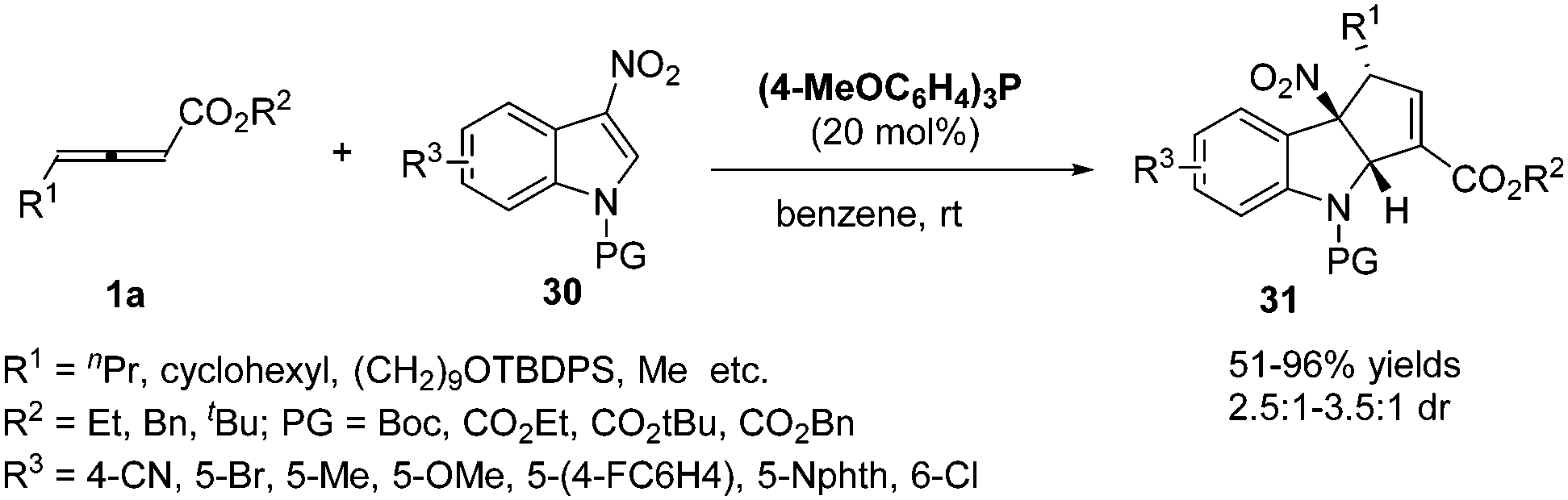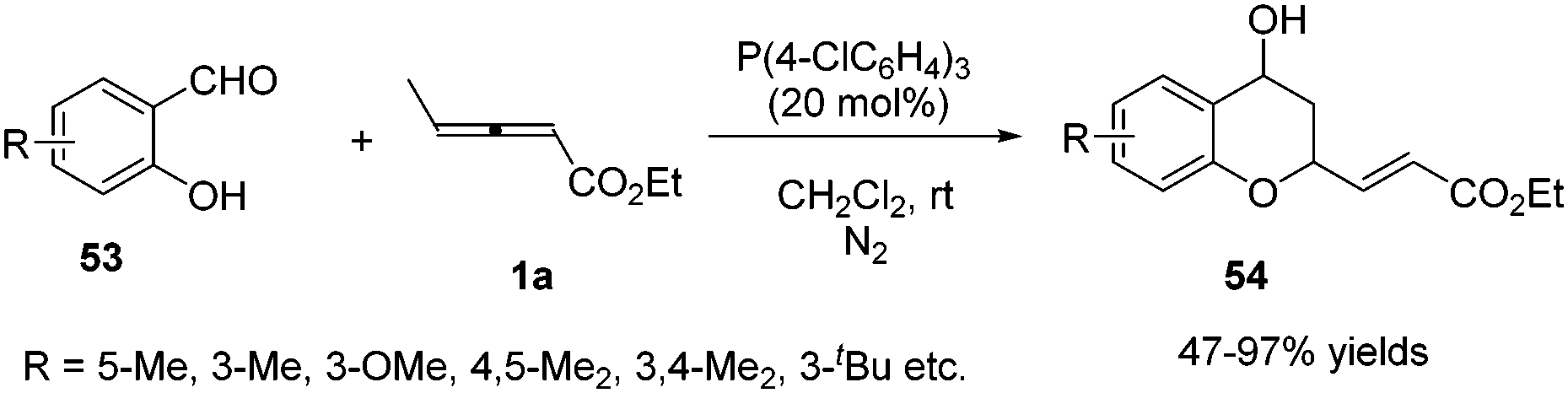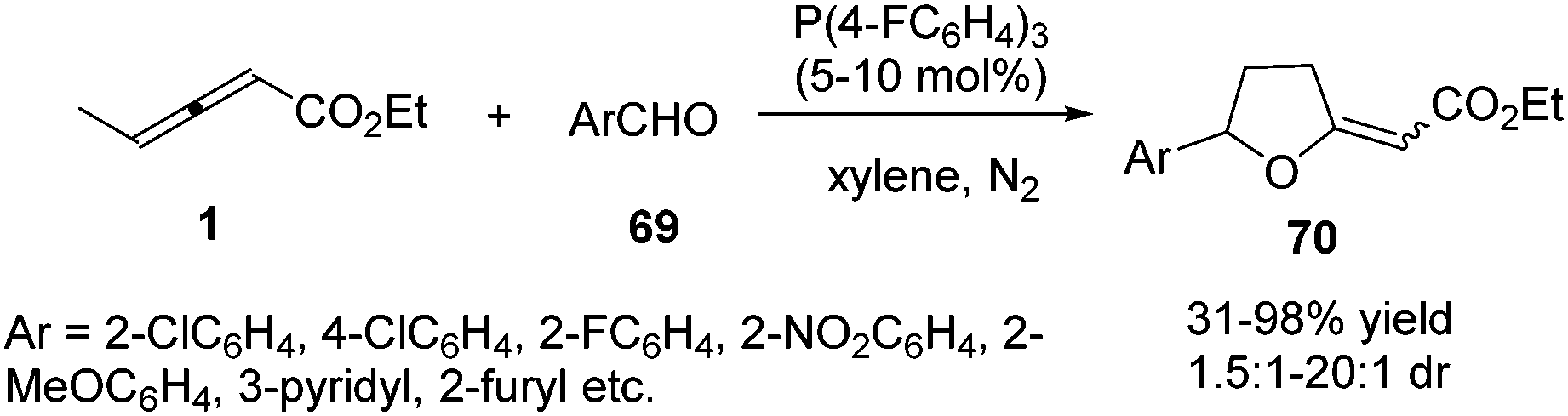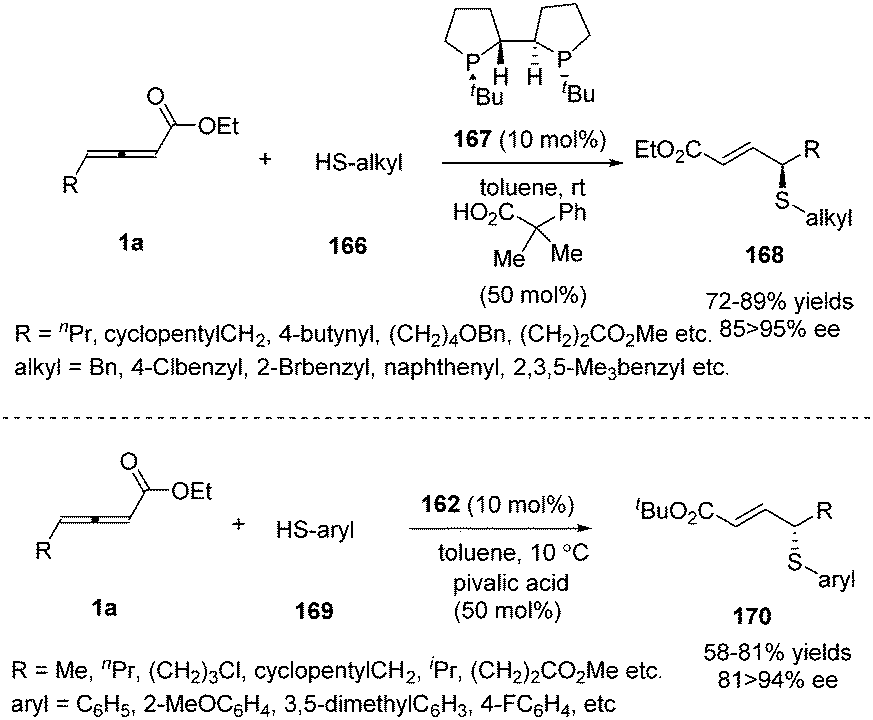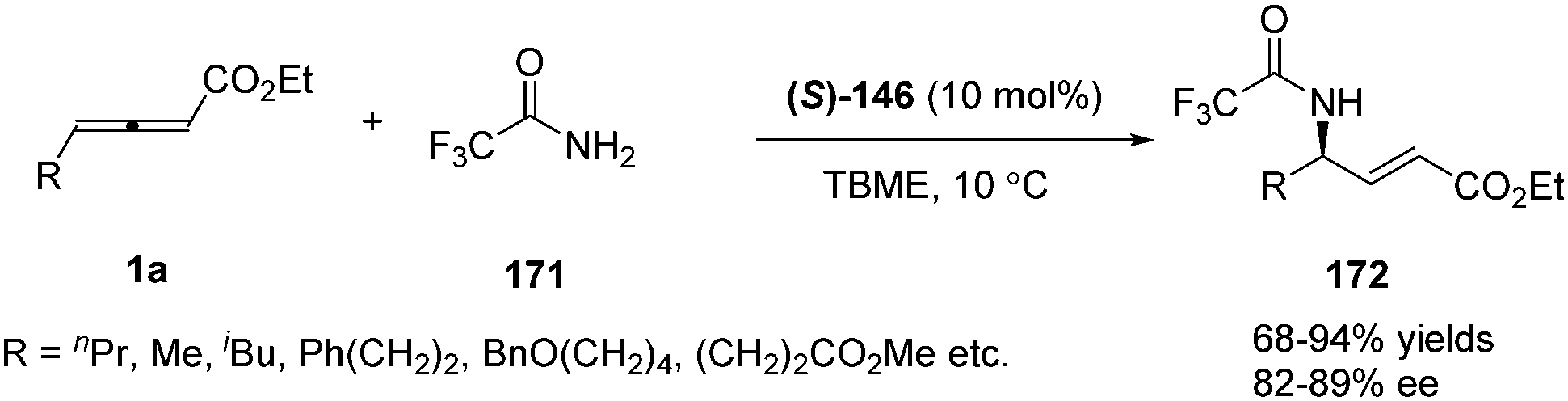DOI:
10.1039/C9CC08241G
(Feature Article)
Chem. Commun., 2020,
56, 680-694
Recent advances in phosphine catalysis involving γ-substituted allenoates
Received
21st October 2019
, Accepted 9th December 2019
First published on 10th December 2019
Abstract
Organophosphine catalysis of allenoates has doubtlessly been one of the most ideal and powerful synthetic strategies for the generation of highly functionalized carbo-/hetero-cycle motifs, which are important structural motifs in biologically active natural products and pharmaceuticals. Because of their diverse and amazing reactivity, chemists usually pay more attention to the study of 2,3-butadienoates and α-substituted allenoates. More recently, there is a growing interest in the study of phosphine catalysis of γ-substituted allenoates, which usually have low reactivity and selectivity. This feature article will describe the selected examples of organophosphine catalysis of γ-substituted allenoates with a wide range of electrophiles.

Er-Qing Li
| Dr Er-Qing Li was born in 1987 in Henan province, China. He received his PhD in 2015 from Nankai University under the guidance of Prof. You Huang. Subsequently he worked as a postdoctoral fellow with Prof. Lutz F. Tietze at Göttingen University, Germany. He then joined Zhengzhou University at the end of 2016 and started his independent research. His major research interests are asymmetric catalysis, design and synthesis of P-stereogenic phosphine ligands and transition metal catalysis. |

You Huang
| Prof. You Huang was born in Henan province, China, in 1962. He received his BS degree in chemistry (1986) and MS degree in organic chemistry (1989) from Henan Normal University in China, and his PhD degree in organic chemistry from Zhejiang (Hangzhou) University, China (1997). After working as a postdoctoral fellow at Nankai University (1997–1999) and Osaka University (1999–2002) he joined Nankai University and is currently a full professor of chemistry. His major research interests include organophosphine chemistry, organocatalysis, asymmetric catalysis and development of new synthetic methodologies. |
1. Introduction
The construction of carbon–carbon and carbon–heteroatom bonds is central to organic synthesis. Despite the enormous progress made over the past few decades, the development of clean, economical and efficient chemical processes is still a perennial quest from the viewpoint of green and sustainable chemistry.1 In this context, organocatalysis, with the advantages of environmental-friendly, metal-free, inexpensive and mild reaction conditions, has lately experienced a remarkable growth. Among the variety of organocatalysts, Lewis base catalysts have received considerable attention and have also emerged as a versatile means for the synthesis of cyclic and heterocyclic compounds, which mainly include tertiary phosphine and amine catalysts. Although both of them are pyramidal, they exhibit different catalytic reactivities. This is because the inversion of amines is rapid in most cases whereas phosphines are configurationally stable. And organophosphines are generally more nucleophilic and less basic than similarly substituted amines. Over the past few decades, organophosphines have proved to be a powerful tool in organic synthesis.2 In particular, phosphine-catalysed domino annulation reactions of allenoates have been extensively investigated since the pioneering works reported by Lu in 1995.3
2,3-Butadienoates are flexible and multifunctional and can be easily transformed to other synthetically interesting products. As illustrated in Scheme 1, nucleophilic attack of an organophosphine catalyst on the electrophilic, β-carbon of an allenoate results in the generation of a zwitterionic intermediate, which can be depicted in several ways, including anion localization at the α-carbon or γ-carbon, as one-, two or three synthons (Scheme 1, (1)).4 Inspired by this result, γ-substituted allenoates, which usually have low reactivity and selectivity as shown in a previous study,5 have begun to attract the attention of chemists. Introduction of δ-methylene leads to the formation of some new zwitterionic intermediates (Scheme 1, (2)). Thus γ-substituted allenoates can show good diversity of reactions by substrate modification. As this is a fast-growing field, a summary of γ-substituted allenoates for organic transformation is highly desired. This review illustrates the recent trend in phosphine-mediated reactions of γ-substituted allenoates. The diverse reactivity, various transformations as well as reaction mechanisms will be described.
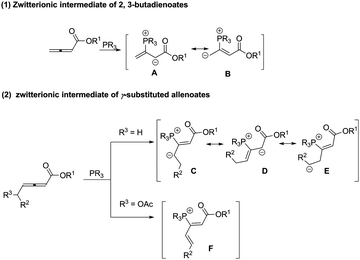 |
| | Scheme 1 The reactivity of allenoates in the presence of organophosphines. | |
2. Phosphine-catalysed classical (3+2) annulations with electrophiles
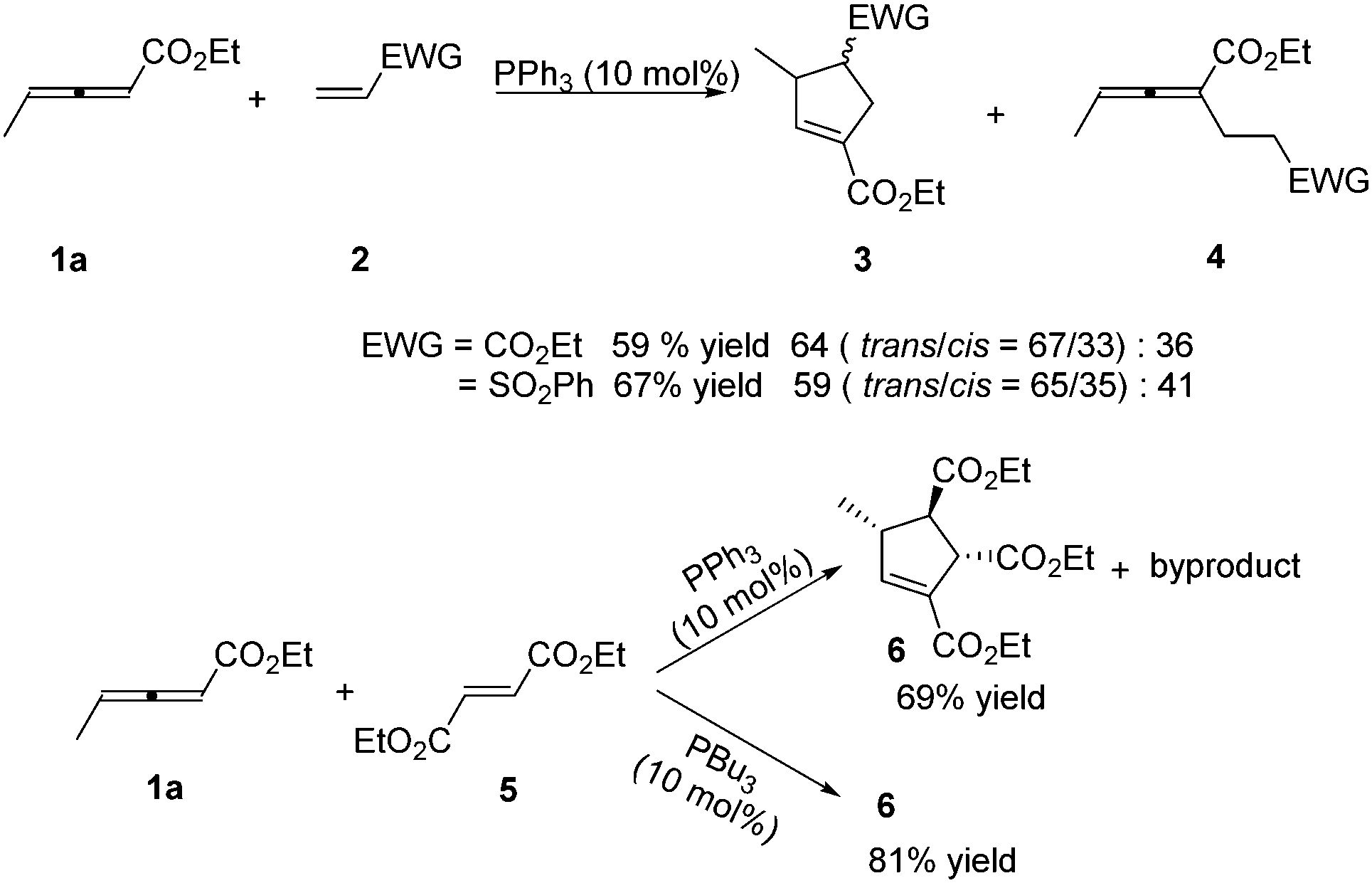
In 1999, Lu reported the first phosphine-catalysed (3+2) annulations of γ-substituted allenoates with electron-deficient olefins.6 It was found that treatment of ethyl 2,3-pentadienoate 1a and ethyl acrylate or vinyl phenyl sulfone 2 in dry benzene with 10 mol% of triphenylphosphine at room temperature resulted in a (3+2) annulation, giving the α-regioisomeric cycloadducts 3 together with noncyclic adducts 4. When the authors used diethyl fumarate 5 as the electron-deficient olefin, high diastereoselective (3+2) annulation adduct 6 was produced with a small amount of an uncharacterized by-product. However, the sole cycloadduct 6 was produced in higher yield by using PBu3 as a catalyst. The result showed that the nucleophilicity of phosphine catalysts had significant impact on this reaction (Scheme 2).
