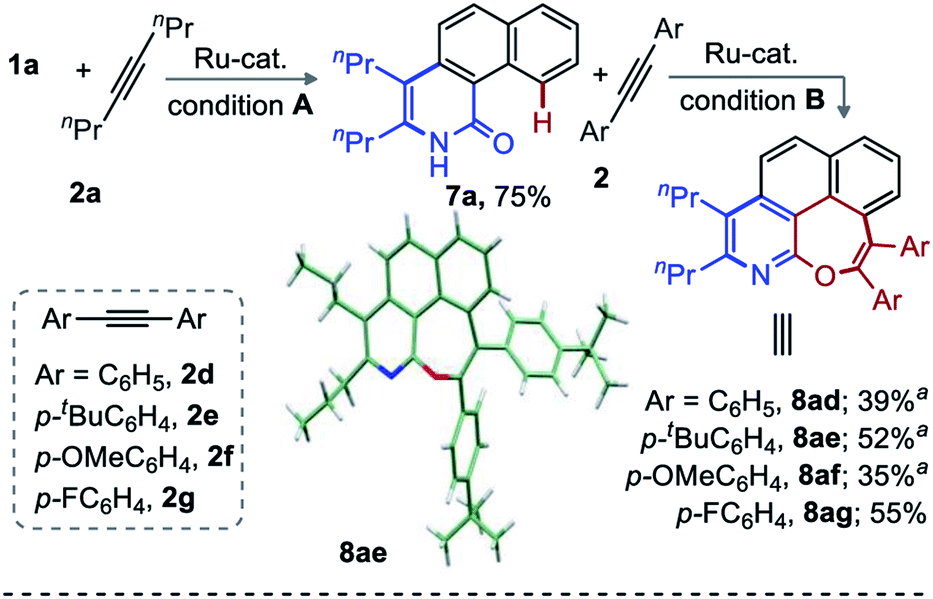 | ||
| Scheme 3 Unsymmetrical double-annulation of arenes with different alkynes. Conditions A: 1 (0.5 mmol), 2a (1.0 mmol), [RuCl2(p-cymene)]2 (5.0 mol%), AgSbF6 (20 mol%), AcOH (4.0 mmol), DCE (2.5 mL) at 120 °C for 20 h. Conditions B: 7a (0.3 mmol), 2 (0.45 mmol), [RuCl2(p-cymene)]2 (7.5 mol%), AgSbF6 (30 mol%), Cu(OAc)2·H2O (0.3 mmol), KH2PO4 (0.6 mmol), 1,4-dioxane (2.0 mL) at 120 °C for 20 h. aIsolation of unreacted mono-annulation product (30–45%). | ||
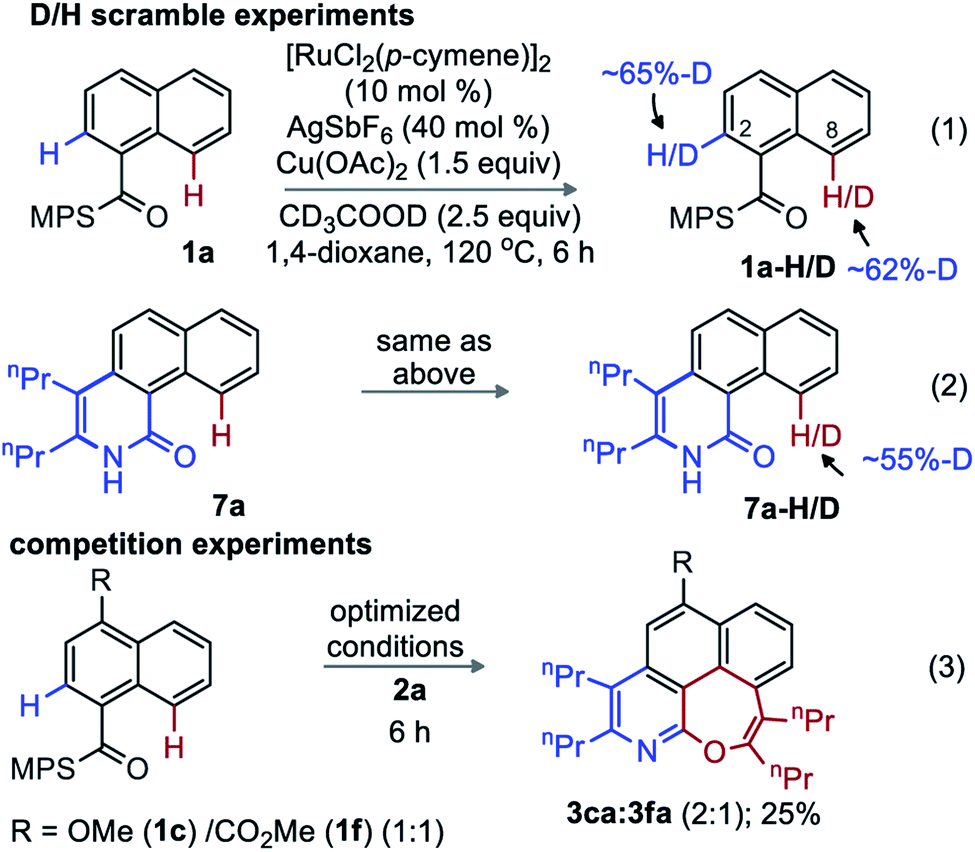 | ||
| Scheme 4 Deuterium scrambling and competition studies. | ||
Exposing 1a to the optimized conditions in presence of CD3CO2D (2.5 equiv.) resulted in D-incorporation at C2 (65%) and C8 (62%) positions (eqn (1)). Similarly, 55% of deuterium incorporation occurred at C8 in an identical experiment with 7a (eqn (2)). Therefore, activation of both the ortho- and peri-C–H bonds of MPS-enabled-1-naphthylamide is reversible. The competitive annulation of an equimolar mixture of 1c and 1f with 2a led to a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 3ca and 3fa; thus, an electron-rich arene reacts faster than an electron-poor one (eqn (3)).
:1 ratio of 3ca and 3fa; thus, an electron-rich arene reacts faster than an electron-poor one (eqn (3)).
In general, the π-conjugated polyfused heteroarenes show interesting photophysical properties. Thus, the absorption and emission spectra of oxepino-pyridines 3nb, 3oa, 3pb, 3qb, 3sa, 4a, and 8ae were measured in dichloromethane (1 × 10−5).14 Of note, compounds 3nb and 3ob show emission maxima at 436–512 nm with broad bandwidths and weak intensities.14