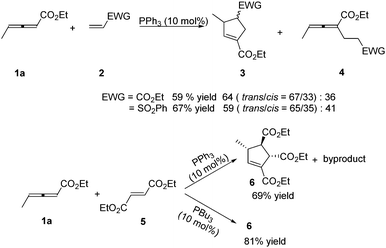 |
| | Scheme 2 Phosphine-catalysed (3+2) cycloaddition reactions of acrylate, vinyl phenyl sulfone or fumarate with γ-methyl allenoate. | |
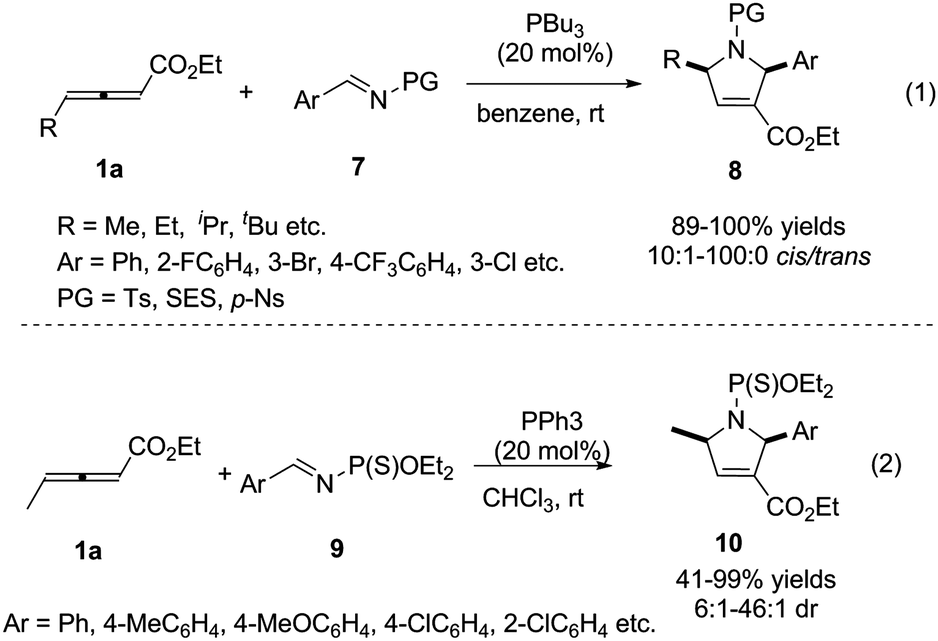
For overcoming the reactive selectivity, Kwon expanded the annulation methodology to N-sulfonylimines 7. The desired pyrroline derivatives 8 were formed in excellent yields with high diastereoselectivity (Scheme 3, (1)).7 In 2008, He also reported a phosphine-catalysed [3+2] cycloaddition reaction of γ-methyl allenoates 1a with imines by employing N-(thio) phosphoryl imines 9 as substrates (Scheme 3, (2)).8
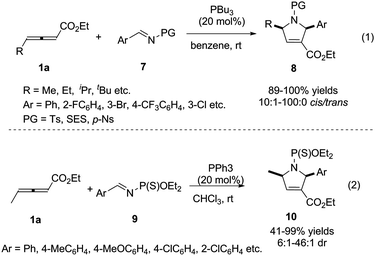 |
| | Scheme 3 Phosphine-catalysed (3+2) cycloaddition reactions of sulfonylimines and N-(thio) phosphoryl imines. | |
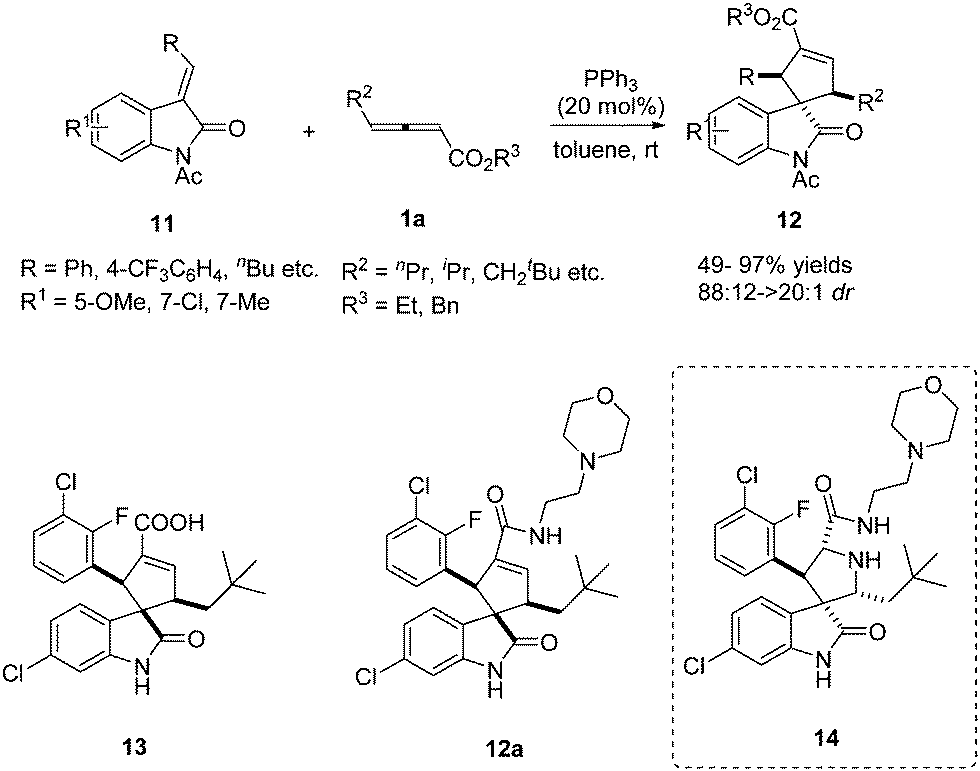
In 2013, Marinetti reported a phosphine-catalysed annulation for the synthesis of 3,3-spirocyclopenteneoxindoles from γ-substituted allenoates.9 This work demonstrated that PPh3 exerted a very efficient control over the relative stereochemistry of the three stereogenic centers of the final products. It should be noted that the authors realized the straightforward synthesis of carbocyclic analogues of an important series of inhibitors of MDM2–p53 interactions with anticancer properties, which were the first representatives of a new class of carbocyclic analogues of the bioactive pyrrolidine spirooxindoles 14. The authors have demonstrated that 13 and 12a displayed antiproliferative activity in the μM range against SJSA-1 osteosarcoma and HCT116 p53-wt cell lines (Scheme 4).10
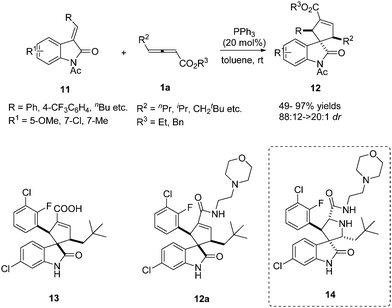 |
| | Scheme 4 Phosphine-catalysed (3+2) cycloaddition reactions of 3-benzylideneindolin-2-ones. | |
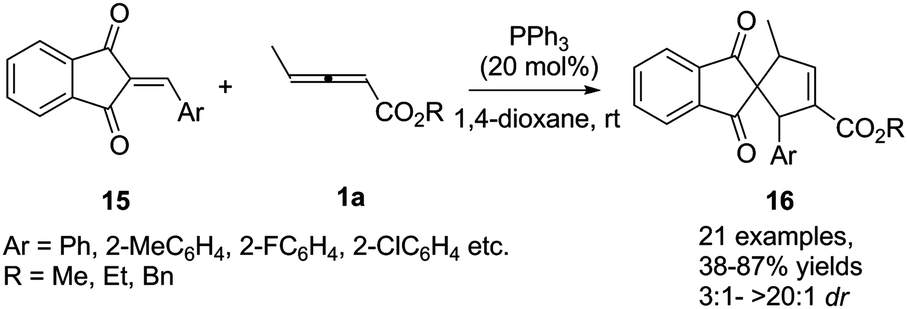
More recently, Duan and Li reported a phosphine-catalysed (3+2) annulation of γ-methyl allenoates 1a with 2-arylidene-1H-indene-1,3(2H)-diones 15. In the reaction, a series of highly functionalized spiro[4.4]dec-6-ene skeletons 16 were obtained in moderate to good yields and high diastereoselectivity. It should be noted that the perfect α-regioselective annulation adducts were obtained with the simple PPh3 catalyst (Scheme 5).11
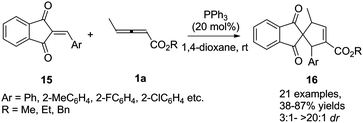 |
| | Scheme 5 Phosphine-catalysed (3+2) annulation of 2-arylidene-1H-indene-1,3(2H)-diones. | |
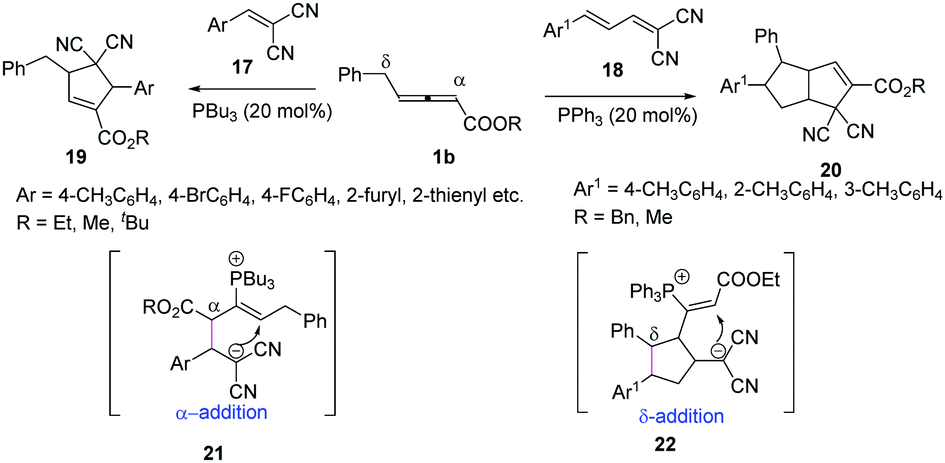
In 2018, Huang and co-workers reported a phosphine-catalysed (3+2) annulation of γ-benzyl allenoates with electron-deficient olefins. The authors found that different substrates could control the selectivity of γ-benzyl allenoates. When the substrates 17 were used, the classic (3+2) annulation products 19 were obtained by α-regioselective addition, while when the substrates 18 were applied, the (2+3)/(3+2) annulation products 20 were obtained with moderate yields and diastereoselectivity by δ-regioselective addition (Scheme 6).12
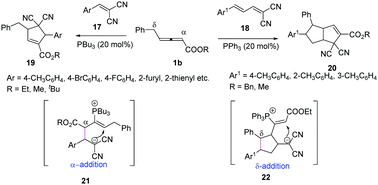 |
| | Scheme 6 Phosphine-catalysed (3+2) annulation of 2-arylidene-malononitriles. | |
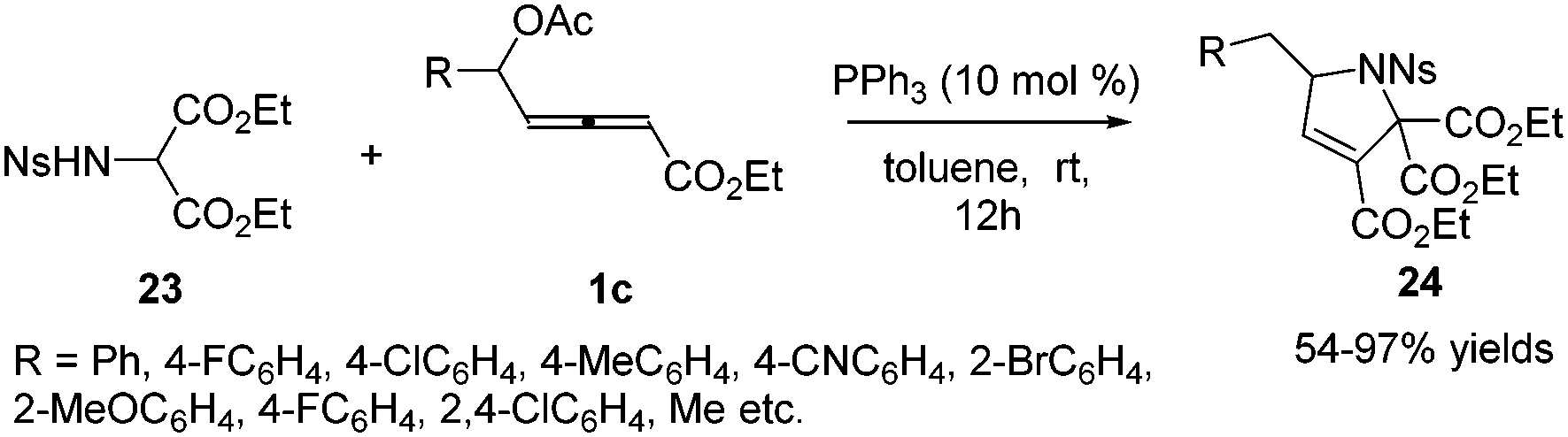
Subsequently in 2018, Tong and co-workers designed a δ-acetoxy allenoates 1c, which were applied to the phosphine-catalysed (3+2) annulations, providing a practical and efficient access to highly substituted 3-pyrrolines 24 in 54–97% yields. The authors found that the chiral phosphine catalyst (R)-SITCP could give good enantioselectivity (up to 83% ee) in the asymmetric version of this reaction (Scheme 7).13
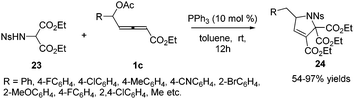 |
| | Scheme 7 Phosphine-catalysed (3+2) annulation of δ-acetoxy allenoates with 2-sulfonamidomalonate. | |
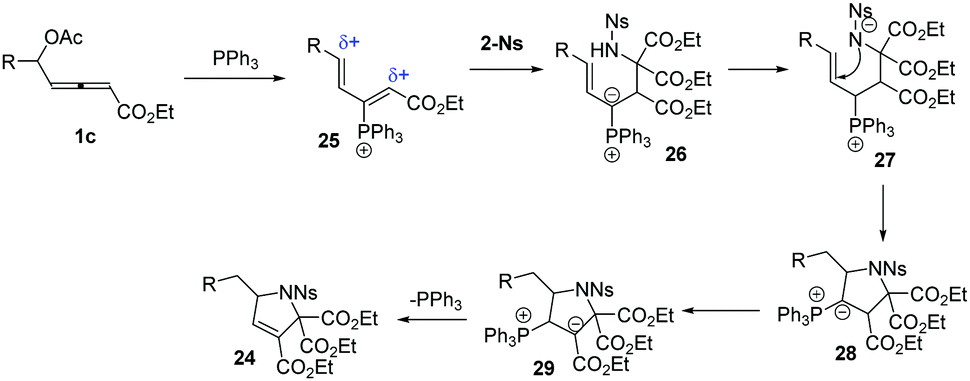
The authors proposed the mechanisms for the reaction of 1 and 2-Ns (23). As shown in Scheme 8, firstly, the δ-acetoxy allenoates underwent a nucleophilic addition and the subsequent elimination of acetate delivered the 3-phosphonium-2,4-dienoate intermediate 25. The authors thought that the compound 2-Ns selectively attacked the αC atom to give the intermediate 26. Next, a proton shift process and the following intramolecular addition resulted in ylide 28. The final proton transfer and 1,2-elimination generated the final products 24 and regenerated the PPh3 catalyst.
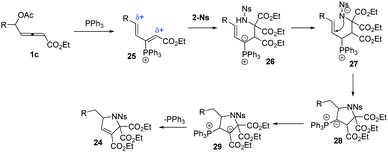 |
| | Scheme 8 Plausible mechanisms. | |
More recently, López and Bandini reported a phosphine-catalysed (3+2) dearomatization of 3-NO2-indoles with γ-substituted allenoates. With (p-MeOC6H4)3P as the catalyst, a range of densely functionalized indolines 31 were obtained in high yields under mild reaction conditions with moderate to good diastereoselectivity. The authors revealed a water-mediated [1,2]-H shift mechanism by DFT calculations and deuterium-labeling experiments, which also accounted for the diastereoselection recorded via DFT calculations (Scheme 9).14
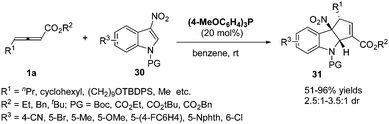 |
| | Scheme 9 Phosphine-catalysed stereoselective dearomatization of 3-NO2-indoles. | |
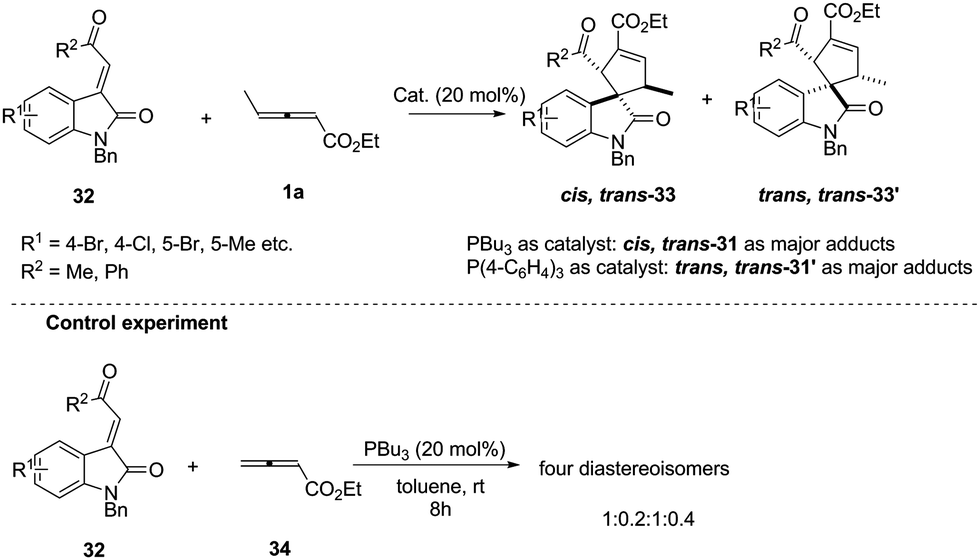
In early research, chemists found that α-addition/annulation usually occurred in the phosphine-catalysed (3+2) annulations of γ-substituted allenoates. In 2011, Shi reported a novel phosphine-catalysed (3+2) annulation by γ-addition/annulation. A control experiment showed that the methyl group on ethyl 2,3-pentadienoate is a key factor to affect the regioselectivity. It should be noted that the nucleophilic phosphine catalyst affected the final product and was proven to be critical for controlling diastereoselectivity (Scheme 10).15
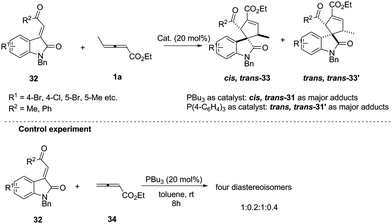 |
| | Scheme 10 Phosphine-catalysed highly diastereoselective (3+2) annulations of isatin derived electron-deficient alkenes. | |
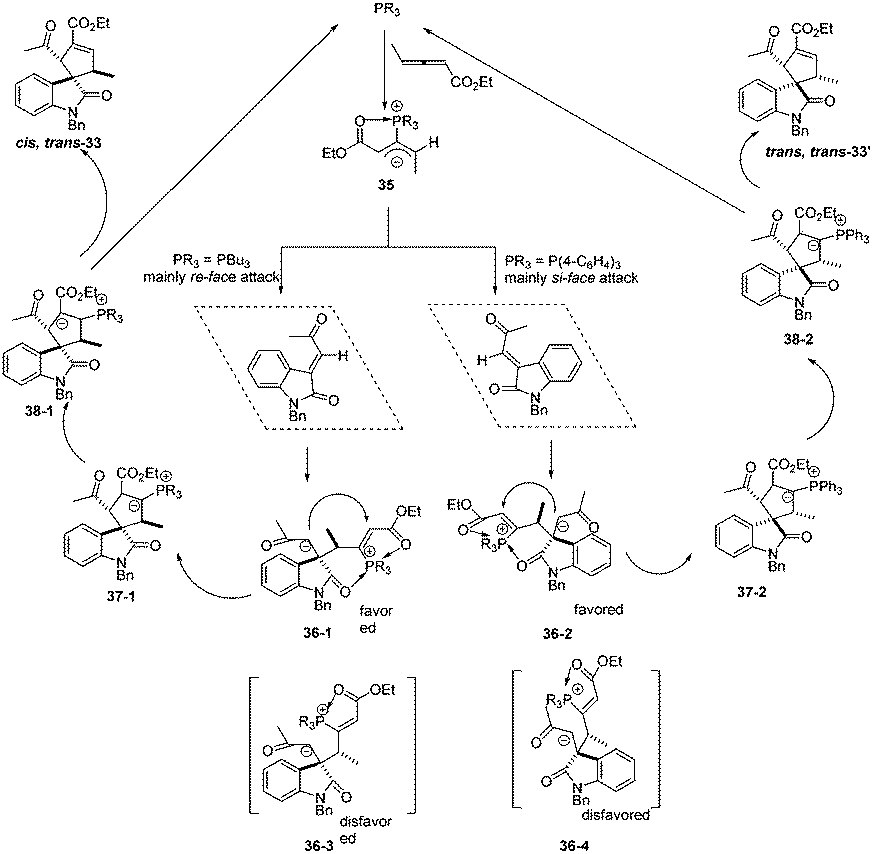
The authors proposed the mechanism to explain how the nucleophilic phosphine catalyst could control reaction diastereoselectivity. As shown in Scheme 11, the nucleophilic phosphine catalyst reacted with ethyl 2,3-pentadienoate 1a to yield the zwitterionic intermediate 35. Subsequently, the intermediate 36-1 or 36-2 was generated through the re-face or si-face's Michael addition. The authors thought that the intermediates 36-1 and 36-2 might be more favored than the intermediates 36-3 and 36-4 due to the internal coordination of oxygen with phosphorus atom, resulting in the methyl group and the carbonyl group on the isatin ring always occupying the trans-position in the final product. Due to the steric reasons, the intermediate 35 was more favored to attack the activated olefin via the re-face if the catalyst was PBu3, while attacking the activated olefin via the si-face if P(4-FC6H4)3 was used as the catalyst. Intermediates 36-1 and 36-2 underwent the ring-closing reaction to produce intermediates 37-1 and 37-2, respectively. The following annulation reaction and proton-transfer process led to the formation of cis, trans-33 and trans, trans-33′ with the regeneration of the catalyst.
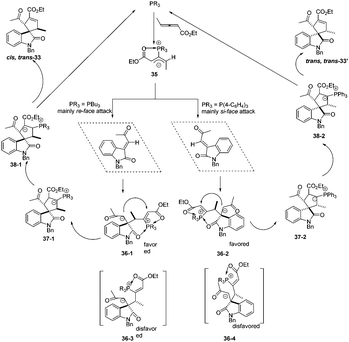 |
| | Scheme 11 The plausible mechanism for phosphine catalysed (3 +2) annulations. | |
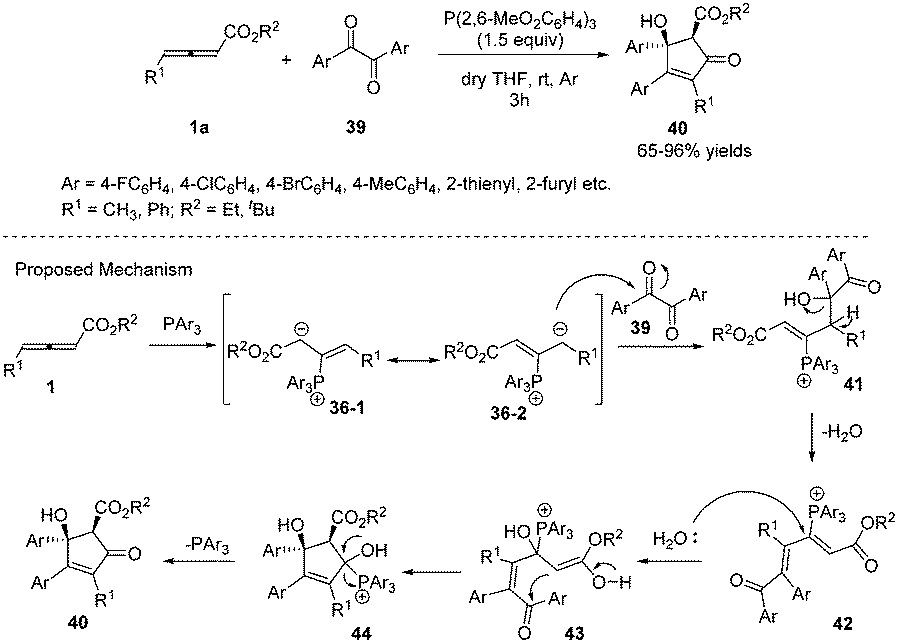
In 2013, Nair and co-workers reported an efficient phosphine-mediated diastereoselective (3+2) annulation for the synthesis of substituted cyclopentenones from γ-alkyl allenoates and diaryl 1,2-diones. The reaction appeared to be working well with respect to different acyclic 1,2-diones and allenoates. The corresponding all-substituted cyclopentenones 40 can be obtained in high yield with high regio- and diastereoselectivity. The authors proposed the mechanism; the first step was the nucleophilic addition of triarylphosphine to allenoates, giving the 1,3-dipolar zwitterions 36-1 and 36-2. The intermediate 36-2 then attacked a carbonyl group of the dione 39 forming 41. This species lost a molecule of water to afford the cationic intermediate 42. Addition of water to the latter followed by cyclization and elimination of phosphine delivered the desired compounds 40 (Scheme 12).16
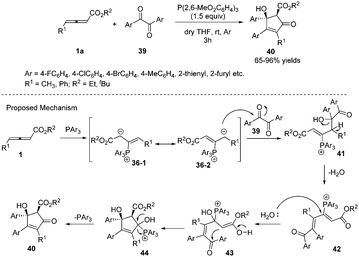 |
| | Scheme 12 Phosphine-mediated reaction of γ-alkyl allenoates and diaryl 1,2-diones. | |
In 2017, Zhong and co-workers developed a phosphine-catalysed (3+2) annulation of γ-substituted allenoates with succinimides by γ-addition/annulation, giving an effective method to obtain functional azaspirane derivatives 46 with good diastereoselectivity and regioselectivity in moderate to good yields. The preliminary asymmetric (3+2) cycloaddition reactions were also developed, affording a moderate yield (40%) and ee value (77% ee) (Scheme 13).17
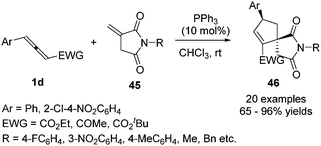 |
| | Scheme 13 Phosphine-catalysed (3+2) annulation reaction of N-methylidenesuccinimides. | |
3. Phosphine-catalysed (m+n) annulations with electrophiles
3.1 Phosphine-catalysed (2+4) annulations with electrophiles
In early research, γ-substituted allenoates often resulted in low yields and selectivity. Thus chemists thought that γ-substituted allenoates weren’t very reactive substrates. In 2009, Huang and co-workers developed novel phosphine-catalysed (2+4) annulations of γ-methyl allenoate 1a with N-thiophosphinylimines 47.18 More importantly, the authors discovered the first example that the γ-CH3 of the allenoates underwent annulation to form the chroman derivatives. Thus the γ-substituted allenoates attracted the interest of chemists, who continued with the studies of γ-substituted allenoates as synthons in the annulation reaction (Scheme 14).
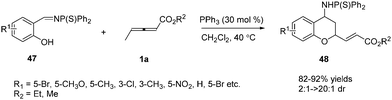 |
| | Scheme 14 Phosphine-catalysed abnormal (2+4) annulations of γ-methyl allenoate with N-thiophosphinylimines. | |
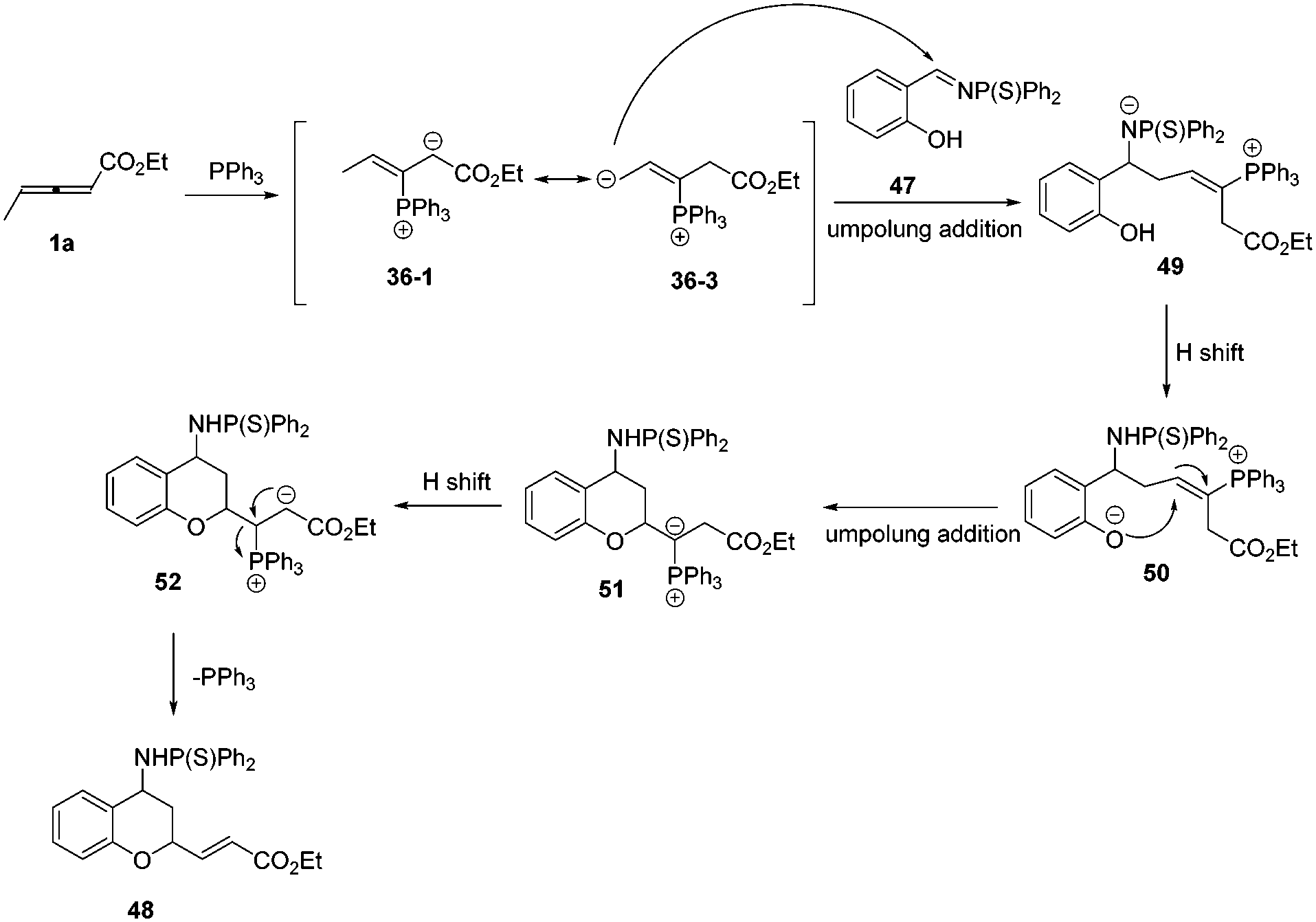
According to these experimental results, the authors proposed a plausible mechanism. First, the phosphine attacked the β carbon of the allenoate to produce the intermediate 36-1, which would transform to intermediate 36-3via 1,4-proton shift. Subsequently, the intermediate 36-3 underwent an umpolung addition with 1a to give the intermediate 49, which formed the intermediate 50via proton transfer from OH to N anion. Finally, the intermediate 51 underwent another umpolung addition of the oxygen anion to the β carbon of 50 to furnish the product and regenerated PPh3 (Scheme 15).
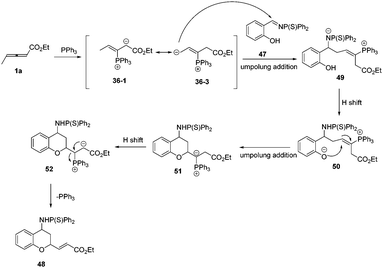 |
| | Scheme 15 The possible mechanism for phosphine catalysed (2+4) annulations. | |
In 2011, He and co-workers reported a phosphine-catalysed (2+4) annulation of γ-methyl allenoate 1a with salicylaldehydes 53, providing highly functionalized chromans 54 in 47–97% yields by applying tris(p-chlorophenyl)phosphine (20 mol%) as a catalyst. The authors thought that the transformation represented a novel reactivity pattern of electron-deficient allenes with aldehydes (Scheme 16).19
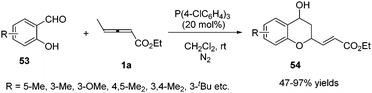 |
| | Scheme 16 Phosphine-mediated diverse reactivity of aldehydes. | |
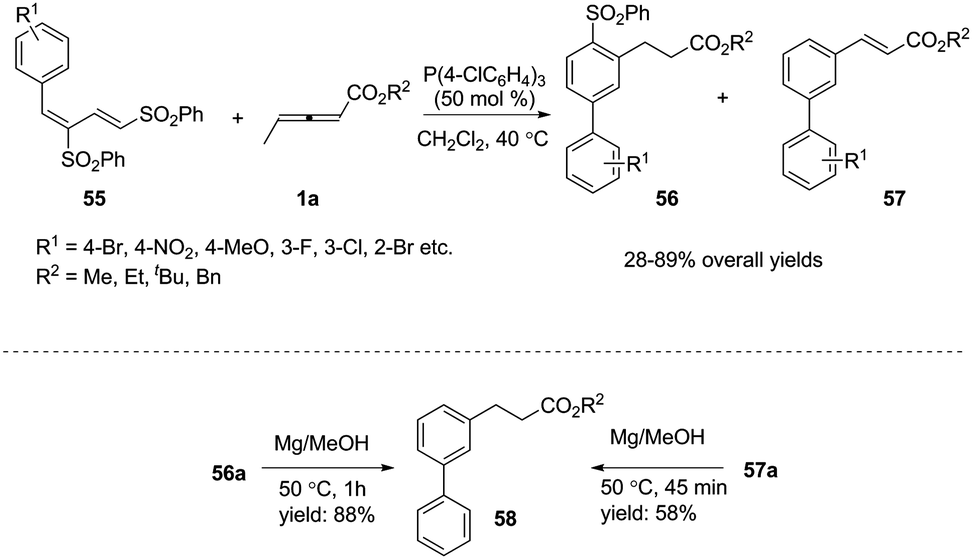
Inspired by this result, Huang and co-workers developed a novel phosphine-catalysed (2+4) benzannulation reaction by applying 1,3-bis(sulfonyl)butadiene as the substrate.20 The reaction provided access to biaryls under mild reaction conditions. Although giving low selectivity in the reactions, biaryls 56 and 57 could be transformed to the same compound 58 under the same reduction conditions. To illustrate the synthetic utility of this method, the corresponding Suzuki, Heck, and Sonogashira products could be obtained in moderate yields through traditional cross-coupling reactions. It should be noted that the authors used γ-methyl allenoates in phosphine-catalysed benzannulation for the first time (Scheme 17).