The mechanism of the title reaction has been studied computationally, employing the Gaussian 09 software package.17 Following a recent report, optimizations were carried out with the M06 functional, the 6-31G(d,p) basis set for all main group elements, and the LANL2DZ+f (ECP)18 basis set for Ru. Single point calculations were conducted at the M06/6-311++G(d,p)-SDD+f(ECP) level of theory. Solvation energies were obtained at the single point level using SMD approach for 1,4-dioxane. The discussed values are solvent-corrected Gibbs free energies at 393.15 K in kcal mol−1 (ΔG393). The molecular system A [1a, 2-butyne (2.0 equiv.), [RuOAcL]+ (L = p-cymene), AcO− ] was used as a reference for the free energies (Fig. 3). Thus, A contains two acetates to ensure two deprotonation of 1a. The complexation of the putative active species [RuOAc(p-cymene)]+ with 1a at first provides B with a release of 20.5 kcal mol−1. Next, C–H metalation occurs through TSBC lying 11.9 kcal mol−1 above B to provide metallacycle C (−23.6 kcal mol−1). Elimination of acetic acid and insertion of 2-butyne delivers the alkyne-complex E (more stable than C by 2.6 kcal mol−1). Alkyne insertion does not yield the proposed metal-alkenyl complex F′, but rather its valence isomer F, which is a metallacyclopropene as witnessed by the distortion of the 7-membered ring and by the short Ru–C distance of 1.85 Å. The formation of F is slightly endergonic by 0.3 kcal mol−1 that requires 14.9 kcal mol−1 of free energy of activation (TSEF). Then, intramolecular nucleophilic addition to the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond gives the annulation intermediate G (see arrows in F′). The conversion of F to G is the rate-determining step with a barrier 25.0 kcal mol−1 (19.6 kcal mol−1 from B), which is consistent with the temperature of the reaction (120 °C). Although the resulting complex G is less stable than F by 3.2 kcal mol−1, the acetate aided dissociation of [Ru(OAc)L]+ promotes spontaneous elimination of PhSOMe from the free ligand to give H, located as low as −69.0 kcal mol−1 on the energy surface. The liberation of PhSOMe, the conjugation of the anion, and the strong H-bond in H assist the loss of the sulfur moiety.