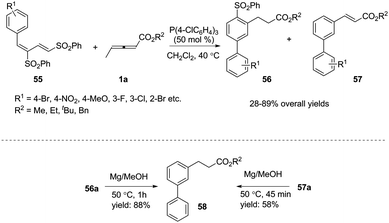 |
| | Scheme 17 Phosphine-catalysed (2+4) benzannulation reaction. | |
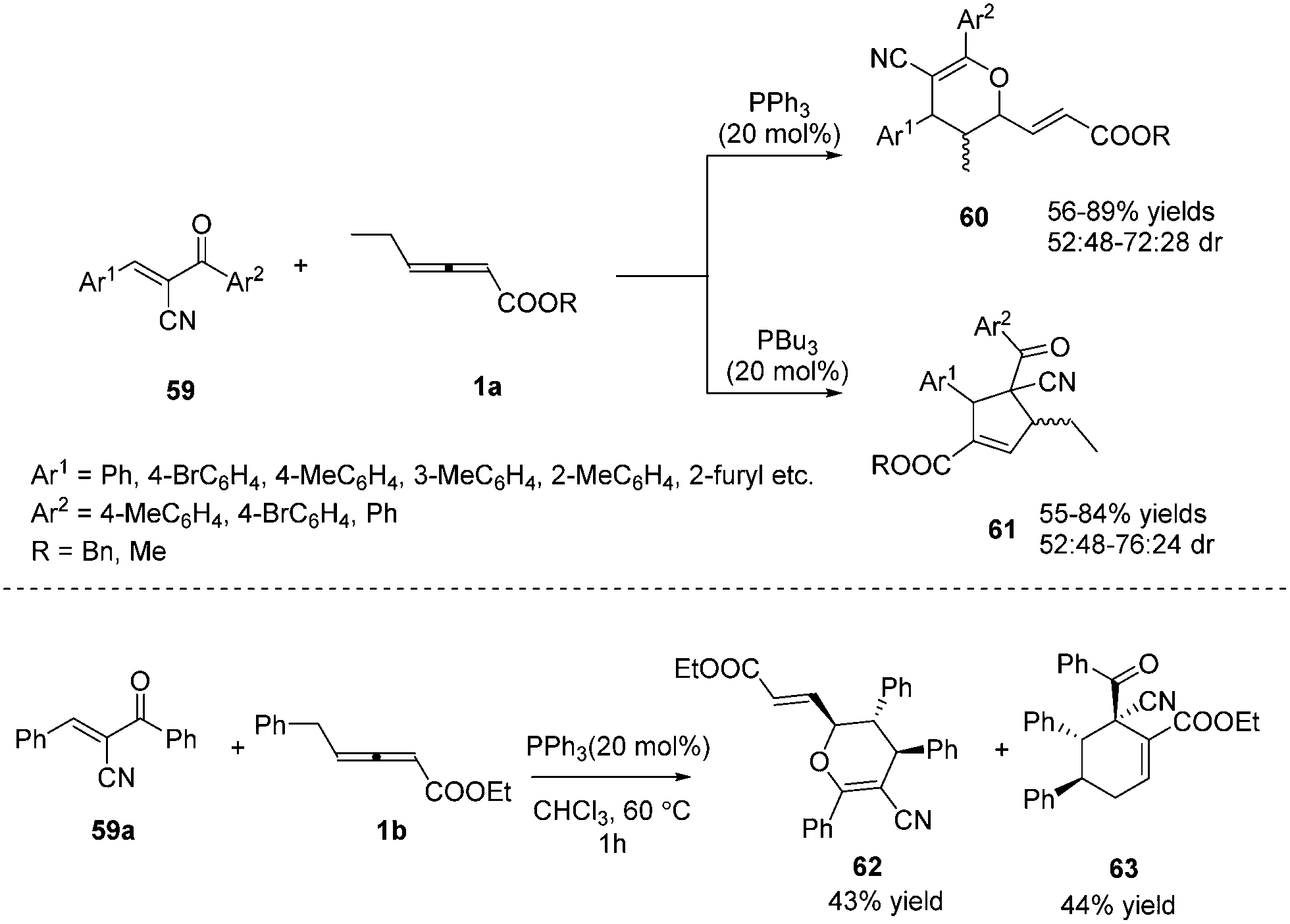
In 2015, Huang and co-workers disclosed a divergent phosphine-catalysed (2+4) or (3+2) cycloaddition reaction of oxadienes. By exploiting the different nucleophilicities of the phosphine catalysts, the γ-substituted allenoates selectively acted as 1,3- or 1,2-dipolar synthons, providing dihydropyran 60 and multifunctional cyclopentene derivatives 61 in moderate to good yields. However, the reaction afforded low diastereoselectivity, and the authors obtained a pair of inseparable diastereoisomers. It should be noted that the γ-ethyl of allenoates played a key role in the reactions, and unidentified side products were obtained if the γ-ethyl was replaced with a methyl, isopropyl, or n-butyl group. When γ-benzyl allenoate was applied as the substrate, the (2+4) annulation product 62 and the (4+2) annulation product 63 were obtained in yields of 43% and 44%, respectively (Scheme 18).21
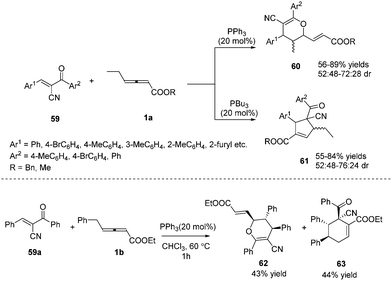 |
| | Scheme 18 Divergent phosphine-catalysed (2+4) or (3+2) cycloaddition reactions of oxadienes. | |
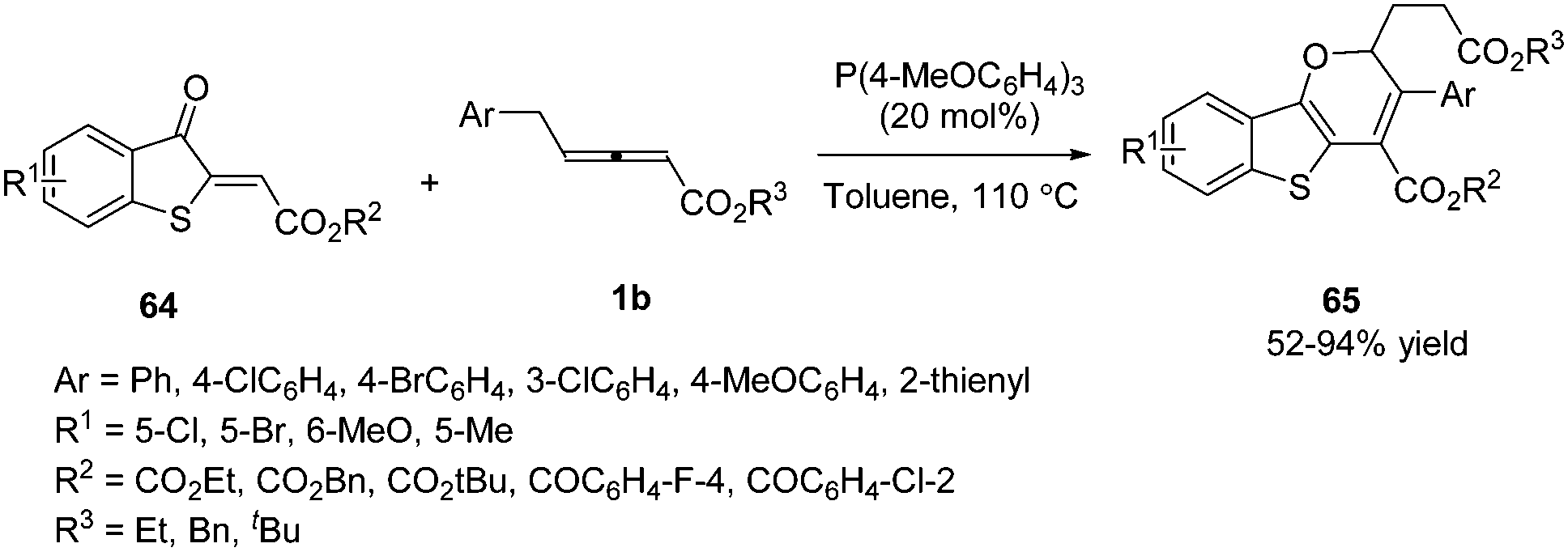
In 2018, Meng and co-workers disclosed a tris(4-methoxyphenyl)phosphine-catalysed (2+4) annulation of γ-benzyl allenoates with benzothiophene, giving a series of 2H-benzo[4,5]thieno[3,2-b]pyran derivatives 65 in high yields. The authors found that the benzyl of allenoates was critical to the reaction. When the γ-methyl substituted allenoate was used in this domino reaction, no desired product was obtained. To test the practicality of this reaction, the authors carried out a gram-scale experiment, giving the corresponding product in 77% yield (Scheme 19).22
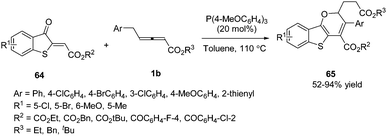 |
| | Scheme 19 Phosphine-catalysed (2+4) annulation of benzothiophen-3(2H)-one. | |
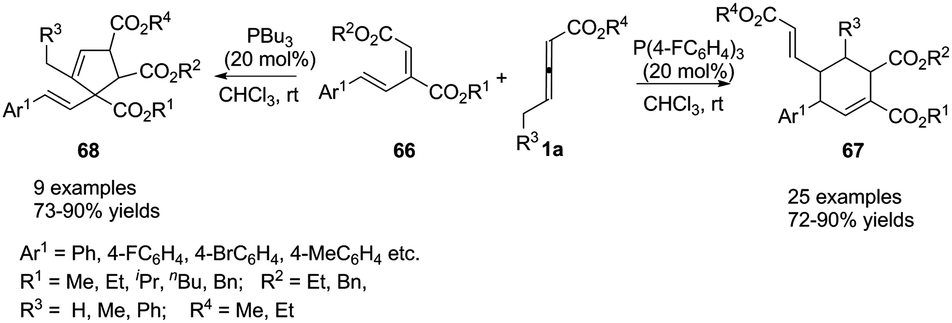
More recently, Duan and Li utilized a molecular engineering approach for a catalytic γ-umpolung addition/annulation, developing a phosphine-catalysed regiodivergent annulation of γ-substituted allenoates with a new diene moiety. The final products suggested that the activity and selectivity could be tuned through the Lewis basicity of the phosphine catalysts. In the presence of (4-FC6H4)3P, the corresponding (2+4) products 67 were isolated in good to excellent yields with high regioselectivity, but the (3+2) products 68 were isolated when PBu3 was used as a catalyst (Scheme 20).23
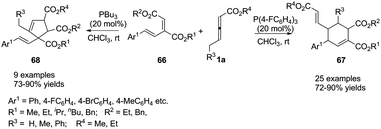 |
| | Scheme 20 Phosphine-catalysed regiodivergent annulations of γ-substituted allenoates with conjugated dienes. | |
3.2 Phosphine-catalyzed non-classical (3+2) annulations with electrophiles
In the aforementioned reaction, the diversity in the reactivity patterns of allenoates with activated alkenes/imines has been well rationalized. Besides, aldehydes could also participate in phosphine-catalysed annulations of nonsubstituted allenoates, which undergo exclusive γ-addition to the allenoate, resulting in the formation of a cyclic adduct.24 Intrigued by this elegant study, He and co-workers reported a phosphine-catalysed (3+2) annulation of γ-substituted allenoates with aldehydes, providing a series of tetrahydrofuran derivatives 70 in moderate to excellent yields and low diastereoselectivity. The γ-substituted allenoates served as a non-classical 1,3-dipolar synthon (δC and βC) in the reaction, which represented an unprecedented reactivity pattern of allenoates with aldehydes under nucleophilic phosphine catalysis. The authors found that the γ-methyl of allenoates plays a key role in the non-classical (3+2) annulations. Compared with γ-methyl allenoate, γ-ethyl allenoate had a significantly decreased reactivity (19% yield). And no desired adducts were obtained when γ-phenyl or γ-tert-butyl allenoates were used (Scheme 21).25
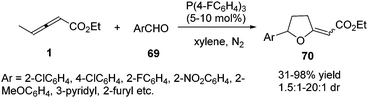 |
| | Scheme 21 Phosphane-catalysed (3+2) annulation of allenoates with aldehydes. | |
In 2013, Zhao and co-workers reported a chemoselective phosphine-catalysed cycloaddition or dienylation reaction between trifluoromethylsubstituted ketones and bis-substituted allenoates. The authors found that the reaction produced a range of trifluoromethylated tetrahydrofurans 72 with broad substrate tolerance and good to excellent stereoselectivity in the presence of triarylphosphine, while CF3-substituted dienyl tertiary alcohols 74 were chemoselectively obtained by the use of trialkylphosphine. And a preliminary study on the asymmetric version was also performed, providing a moderate ee value (up to 52%) (Scheme 22).26
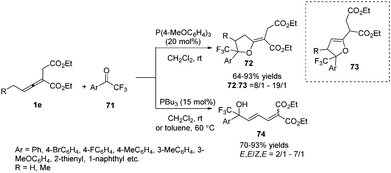 |
| | Scheme 22 Phosphine catalyst-controlled cycloaddition or dienylation reactions of trifluoromethyl aryl ketones. | |
In 2015, Nair and co-workers reported a phosphine mediated (3+2) annulation reaction of γ-methyl allenoates with isatins in the presence of 1.5 equiv. of PPh3, providing a series of spirofuran oxindoles 76 in 32–88% yields under mild conditions (Scheme 23).27
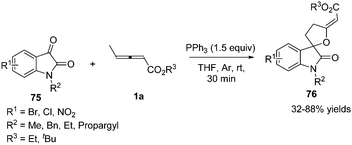 |
| | Scheme 23 Phosphine-mediated reaction of γ-methyl allenoates and isatins. | |
In 2018, Xiao and co-workers reported a phosphine-mediated (3+2) cycloaddition of bis-substituted allenoates with aldimines. The authors found the reaction was applicable to various aryl-substituted N-tosylaldimines, regardless of the substitution patterns and electronic properties of the aromatic substitutes, and a range of functionalized 2-pyrrolines 77 were attainable in moderate yields albeit with random diastereoselectivity in the presence of PPh3 (60 mol%) (Scheme 24).28
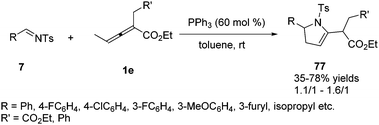 |
| | Scheme 24 PPh3-Mediated (3+2) cycloaddition reaction between bis-substituted allenoate and N-tosylaldimines. | |
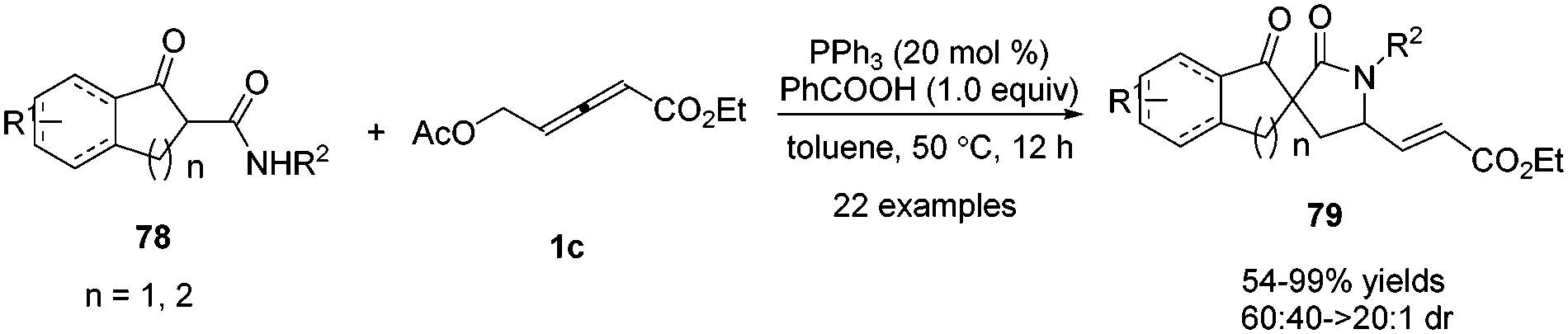
In 2017, Shi and co-workers disclosed a phosphine catalysed (3+2) annulation by applying α-substituted secondary β-ketoamides 78 and δ-acetoxy allenoates 1c as substrates. The authors found that tertiary nitrogen (NR3) also promoted the reaction besides tertiary phosphine (PR3). In addition, the addition of PhCOOH may be beneficial to the H-transfer processes. And when β-ketoamides were applied to the bis-nucleophilic partner, the δ,γ-carbon of δ-acetoxy allenoates participated as a 1,2-dipole synthon, affording the desired functionalized (3+2) adducts 79 in moderate to excellent yields and diastereoselectivity in this spiroannulation reaction (Scheme 25).29
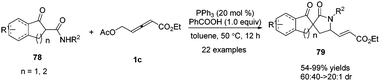 |
| | Scheme 25 PPh3-Catalysed (3+2) spiroannulation of α-substituted secondary β-ketoamides with δ-acetoxy allenoates. | |
3.3 Phosphine-catalysed (4+2) annulations with electrophiles
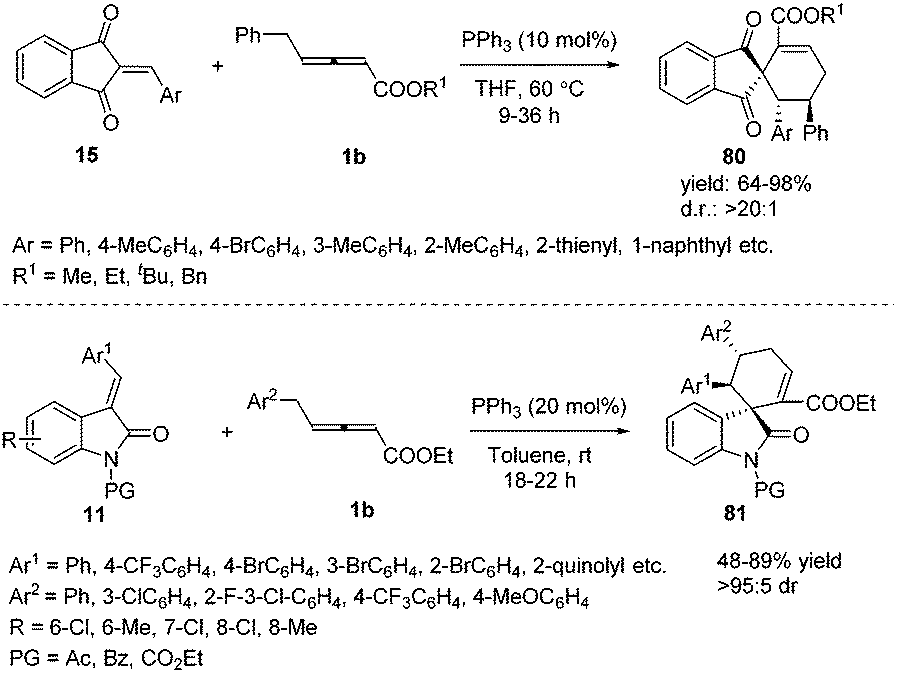
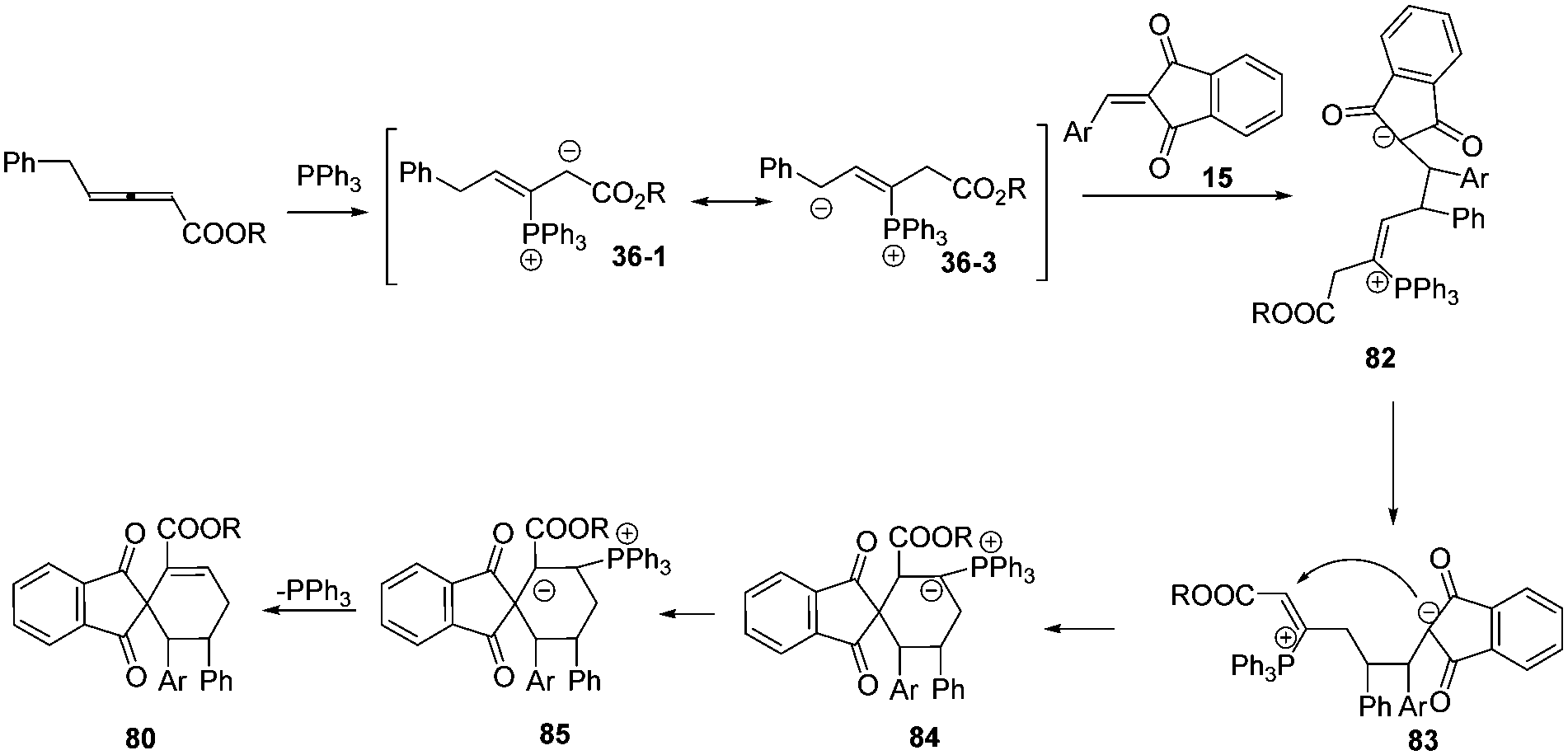
Prior to these studies, only α-substituted allenoates were used as a 1,4-dipolar synthon in phosphine promoted annulation reactions,30 and these reactions involved the β′-carbon atom of the allenoates. On the other hand, γ-substituted allenoates were usually applied as 1,2- or 1,3-dipolar synthons. In 2013, Huang and co-workers reported a novel phosphine-catalysed (4+2) annulation, in which γ-substituted allenoates for the first time served as a new type of 1,4-dipolar synthon. This reaction offered a powerful approach to the construction of highly substituted spiro[4.5]dec-6-ene skeletons 80 in excellent yields and with complete regioselectivity and high diastereoselectivity. In addition, the experiment demonstrated the practicality of the strategy.31 During the same time period, Marinetti and Voituriez also found the new (4+2) annulation process by applying 3-arylideneoxindoles as electron-deficient olefins (Scheme 26).32
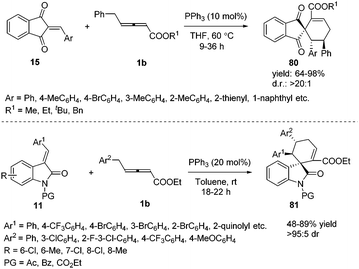 |
| | Scheme 26 Phosphine-catalysed (4+2) annulation of 2-arylidene-1H-indene-1,3(2H)-diones or 3-arylideneoxindoles. | |
Based on the experimental results, the authors proposed a possible mechanism involving addition of the phosphine to the β-position of the allenoates to form 1,3-dipolar zwitterion 36-1. The latter underwent a reversible equilibrium overall proton shift, giving the intermediate 36-3. The authors thought that the key role of the phenyl group here was to favor this rearrangement by stabilizing the intermediate 36-3. Next the latter underwent δ-addition to the olefinic substrate, resulting in the formation of intermediate 82. Then, an isomerization to give intermediate 83, an umpolung addition to give 84, an H-shift, and release of the phosphorus catalyst constituted the reaction pathway (Scheme 27).
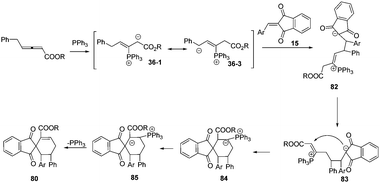 |
| | Scheme 27 A possible mechanism. | |
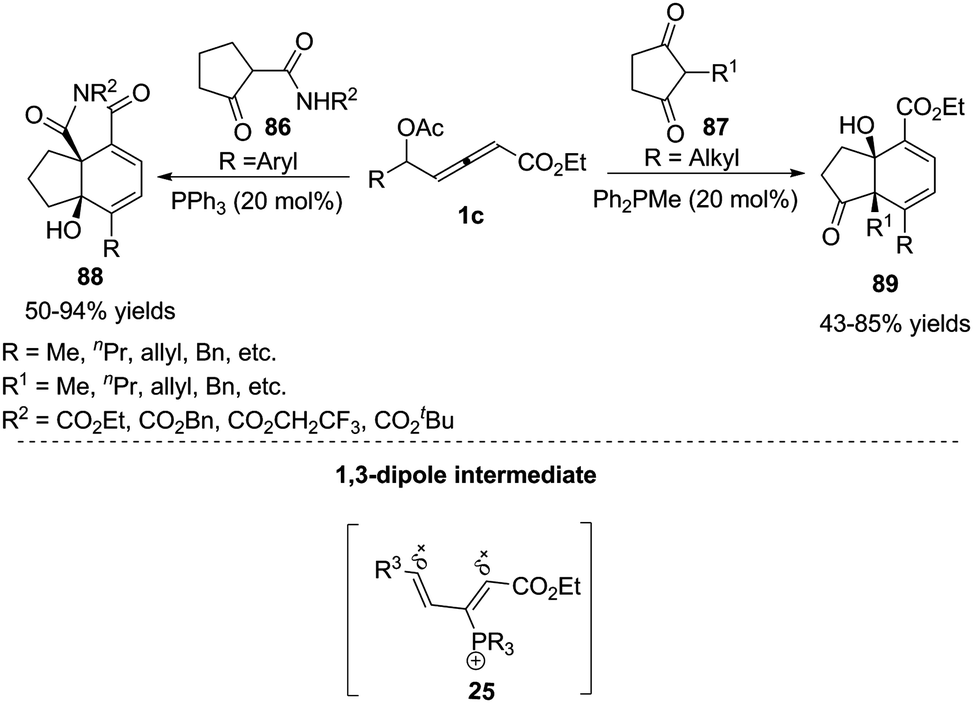
Inspired by the result of the phosphine-catalysed annulations of β′-acetoxy allenoates,33 Tong and co-workers designed the novel δ-acetoxy allenoates, which were successfully applied to phosphine-catalysed substrate-dependent (4+2) annulations of ketones, giving the corresponding products 88 or 89 in good yields.34 Mechanistically, the authors thought that allenoates with an alkyl group at δC exhibited δC electrophilicity and αC nucleophilicity, while opposite reactivity was observed for the ones bearing an aryl group at δC. In a preliminary attempt, the asymmetric (4+2) annulation was examined with several chiral phosphine catalysts, affording 88a in 78% yield and 45% ee (Scheme 28).
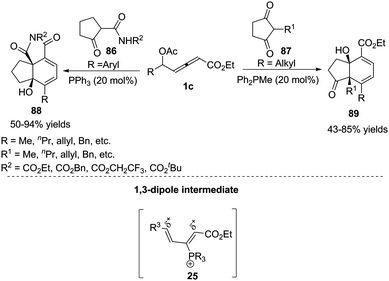 |
| | Scheme 28 Phosphine-catalysed (4+2) annulations of δ-acetoxy allenoates and ketones. | |
4. Phosphine-catalysed sequential annulation reaction of γ-substituted allenoates
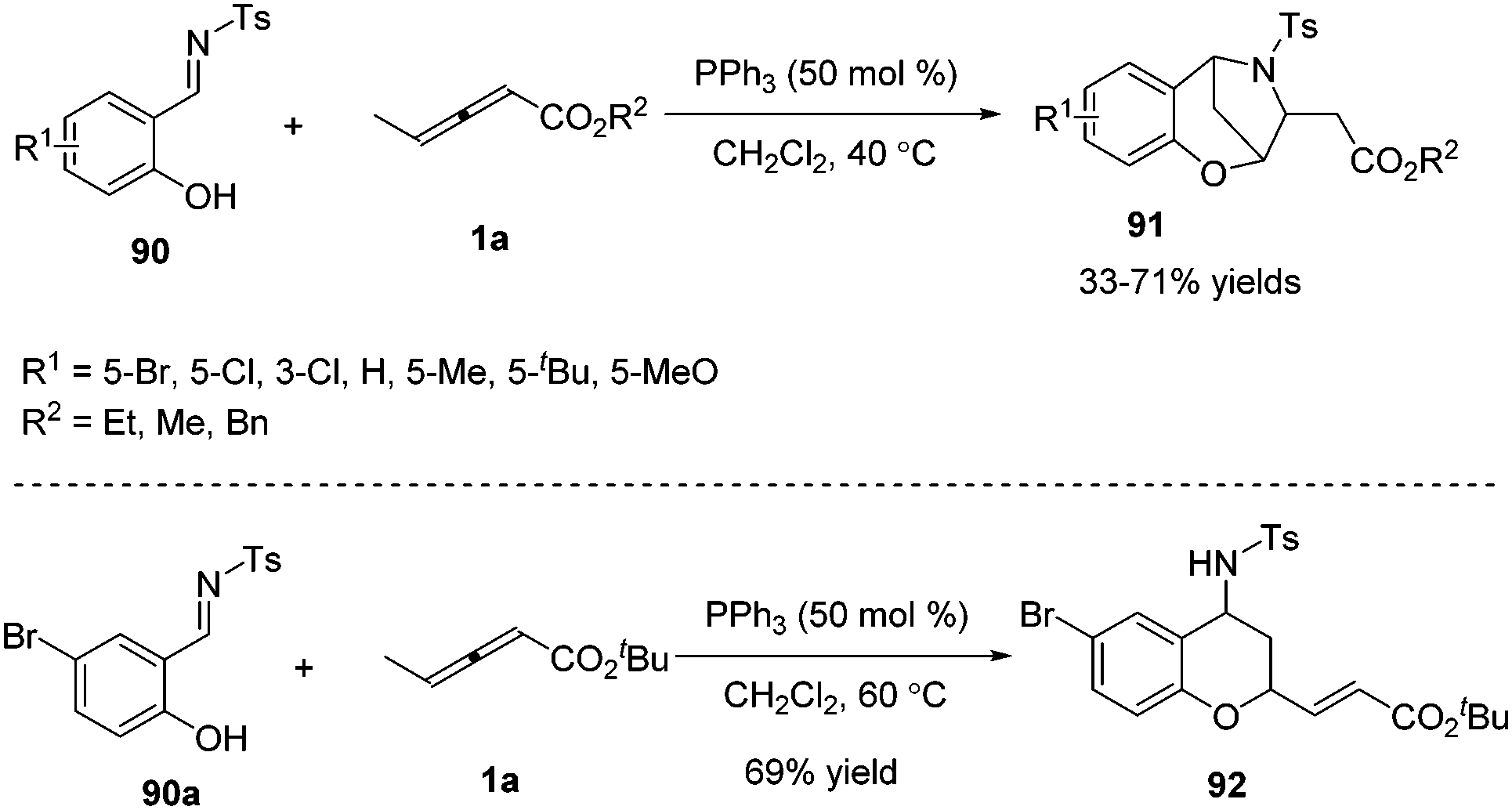
γ-Substituted allenoates have proven to function as 1,2-, 1,3-, or 1,4-dipole synthons when reacting with a variety of electrophilic coupling partners (including imines, aldehydes, or alkenes). While only two chemical bonds are generated in usual reactions, owing to the presence of multiple reactive sites in γ-substituted allenoates, it was possible that more sites took part in the formation of chemical bonds. In this context, Huang and co-workers first developed a phosphine-catalysed sequential (2+4) and (2+3) annulation reaction of γ-methyl allenoates 1a and salicyl N-tosylimines 90, forming the benzoxazepine derivatives 91 in moderate to good yields. It should be noted that three sites (β, γ, δ sites) participated in the formation of chemical bonds, forming C–C, C–O and C–N bonds in one process. The authors found that the steric effect of substituted allenoates had a significant impact on the reaction; when R2 was changed to a tBu group, no benzoxazepine derivative 91 was produced, and instead chroman derivative 92 was obtained in 69% yield (Scheme 29).35
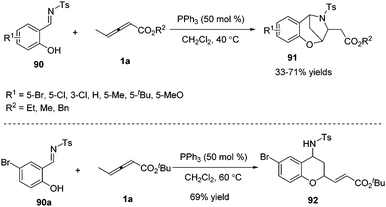 |
| | Scheme 29 PPh3-Catalysed sequential annulation reactions between salicyl N-tosylimines and allenoates. | |
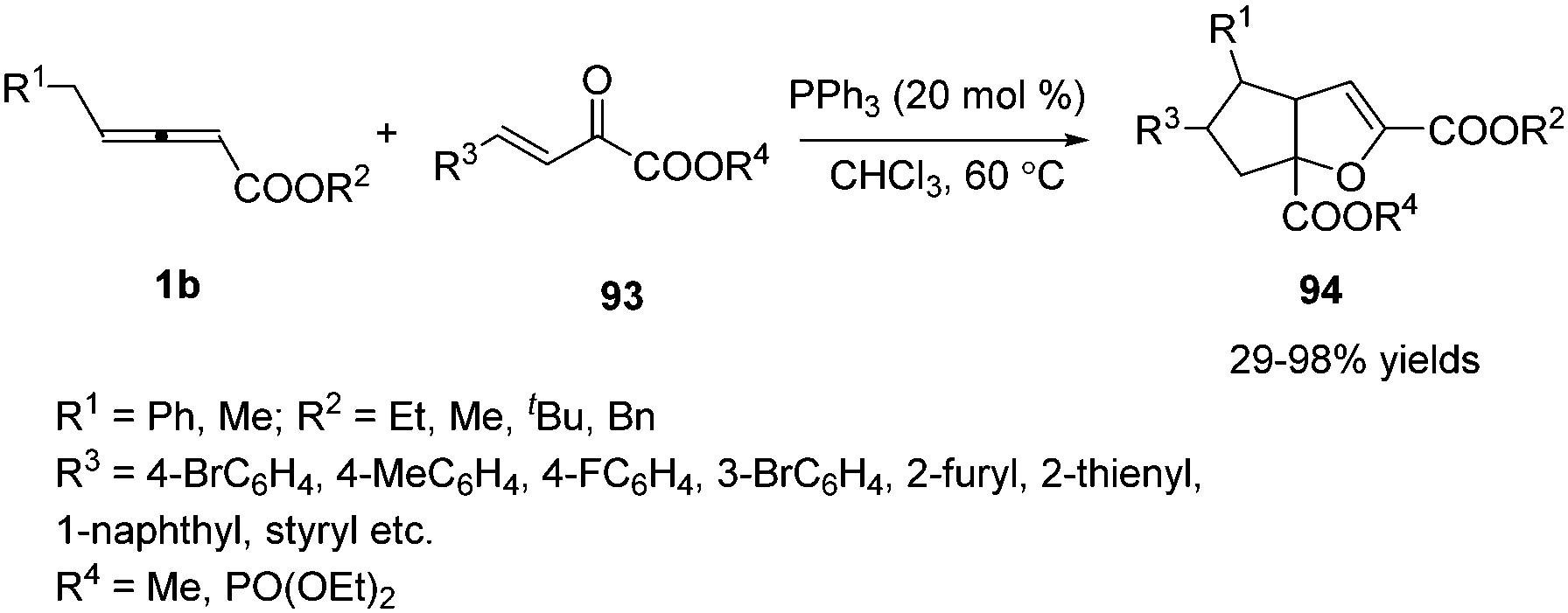
Encouraged by this result, Huang and co-workers discovered a phosphine-catalysed sequential (2+3) and (3+2) annulation reaction for the construction of cyclopenta[b]dihydrofuran derivatives. The α, γ, δ sites of allenoates participated in the formation of chemical bonds, forming two C–C bonds and one C–O bond in one process. The authors found that the reaction showed good tolerance towards the electronic properties of the aromatic substitutes, and high to excellent yields were achieved in the presence of PPh3 (20 mol%). In addition, chiral phosphine catalysis was also employed in this reaction and moderate enantioselectivity (40% ee) was obtained (Scheme 30).36
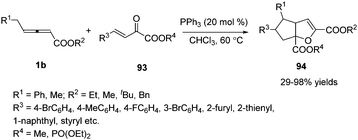 |
| | Scheme 30 Phosphine-catalysed sequential (2+3) and (3+2) annulation reaction of a β, γ-unsaturated α-ketoester. | |
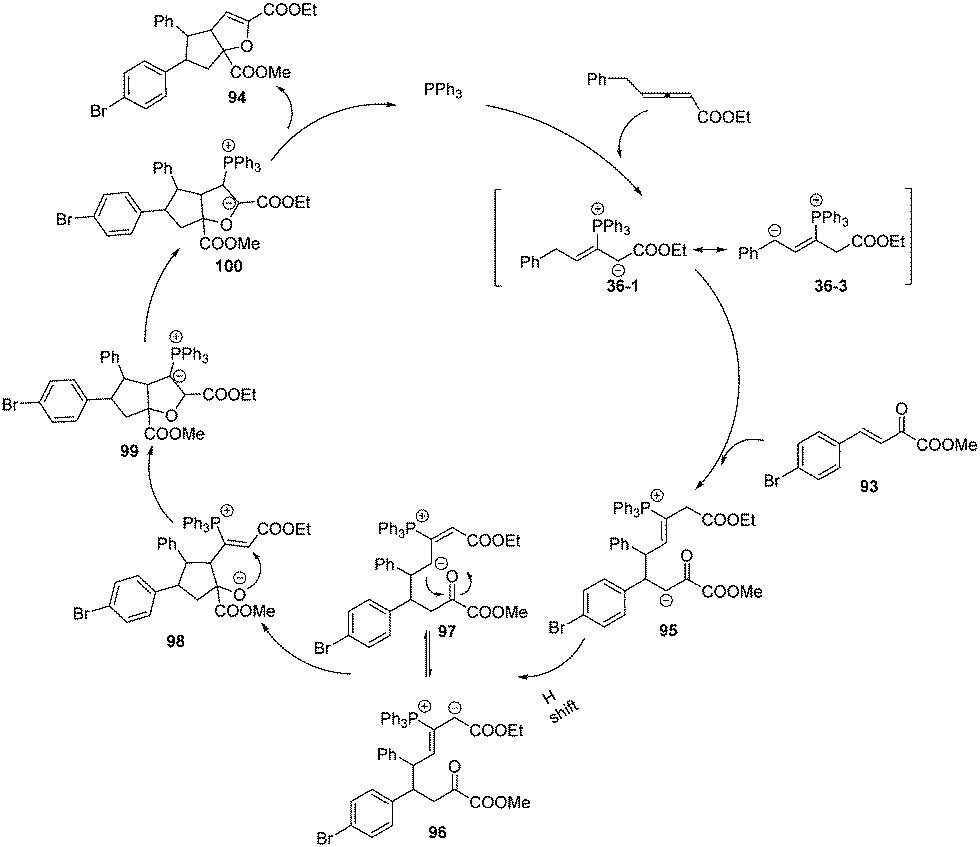
The authors proposed a possible mechanism for the sequential annulation reaction. As outlined in Scheme 31, nucleophilic addition of phosphine to the γ-benzyl allenoates 1b gave the intermediate 36-1, which could isomerize to the crucial zwitterionic intermediate 36-3. A nucleophilic addition reaction yielded the intermediate 95. Next a H-shift occurred to give intermediates 96 and 97 by a reverse equilibrium, which underwent a nucleophilic addition to give the intermediate 98. Again nucleophilic addition reaction yielded the intermediate 99. Proton transfer and subsequent β-elimination of the catalyst phosphine led to the formation of the corresponding adducts 94.
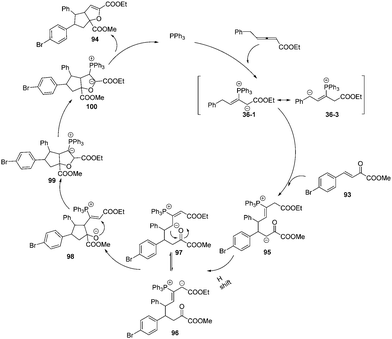 |
| | Scheme 31 The reaction mechanism for phosphine-catalysed sequential annulations. | |
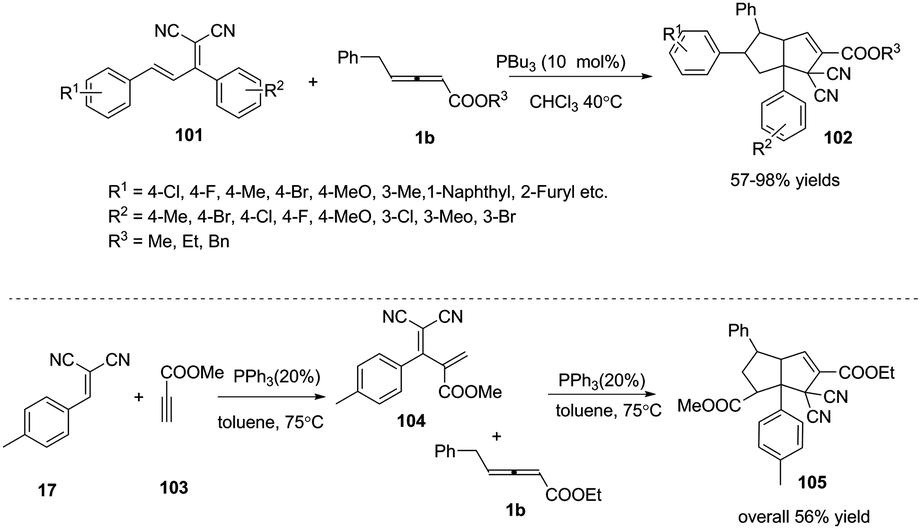
Next, Huang and co-workers applied (E)-2-(1,3-diphenylallyli-dene)malononitrile 101 as an electron-deficient olefin, in the presence of PBu3 (10 mol%), developing a phosphine catalysed sequential (2+3) and (3+2) annulation reaction under mild conditions. These reactions provided efficient syntheses of highly functionalized bicyclic[3,3,0]octene derivatives 102 in good to excellent yields and complete diastereoselectivity. The authors envisioned that the domino reaction could proceed in one pot. When 2-(4-methylbenzylidene)-malononitrile 17, methylpropiolate 103 and γ-substituted allenoates 1b were applied as substrates, the corresponding adduct 105 was obtained in one pot in 56% yield (Scheme 32).37
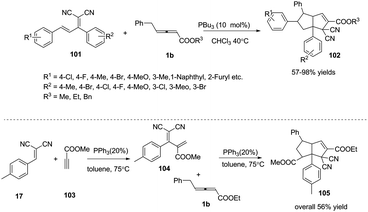 |
| | Scheme 32 Phosphine-catalysed sequential (2+3) and (3+2) annulation reactions of γ-substituted allenoates with conjugate dienes. | |
Aza-bicyclo[3,3,0]octane derivatives are important structural motifs present in numerous biologically active natural products.38 Thus, the development of efficient synthetic methods for accessing these useful aza-bicyclo compounds has been an attractive field for organic chemists.