S bond gives the annulation intermediate G (see arrows in F′). The conversion of F to G is the rate-determining step with a barrier 25.0 kcal mol−1 (19.6 kcal mol−1 from B), which is consistent with the temperature of the reaction (120 °C). Although the resulting complex G is less stable than F by 3.2 kcal mol−1, the acetate aided dissociation of [Ru(OAc)L]+ promotes spontaneous elimination of PhSOMe from the free ligand to give H, located as low as −69.0 kcal mol−1 on the energy surface. The liberation of PhSOMe, the conjugation of the anion, and the strong H-bond in H assist the loss of the sulfur moiety.
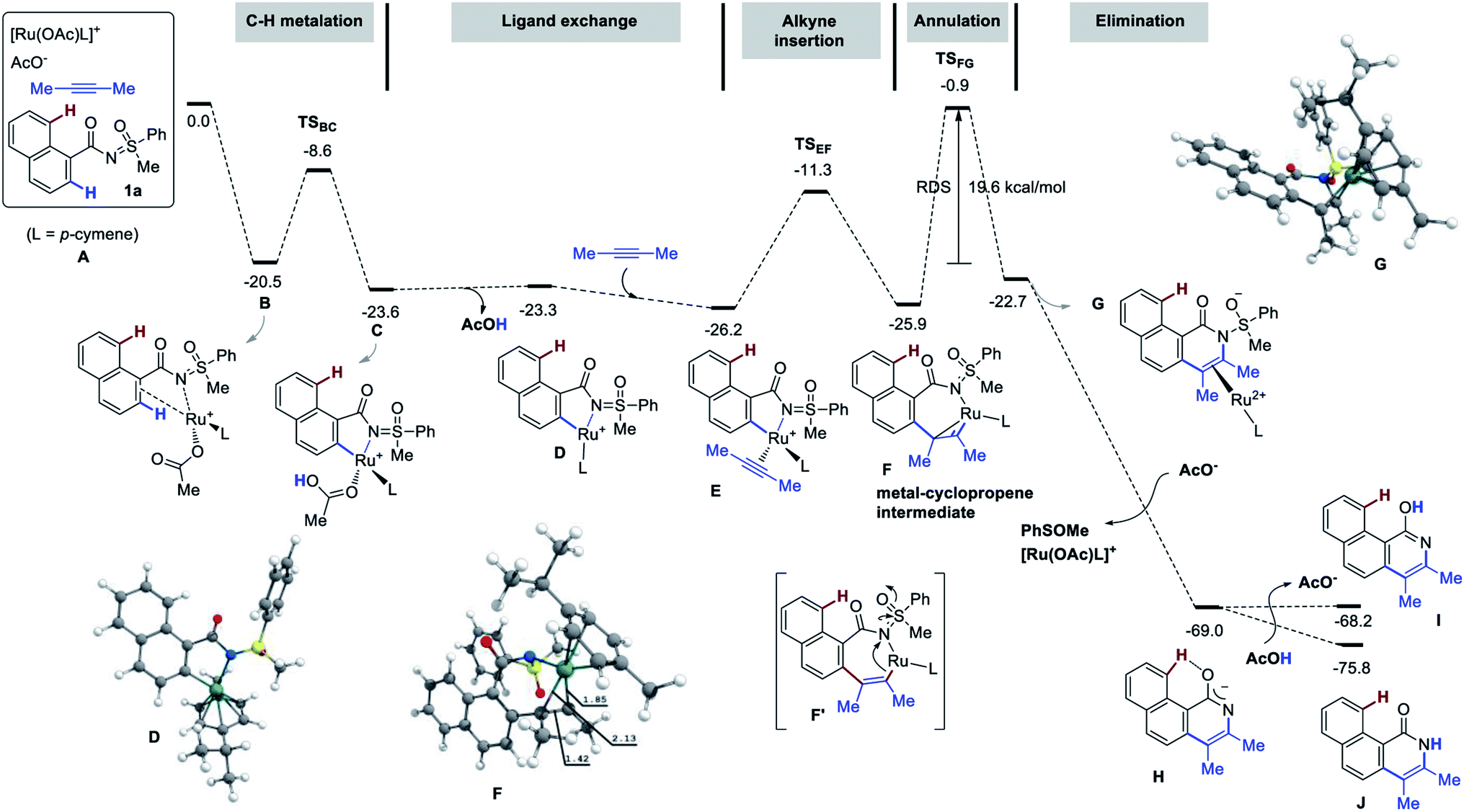 | ||
| Fig. 3 Free energy profile (ΔG393, kcal mol−1), part 1 (first annulation). | ||
Finally, protonation of H by AcOH produces pyridine I or the pyridone species J. In line with the experimental observations, J is significantly more stable. The mechanistic insight directed towards the second annulation for the construction of pyridine-fused 7-membered oxepine ring is depicted in Fig. 