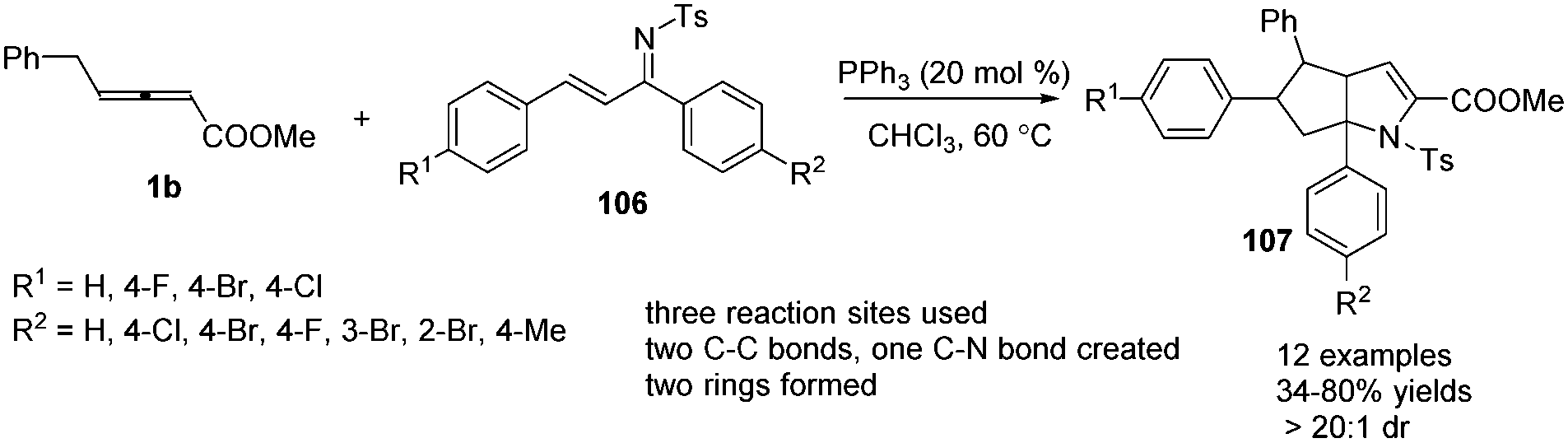
In 2014, Huang and co-workers disclosed a phosphine-catalysed sequential (2+3) and (3+2) annulation reaction of γ-benzyl allenoates with α,β-unsaturated ketimines. By applying PPh3 (20 mol%) as a catalyst in CHCl3 at 60 °C, the reaction could produce a series of the corresponding aza-bicyclo-[3,3,0]octane derivatives 107 in moderate to good yields and excellent diastereoselectivity (Scheme 33).39
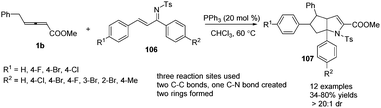 |
| | Scheme 33 Phosphine-catalysed sequential annulation reactions of α,β-unsaturated ketimines. | |
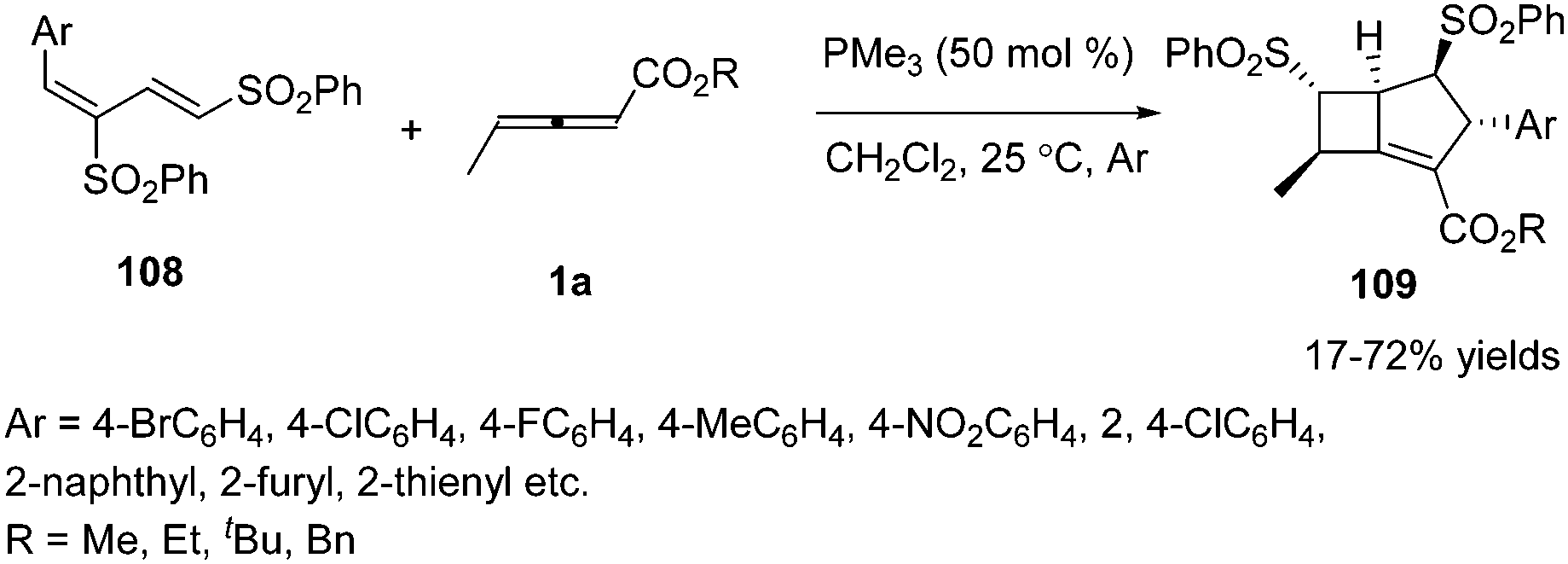
Owing to the multiple reactive sites of γ-substituted allenoates, chemists wished that more reactive sites could participate in the formation of chemical bonds in one step. In 2013, Huang and co-workers disclosed a phosphine catalysed sequential annulation of γ-methyl allenoates 1a with the dienic sulfones 108, in which three (α, β, γ) sites of γ-methyl allenoates simultaneously participated in the formation of chemical bonds. By optimizing the reaction condition, the desired bicyclo-[3.2.0]heptene derivatives 109 could be obtained in moderate to good yields. And the authors found that the esters of allenoates had a significant influence on the yield when changing the substituent on the dienic sulfone from 2-naphthyl to 4-bromophenyl (Scheme 34).40
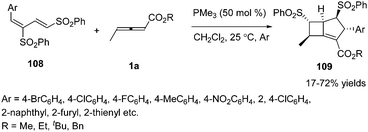 |
| | Scheme 34 Phosphine-catalysed sequential annulation reactions of dienic sulfones. | |
A plausible mechanism is presented in Scheme 35. The reaction was first triggered by nucleophilic addition of phosphine (PMe3) to allenoates 1a. The α-regioselective addition of the zwitterionic intermediate 36 to the dienic sulfone 108 gave the intermediate 110, which underwent an intramolecular nucleophilic addition to give another zwitterionic intermediate 111. The authors thought that the stable configuration resulted in high diastereoselectivity. Next, another intramolecular addition followed by proton transfer and elimination of the catalyst afforded the corresponding adduct 109.
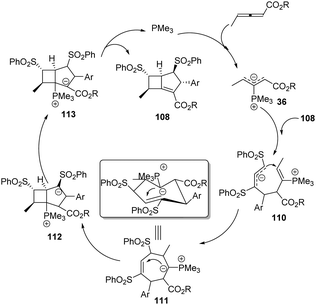 |
| | Scheme 35 The proposed mechanism for the synthesis of bicyclo-[3.2.0]heptene derivatives. | |
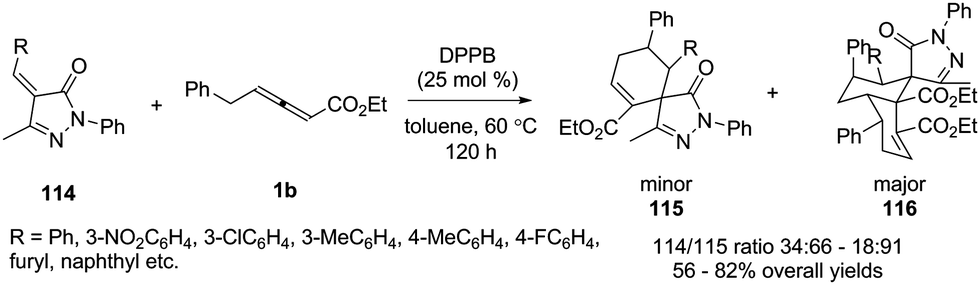
In 2015, Miao and co-workers reported an efficient bisphosphine-catalysed sequential annulation reaction of γ-benzyl allenoates 1b with benzylidenepyrazolones 114. The authors found that the catalysts played a key role in the product. By using bisphosphine (DPPB) as a catalyst, the sequential (4+2)/(4+2) annulation adducts 116 were obtained as major products in moderate to good yields and chemoselectivity (Scheme 36).41
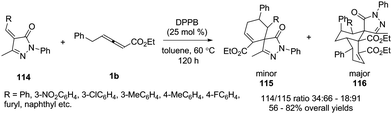 |
| | Scheme 36 Bisphosphine-catalysed sequential annulation of an allenoate with benzylidenepyrazolones. | |
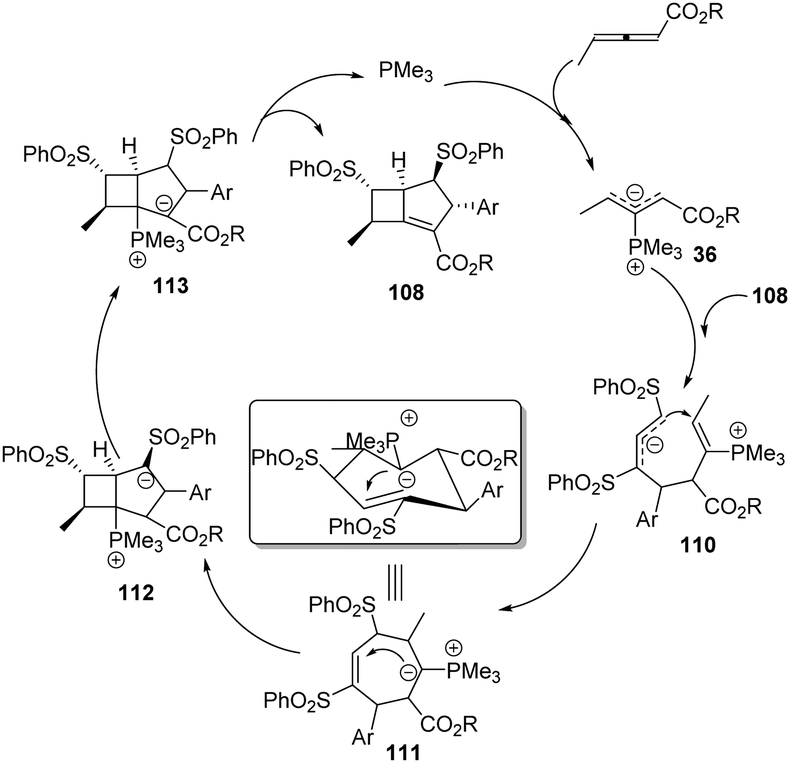
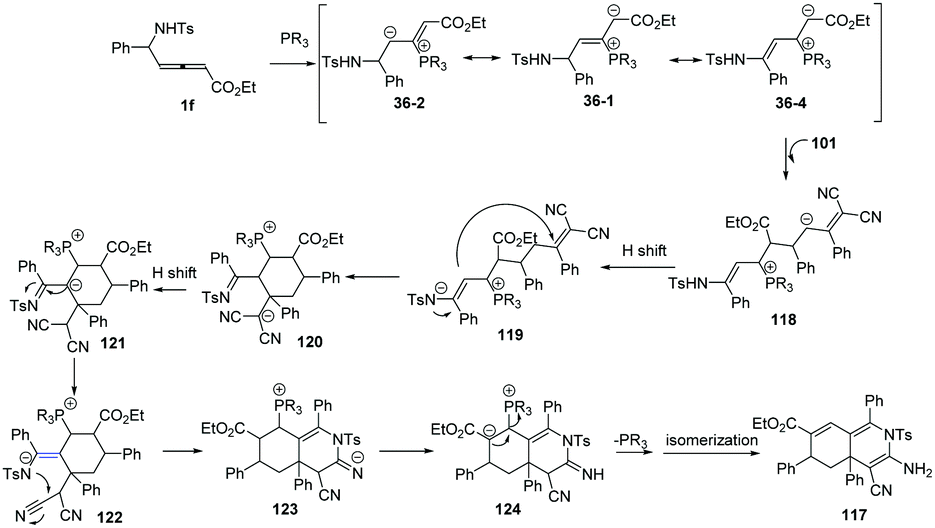
More recently, Huang and co-workers disclosed a phosphine-catalysed sequential (3+3)/(3+3) annulation of δ-sulfonamido-allenoates, providing a facile access to highly functionalized hydroisoquinoline derivatives 117 in 49–97% yields and moderate diastereoselectivity. Three novel sites (α, γ, ε) of allenoates simultaneously participated in the formation of chemical bonds in one process (Scheme 37).42
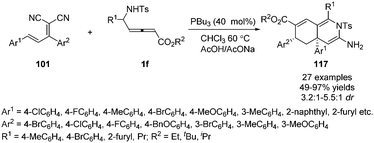 |
| | Scheme 37 Phosphine-catalysed sequential (3+3)/(3+3) annulations of δ-sulfonamido-allenoates. | |
The authors proposed the mechanism as outlined in Scheme 38. β-Addition of phosphine to δ-sulfonamido-allenoates 1 generated resonance zwitterion 36-1, 36-2 and 36-4. Addition of 36-4 to 101 produced 118. Next, the [3+3] annulation intermediate 120 was obtained by H-shift and subsequent Michael addition driven by the enamine anion from 119. The authors thought that another H-shift and imine–enamine tautomerism of 120 made it possible the subsequent aza-intramolecular nucleophilic addition to the cyano group of 122, which completed the second (3+3) annulation process. H-Shift of 123 gave rise to the intermediate 124. The elimination of phosphine and the following isomerization led to the final product 117.
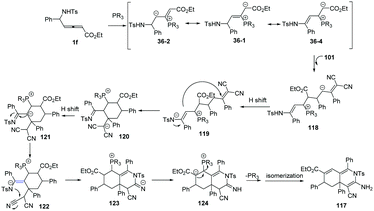 |
| | Scheme 38 Proposed mechanism. | |
5. Phosphine-catalysed other reactions of γ-substituted allenoates
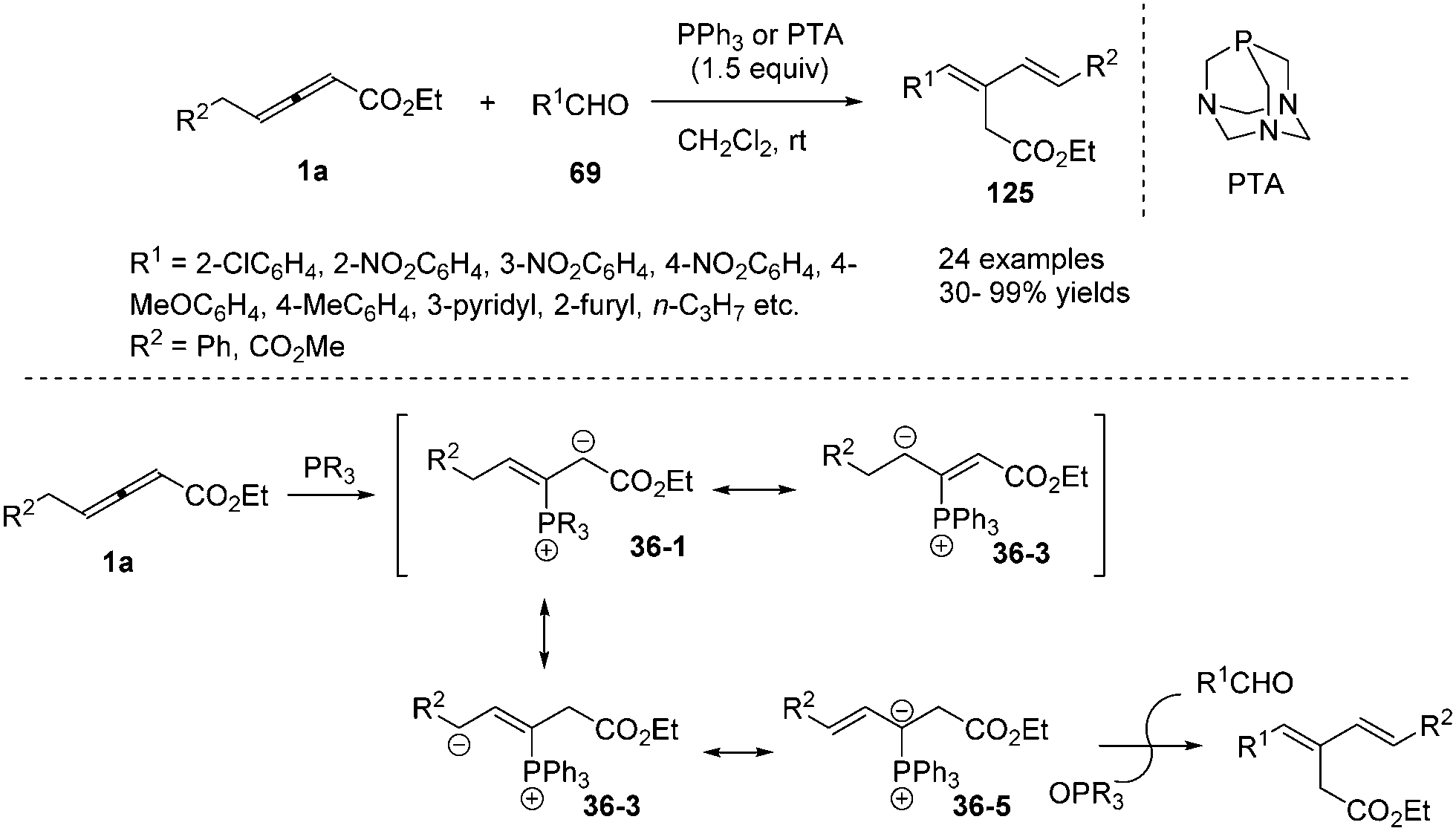
In 2009, He and co-workers reported a phosphine-mediated olefination between aldehydes 69 and γ-benzyl allenoates 1a, providing a highly stereoselective synthetic method for preparing 1,3-conjugated dienes 125 in good to excellent yields.43 For the olefination reaction, a few tertiary phosphines (PPhMe2, PPh2Me and PBu3) with relatively strong nucleophilicity compared to PPh3 were explored to furnish the olefination product in more yield. So triaza-7-phosphaadamantane (PTA), which was a readily available, air-stable, and water-soluble phosphine with comparable nucleophilicity to trialkylphosphines, was chosen as the preferable phosphine for the less reactive aldehydes. In the mechanism, the authors thought that the phosphine acted as a nucleophilic promoter to generate in situ an active phosphorus ylide which mediated the intermolecular olefination (Scheme 39).
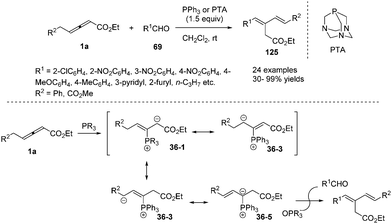 |
| | Scheme 39 Phosphine-mediated olefination between aldehydes and γ-benzyl allenoates. | |
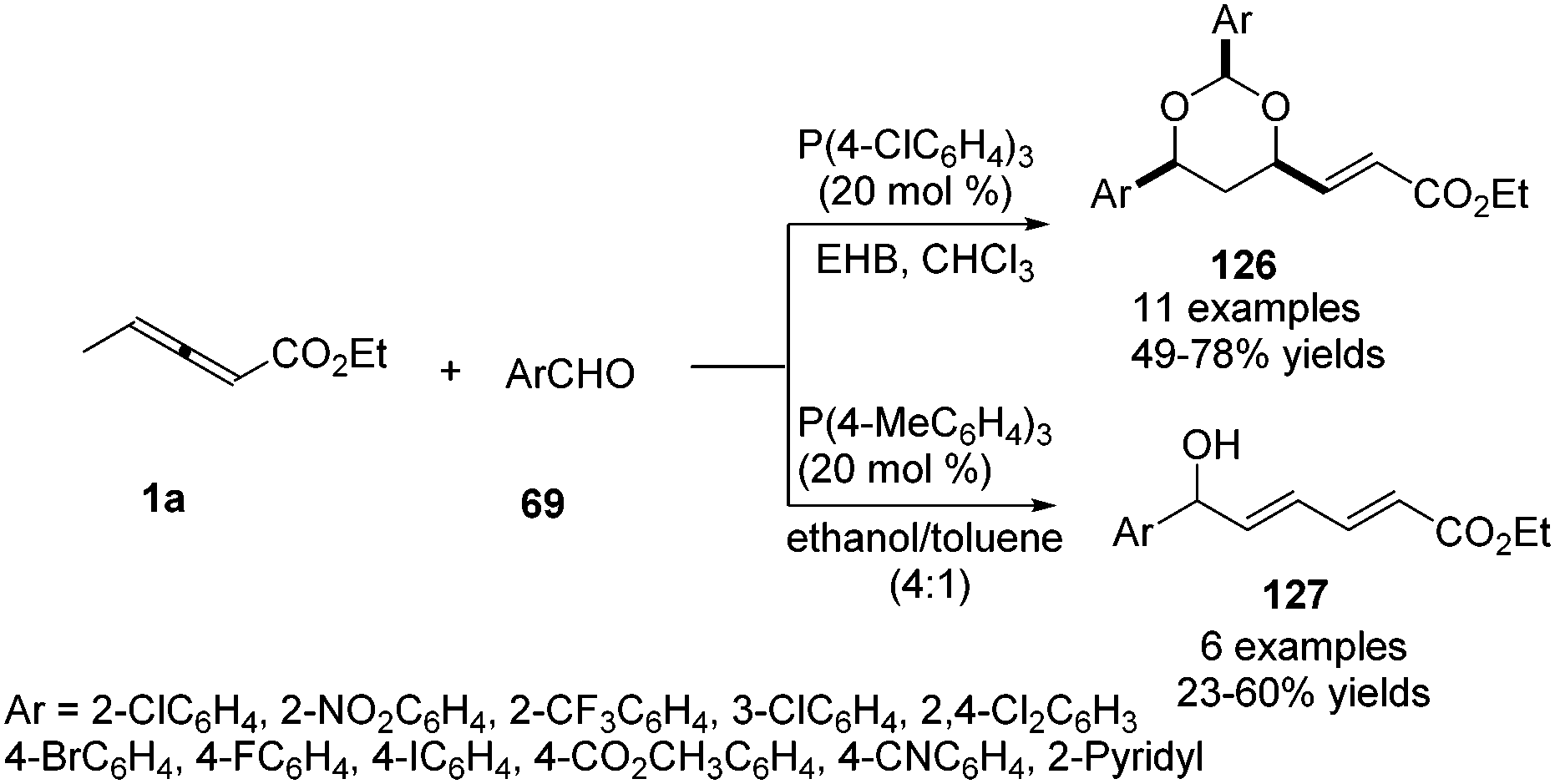
He and co-workers applied γ-methyl allenoate 1a and aldehydes 69 as substrates and developed a phosphine-catalysed formal vinylogous aldol reaction. Under the catalysis of P(4-ClC6H4)3 (20 mol%) and in the presence of ethyl 4-hydroxybenzoate (EHB), the (2+2+2) annulation products 126 were obtained in modest to good yields. On the other hand, γ-addition products 127 were obtained by using P(4-MeC6H4)3 (20 mol%) and an alcoholic solvent. The authors found that electron-poor aromatic aldehydes were effective in the reaction, but relatively electron-rich benzaldehydes, as well as aliphatic aldehydes, proved to be ineffective (Scheme 40).44
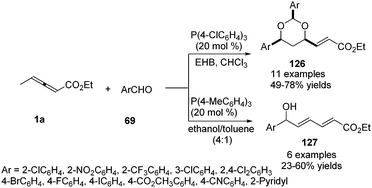 |
| | Scheme 40 Phosphine-catalysed formal vinylogous aldol reaction of γ-methyl allenoate with aldehydes. | |
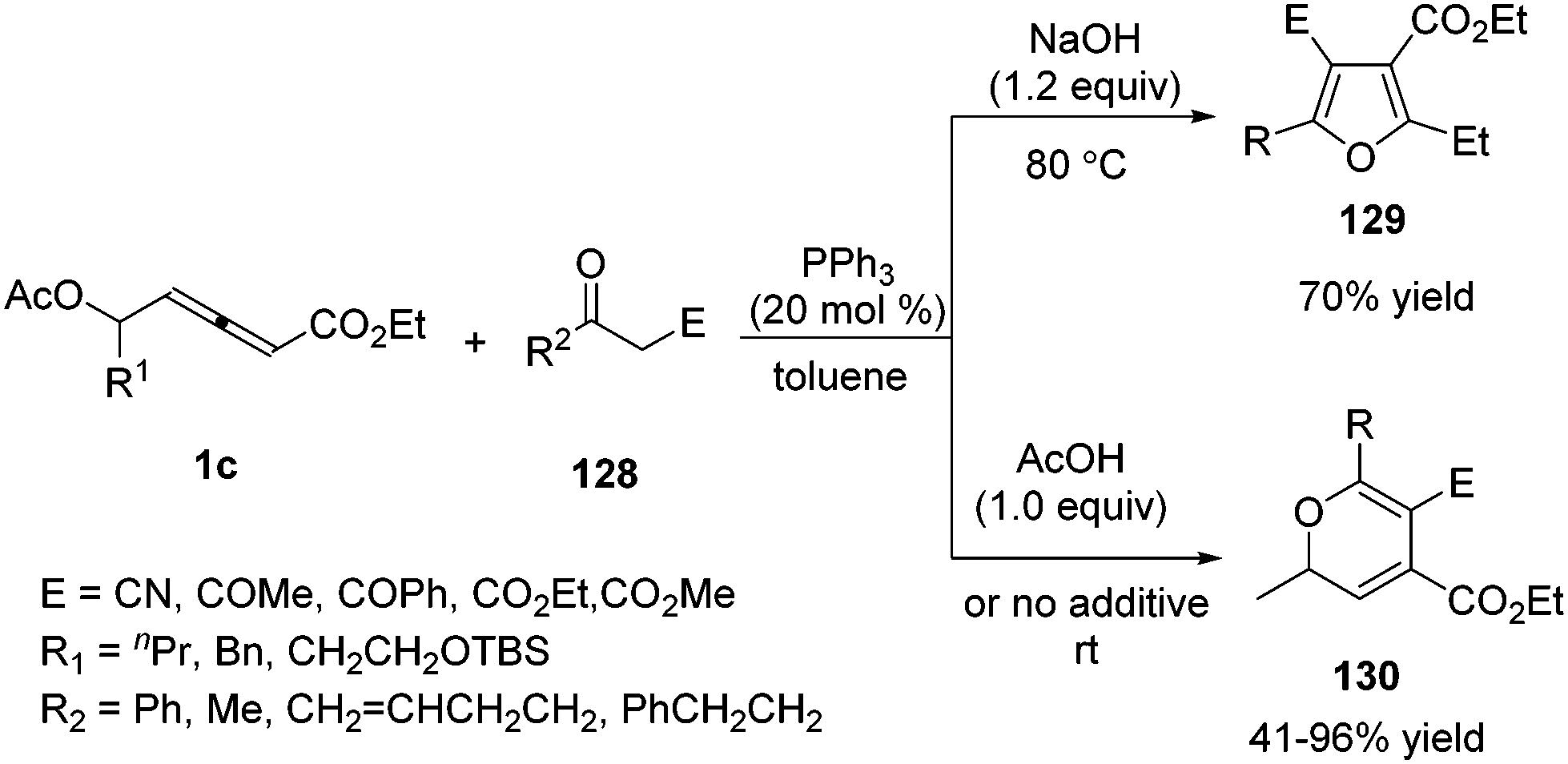
In 2012, Tong and co-workers reported a phosphine-catalysed (3+2) and (3+3) annulation between δ-acetoxy allenoates and 1C, 3O-bisnucleophiles.45 The authors found that the (3+2) adduct 129 was favoured with the assistance of a base additive while (3+3) adducts 130 were achieved under acidic reaction conditions. A series of deuterium-labeling experiments disclosed that the divergence in annulation might be strongly dependent on the involved proton transfer processes (Scheme 41).
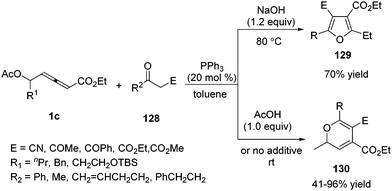 |
| | Scheme 41 Phosphine-catalyzed (3+2) or (3+3) annulations between δ-acetoxy allenoates and 1C, 3O-bisnucleophiles. | |
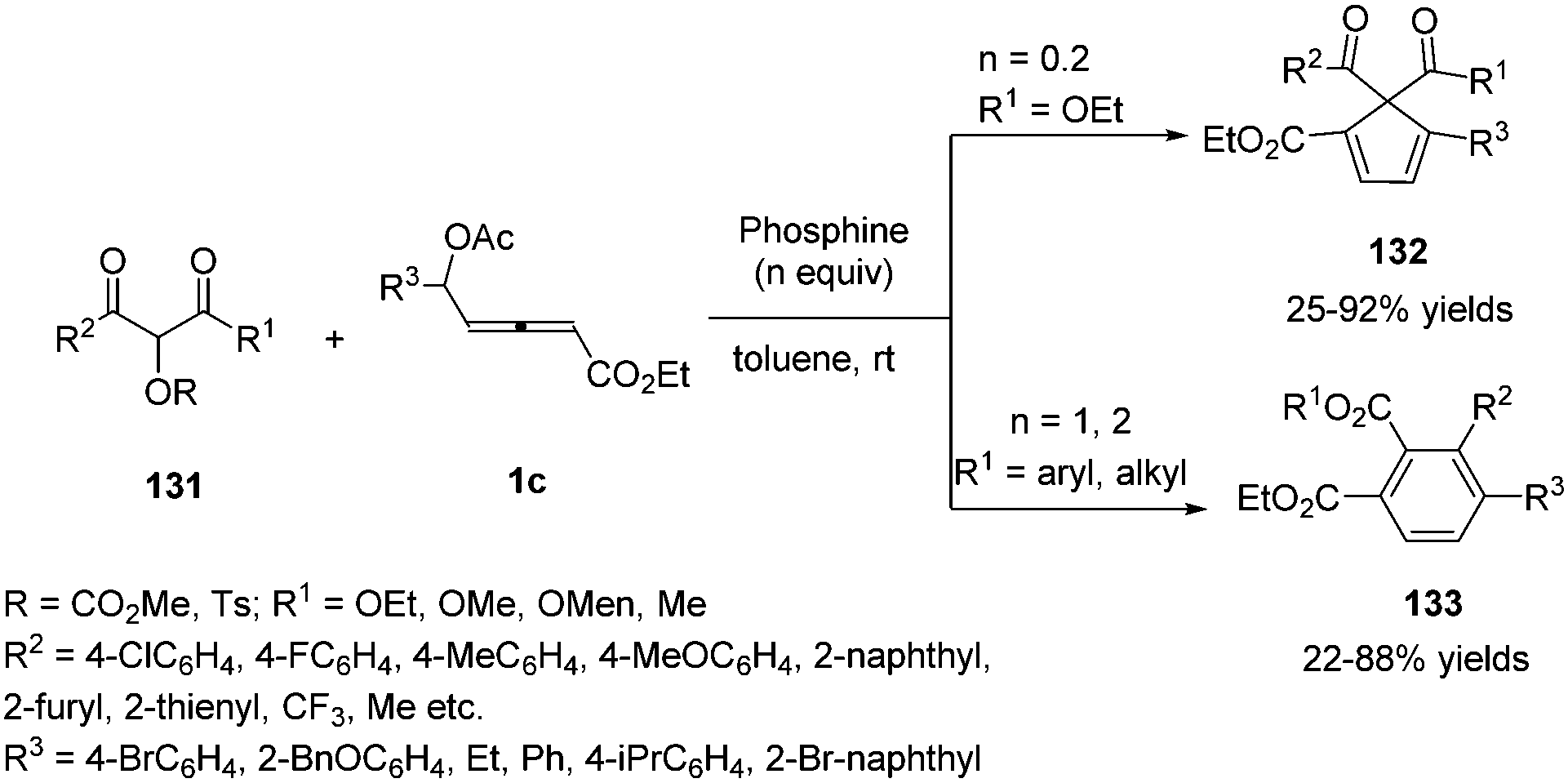
Very recently, Tong and co-workers disclosed a phosphine-promoted substrate-dependent divergent annulation of δ-acetoxy allenoates 1c with α-hydroxy-β-carbonyl ester derivatives 131. The authors realized that 2-[(methoxycarbonyl)oxy]malonate 131 as a C1 partner was able to undergo (4+1) annulations with allenoates in the presence of a catalytic amount of phosphine. But 2-(tosyloxy)-3-ketoesters as a C2 partner were able to deliver (4+2) annulations with a stoichiometric amount of phosphine and base (Scheme 42).46
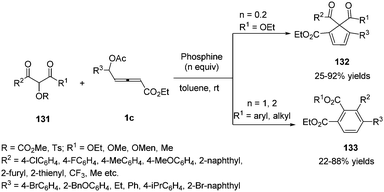 |
| | Scheme 42 Phosphine-promoted divergent annulations of δ-acetoxy allenoates with α-hydroxy-β-carbonyl ester derivatives. | |
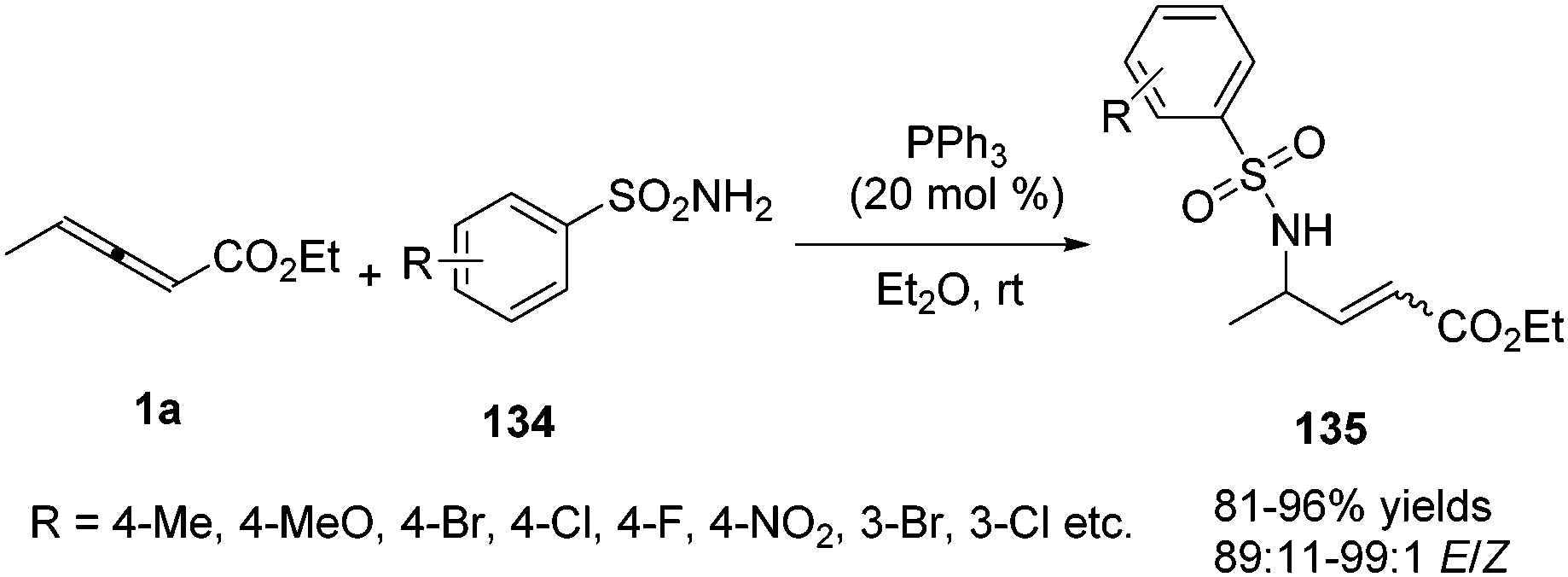
In 2015, Kwon and co-workers disclosed a phosphine catalysed γ-umpolung addition of sulphonamides (134) to γ-substituted allenoates 1a, providing a series of α,β-unsaturated γ-amino esters 135 in high yields (up to 96% yield) and with high stereoselectivity (89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11–99:1 E/Z) (Scheme 43).47
:11–99:1 E/Z) (Scheme 43).47
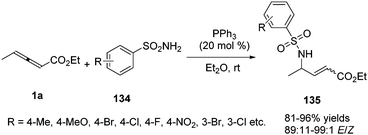 |
| | Scheme 43 Phosphine-catalysed γ-umpolung additions of sulfonamides to c-substituted allenoates. | |
6. Chiral phosphine catalysed reaction of γ-substituted allenoates
In 1997, Zhang and co-workers first reported chiral phosphine-catalysed asymmetric (3+2) cycloaddition of 2,3-butadienoates with electron-deficient olefins.48 Following Zhang's work, a diversity of chiral phosphine catalysts were synthesized and applied in enantioselective annulations of this type with allenoates.49 In this context, we have summarized some examples of chiral phosphine catalysed asymmetric reactions of γ-substituted allenoates in subsequent sections.
6.1 Chiral phosphine catalysed annulations of γ-substituted allenoates
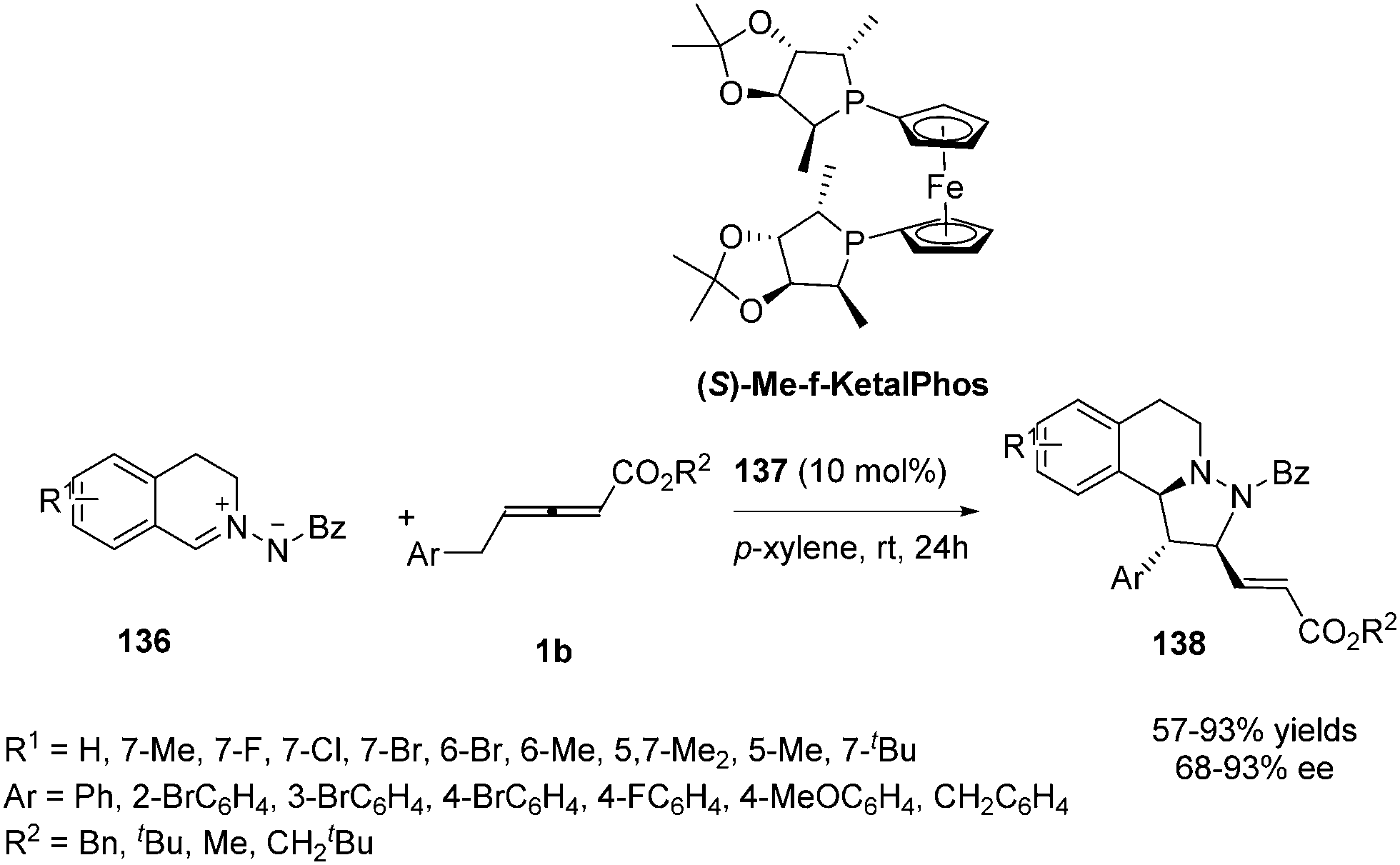
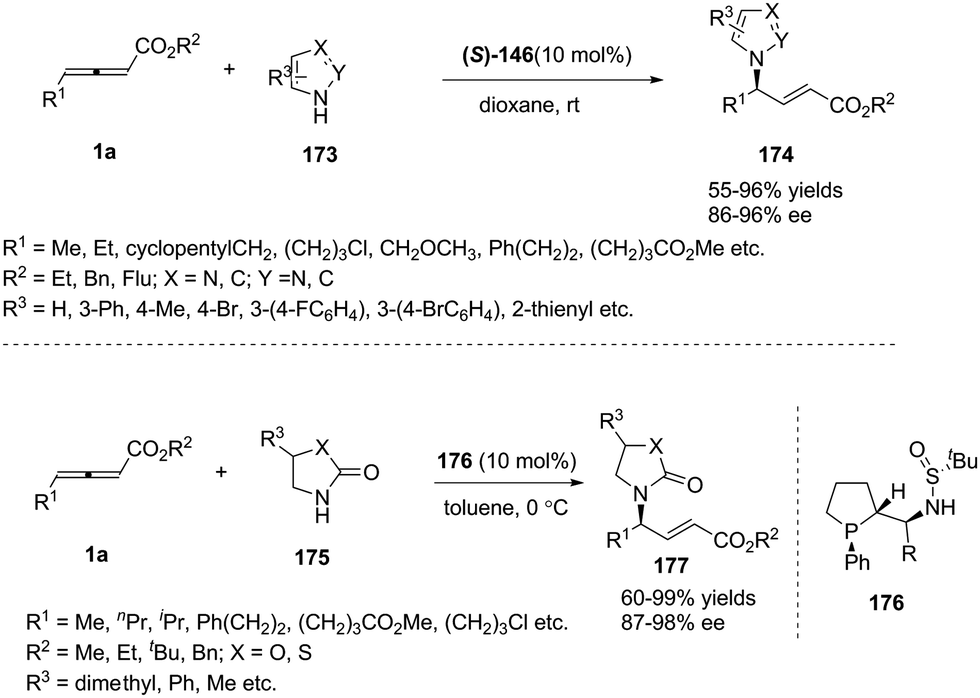
In 2014, Shi and co-workers investigated the propensity for known, readily available phosphine catalysts to serve as Lewis base catalysts for (3+2) cycloadditions.50 When screening chiral phosphines, catalyst 137 [(S)-Me-f-KetalPhos] gave the highest enantioselectivity (87%) in this reaction compared to other catalysts. The enantioselectivity of the reaction was improved by screening of the solvent, reaction temperature and additive. The outcome established that the use of 137 (10 mol%) as the catalyst and carrying out the reaction in p-xylene at room temperature served as the best condition (93% ee). With the identification of the optimal reaction conditions, a variety of C,N-cyclic azomethine imines (136) participated in the annulations with 1b providing products 138 in high yields (57–93%) and high ee's (68–93%) (Scheme 44).