4. The complexation of H (at −69.0 kcal mol−1) with [Ru(OAc)L]+ is exergonic by 56.4 kcal mol−1 and yields K at −125.4 kcal mol−1. Intermediate K shows a H-bond between the acetate ligand and the peri-H of the naphthalene moiety. The Ru–C bond is short (2.36 Å), due to the coordination of Ru to the ipso-carbon and makes the peri-H acidic. The C–H metalation of the pre-organized complex K provides L (at −137.5 kcal mol−1 on the energy surface). This step requires 6.5 kcal mol−1 free energy of activation (TSKL). Next, the substitution of acetic acid with second alkyne equivalent is endergonic by 10.9 kcal mol−1 to afford N (−126.6 kcal mol−1). Of particular interest, the formation of 7-membered ring does not arise from the reductive elimination of a simple 8-membered metallacycle (O′′). Instead, at the expense of 15.9 kcal mol−1 of free energy of activation, the ruthena-oxabicyclooctene complex O, located at −132.7 kcal mol−1, is achieved from NviaTSNO. Among the Lewis depiction of O and O′, the structure O is supported by the Ru–Cipso distance of 2.35 Å and other geometrical parameters. Its formation can be understood as an intramolecular [2 + 2] cycloaddition between the alkyne and a RuC bond as shown in N′ (a fictive valence isomer of N). This process eventually avoids the participation of a highly strained phenanthrene-containing 8-membered ring (O′′). Then, the reductive elimination of O demands 25.2 kcal mol−1 free energy of activation to give P. This process is slightly endergonic and is the rate-determining step of this second annulation process. The transfer of the RuL moiety from P to the precursor 1a produces the desired [6,7]-fused oxepino-pyridine skeleton Q and chelate R. This step is exergonic by 7.9 kcal mol−1. Finally, as it is generally accepted, one can then propose that complex R transforms into B by Cu(OAc)2 mediated oxidation. Based on the experimental observations and insightful computational data, the mechanism of this double annulation is sketched in Fig. 5.4
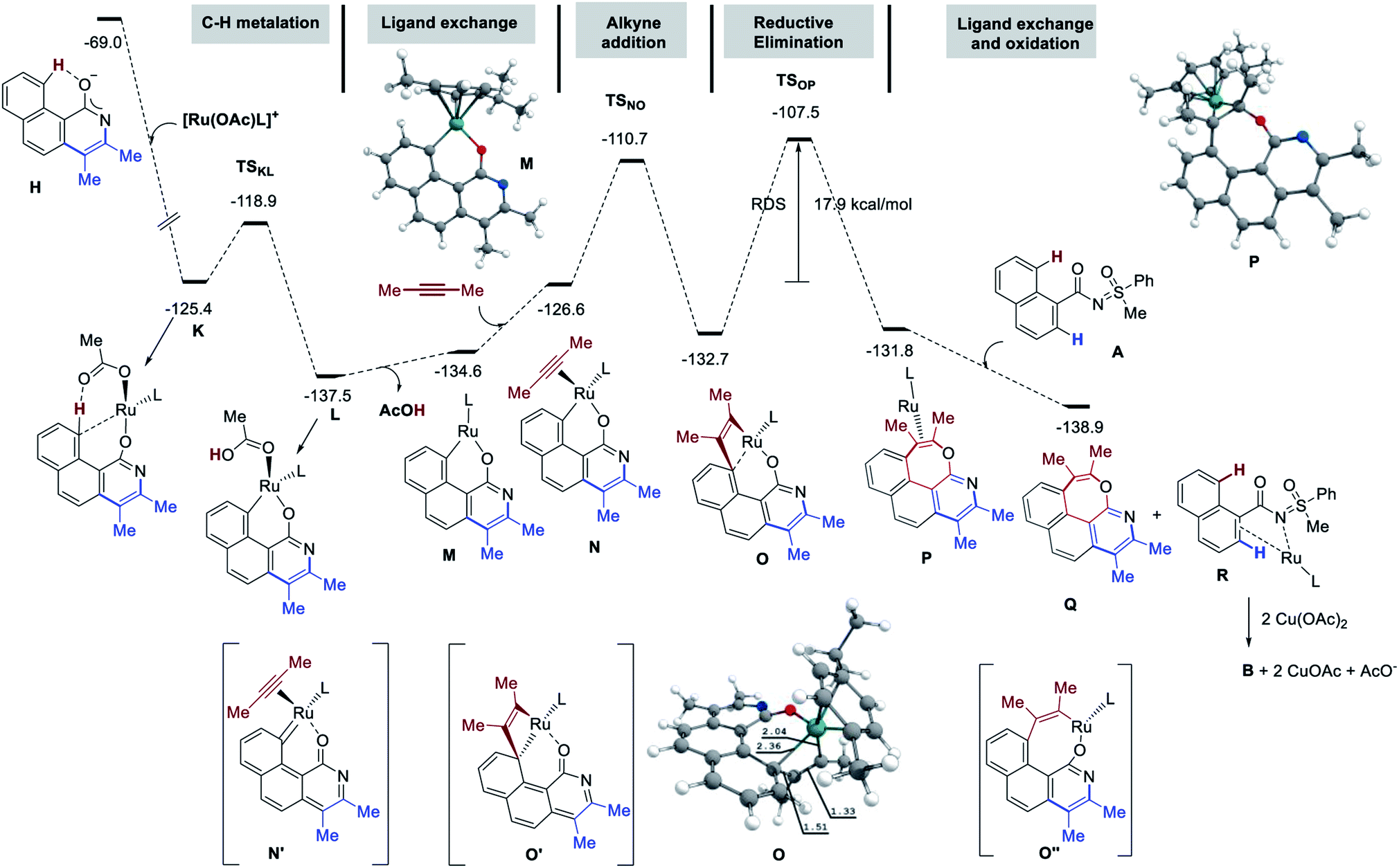 | ||
| Fig. 4 Free energy profile (ΔG393, kcal mol−1), part 2 (second annulation). | ||
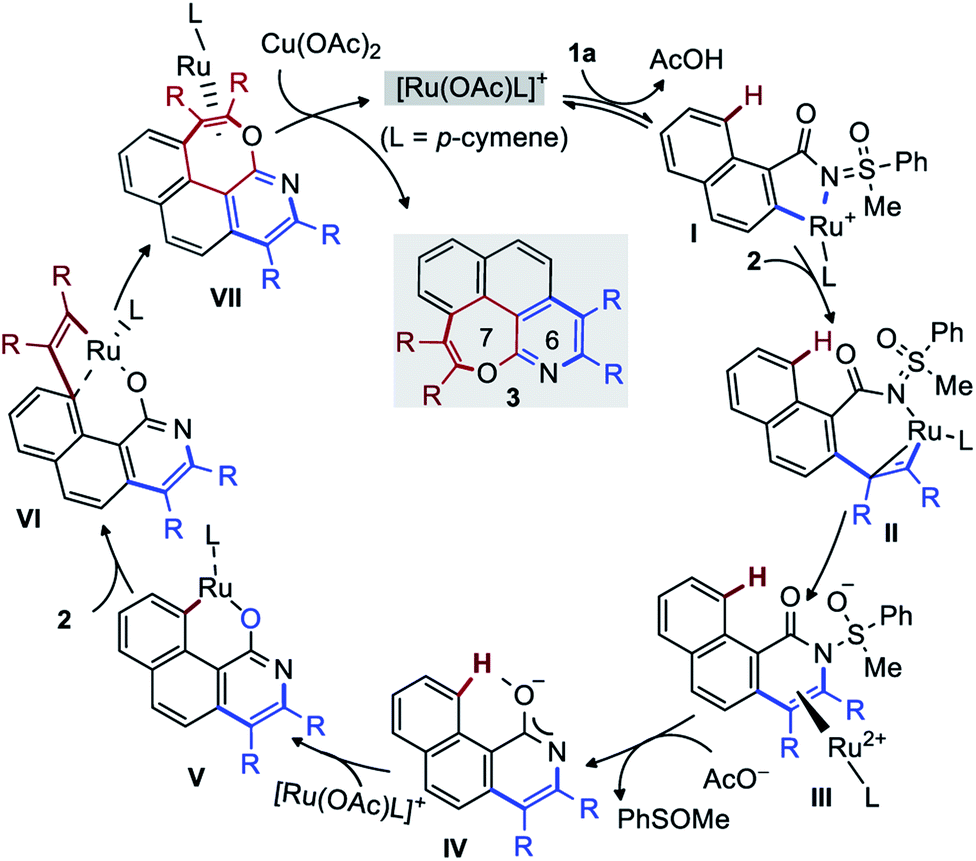 | ||
| Fig. 5 Plausible catalytic cycle. | ||
The active Ru-catalyst {generated from [Ru(p-cymene)Cl2]2, AgSbF6, and AcO−} first coordinates to MPS and activates the C(2)–H bond of 1a to form I (D in Fig. 3). The coordination of alkyne to I and its migratory insertion leads to II (F in Fig. 3). Next, the intramolecular nucleophilic addition to the NS bond provides III (G in Fig. 3), which is the rate-determining step of the mono-annulation. The acetate-aided expulsion of [Ru(OAc)L]+ and elimination of PhSOMe leads to pyridone species IV (H in Fig. 3). Next, direct C(8)–H ruthenation of IV affords V (M in Fig. 4). Then, alkyne insertion into V generates the unusual ruthena-oxabicyclooctene complex VI (O in Fig. 4). The reductive elimination of VI gives VII (P in Fig. 4) and is the rate-determining step of the second annulation. Finally, Cu(OAc)2 mediated transfer of RuL moiety to 1a liberates the desired [6,7]-fused oxepino-pyridine skeleton.
Conclusion
In summary, we have developed an unprecedented Ru-catalyzed sulfoximine-directed one-pot domino {[4 + 2] & [5 + 2]} double annulation of 1-naphthoic acid derivatives with alkynes for the synthesis of unique [6,7]-fused oxepino-pyridine motifs. This transformation functionalizes both chemically distinct ortho- and peri-C–H bonds of fused-hetero(arenes) through double annulation, making four (C–C & C–N and C–C & C–O) bonds in a single operation. In addition, two-step unsymmetrical annulations with different alkynes are also shown. The detailed DFT calculations endorse the participation of metal-cyclopropene and ruthena-oxabicyclooctene intermediates. The construction of biologically relevant drugs anchored oxepino-pyridine scaffolds, broad scope, and gram scale synthesis make the transformation synthetically viable.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the SERB-India (grant no: EMR/2014/385). We thank University of Hyderabad (UoH; UPE-CAS and PURSE-FIST) for overall facility. M. S, S. S, and K. M thank CSIR, India, for fellowship. We thank Piyas Saha for his help. We thank Arijit Saha and Anilkumar Gunnam (UoH) for fluorescence measurement and crystallographic studies. VG thank CNRS, UPSaclay, Ecole Polytechnique and ANR-18-CE07-0012-02 for financial support.Notes and references
-
(a) M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46 CrossRef PubMed
; (b) W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 CrossRef PubMed
-
(a) K. Narita, K. Nakamura, Y. Abe and T. Katoh, Eur. J. Org. Chem., 2011, 4985 CrossRef CAS
-
(a) S. K. Sinha, G. Zanoni and D. Maiti, Asian J. Org. Chem., 2018, 7, 1178 CrossRef CAS
-
(a) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212 CrossRef CAS PubMed
-
(a) Y. Su, M. Zhao, K. Han, G. Song and X. Li, Org. Lett., 2010, 12, 5462 CrossRef CAS PubMed
-
(a) S. Mochida, M. Shimizu, K. Hirano, T. Satoh and M. Miura, Chem.–Asian J., 2010, 5, 847 CrossRef CAS PubMed
-
(a) J. Yin, M. Tan, D. Wu, R. Jiang, C. Li and J. You, Angew. Chem., Int. Ed., 2017, 56, 13094 CrossRef CAS PubMed
-
(a) A. Seoane, N. Casanova, N. Quiñones, J. L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2014, 136, 834 CrossRef CAS PubMed
- G. Favini, J. Mol. Struct., 1983, 93, 139 CrossRef
-
(a) X. Tan, B. Liu, X. Li, B. Li, S. Xu, H. Song and B. Wang, J. Am. Chem. Soc., 2012, 134, 16163 CrossRef CAS PubMed
-
(a) S. Mochida, N. Umeda, K. Hirano, T. Satoh and M. Miura, Chem. Lett., 2010, 39, 744 CrossRef CAS
-
(a) D. Sarkar, F. S. Melkonyan, A. V. Gulevich and V. Gevorgyan, Angew. Chem., Int. Ed., 2013, 52, 10800 CrossRef CAS PubMed
- The release of sulfoxide from the sterically encumbered metallacyclic RuL-MPS moiety (Int-II or Int-III; Fig. 5) and the direct participation of Int-IV presumably helps the annulation.
- See the ESI.†.
- CCDC 1979334 (3ha), CCDC 1979335 (3ja), and CCDC 1979336 (8ae) contain the supplementary crystallographic data for this paper.†.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachael and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed
-
(a)
T. H. Dunning Jr and P. J. Hay, Modern Theoretical Chemistry, ed. H. F. Schaefer III, Plenum, New York, 1997, vol. 3 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Copies of the 1H NMR, 13C NMR and HRMS data for all products. CCDC 1979334–1979336. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01373k |
| This journal is © The Royal Society of Chemistry 2020 |