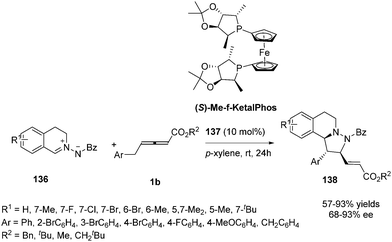 |
| | Scheme 44 Chiral phosphine-catalysed asymmetric (3+2) annulations of γ-substituted allenoates. | |
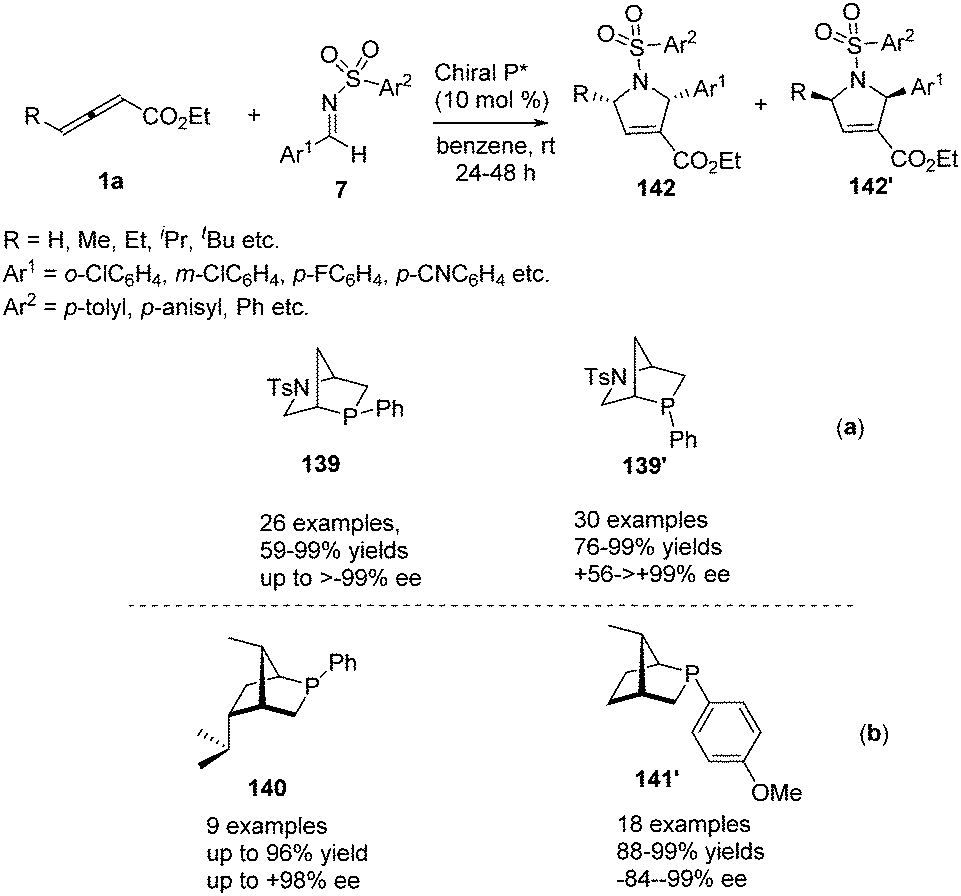
Simultaneously, Kwon reported a chiral phosphine-catalysed (3+2) annulation of γ-substituted allenoates with imines by using their two new diastereoisomeric 2-aza-5-phosphabicyclo-[2.2.1]heptanes 139 and 139′, providing enantiomerically enriched 1,2,3,5-substituted pyrrolines 142 and 142′ in good yields with excellent diastereoselectivity.51 It should be noted that the two diastereoisomeric phosphines function as pseudoenantiomers, producing their pyrrolines in opposite enantiomeric forms (Scheme 45, a). Recently, Kwon have prepared a new family of P-stereogenic[2.2.1]bicyclic chiral phosphines through straightforward syntheses starting from the natural product carvone, which was applied to γ-substituted allenoates-imine (3+2) annulation, producing a series of high enantioselective 1,2,3,5-tetrasubstituted pyrrolines, including the biologically active molecule efsevin (Scheme 45, b).52
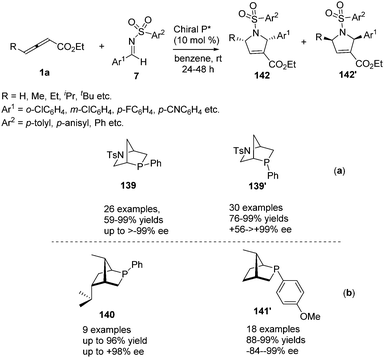 |
| | Scheme 45 Hydroxyproline-derived pseudoenantiomeric[2.2.1]bicyclic phosphines: asymmetric synthesis of (+)- and (−)-pyrrolines. | |
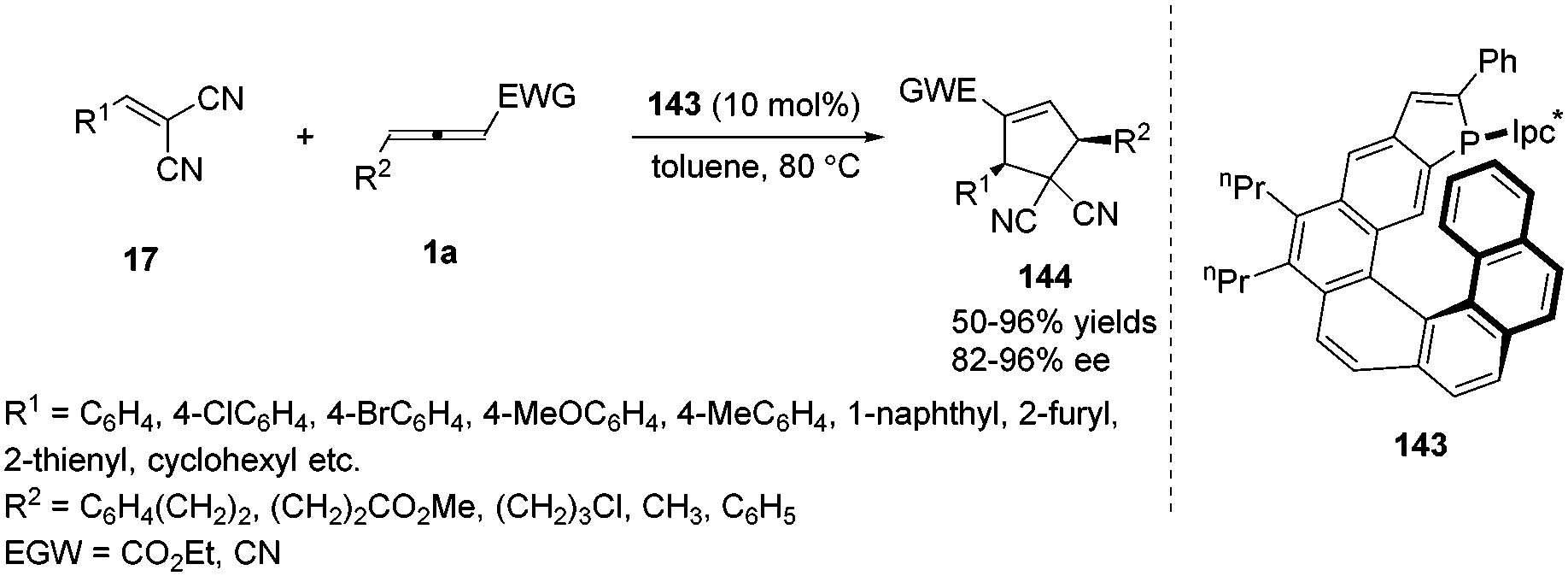
The Marinetti group was devoted to design a series of phosphahelicenes with phosphole units embedded at the end of a helical sequence of aromatic rings.53 In 2015, Marinetti and co-workers disclosed the novel P-Ipc*-substituted phosphahelicene 143, which was applied to highly enantioselective (3+2) cyclization reactions between activated olefins and γ-substituted allenoates giving ee values of up to 97%.54 It was noted that the first examples of highly enantioselective phosphine-catalysed cyclizations on cyanoallenes were realized in the reaction. And the results afforded the first evidence for efficient stereochemical control of organocatalytic processes induced by helically chiral phosphines (Scheme 46).
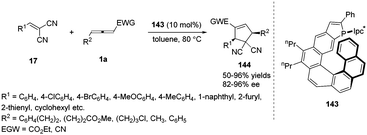 |
| | Scheme 46 Phosphahelicene catalysed (3+2) cyclizations of γ-substituted allenes and electron-poor olefins. | |
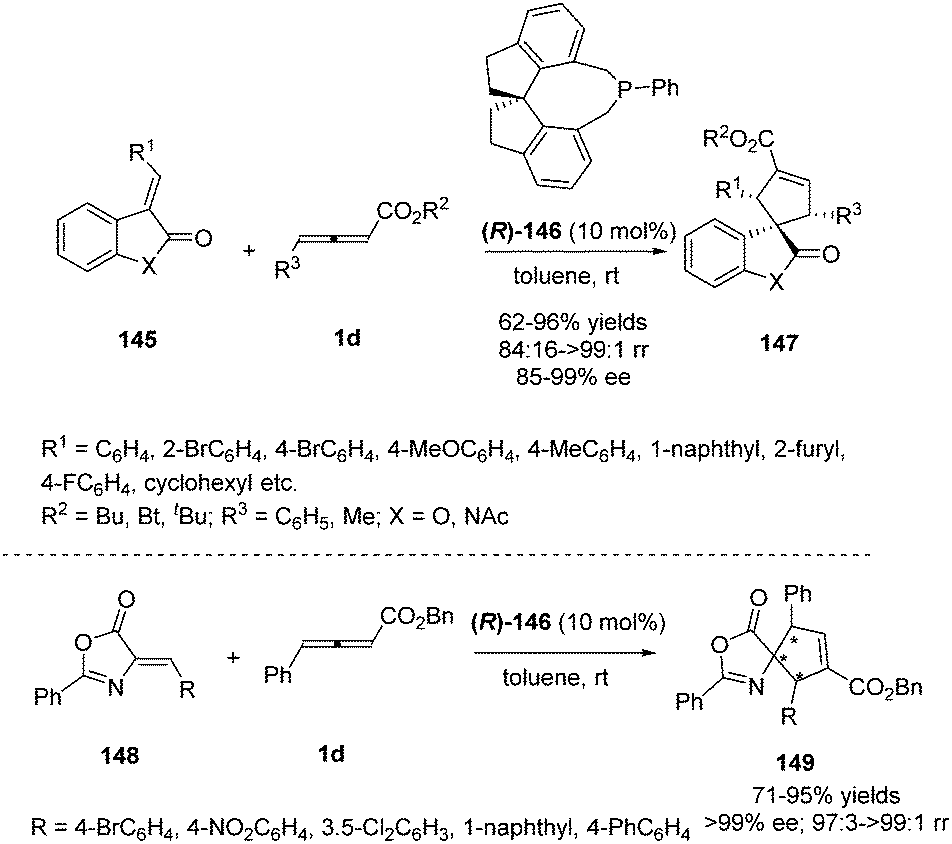
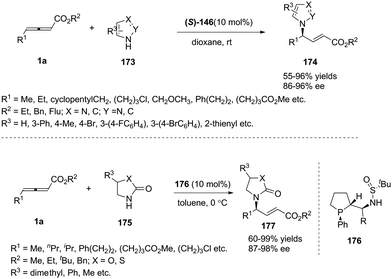
Simultaneously, Shi and Zhou reported a chiral phosphine catalysed asymmetric (3+2) cycloaddition of benzofuranone derived olefins with γ-substituted allenoates.55 The authors initially screened a variety of chiral phosphines using (E)-3-(2-bromobenzylidene)benzofuran-2(3H)-one 145 and γ-substituted allenoates 1d, and found that the catalyst (R)-SITCP (R)-146 gave the highest yield and regio- and enantioselectivity. Moreover, the DFT studies disclosed the origin of the regioselective outcomes for this phosphine-catalyzed (3+2) reaction (Scheme 47).
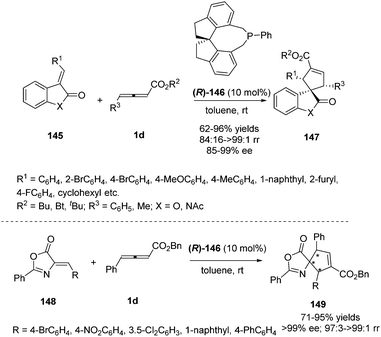 |
| | Scheme 47 Chiral phosphine-catalysed cycloaddition reactions of allenoates with benzofuranone derived olefins. | |
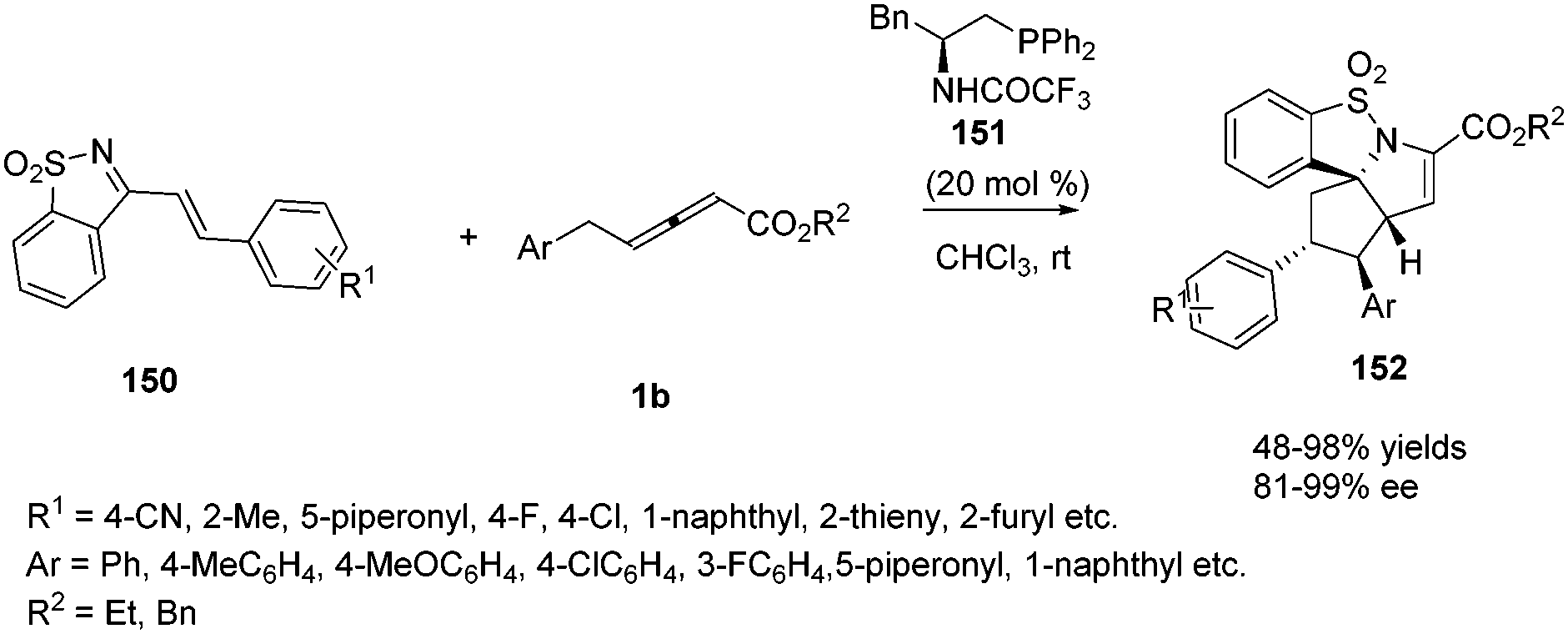
In recent years, a variety of primary amino acid based phosphine catalysts have been designed by Lu,56 Zhao57 and Huang58 groups respectively. They have been proven to be effective bifunctional phosphine catalysts in asymmetric synthesis. In 2016, Huang and co-workers disclosed that a chiral phosphine catalysed highly enantioselective sequential (2+3)/(3+2) annulation reaction of 1-azadiene 150 with γ-benzyl allenoates 1b. A variety of chiral phosphine catalysts, derived from natural amino acids, were screened and compound 151 was identified as the best catalyst. In the presence of 20 mol% of 151, functionalized poly-heterocyclic products 152 were obtained in 48–98% yields, with complete diastereoselectivity and nearly perfect enantioselectivity (up to 99% ee) (Scheme 48).59
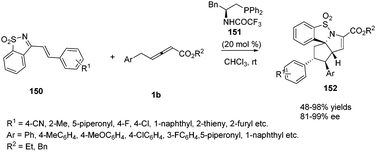 |
| | Scheme 48 Bifunctional-phosphine-catalysed sequential annulations of 1-azadiene with γ-benzyl allenoates. | |
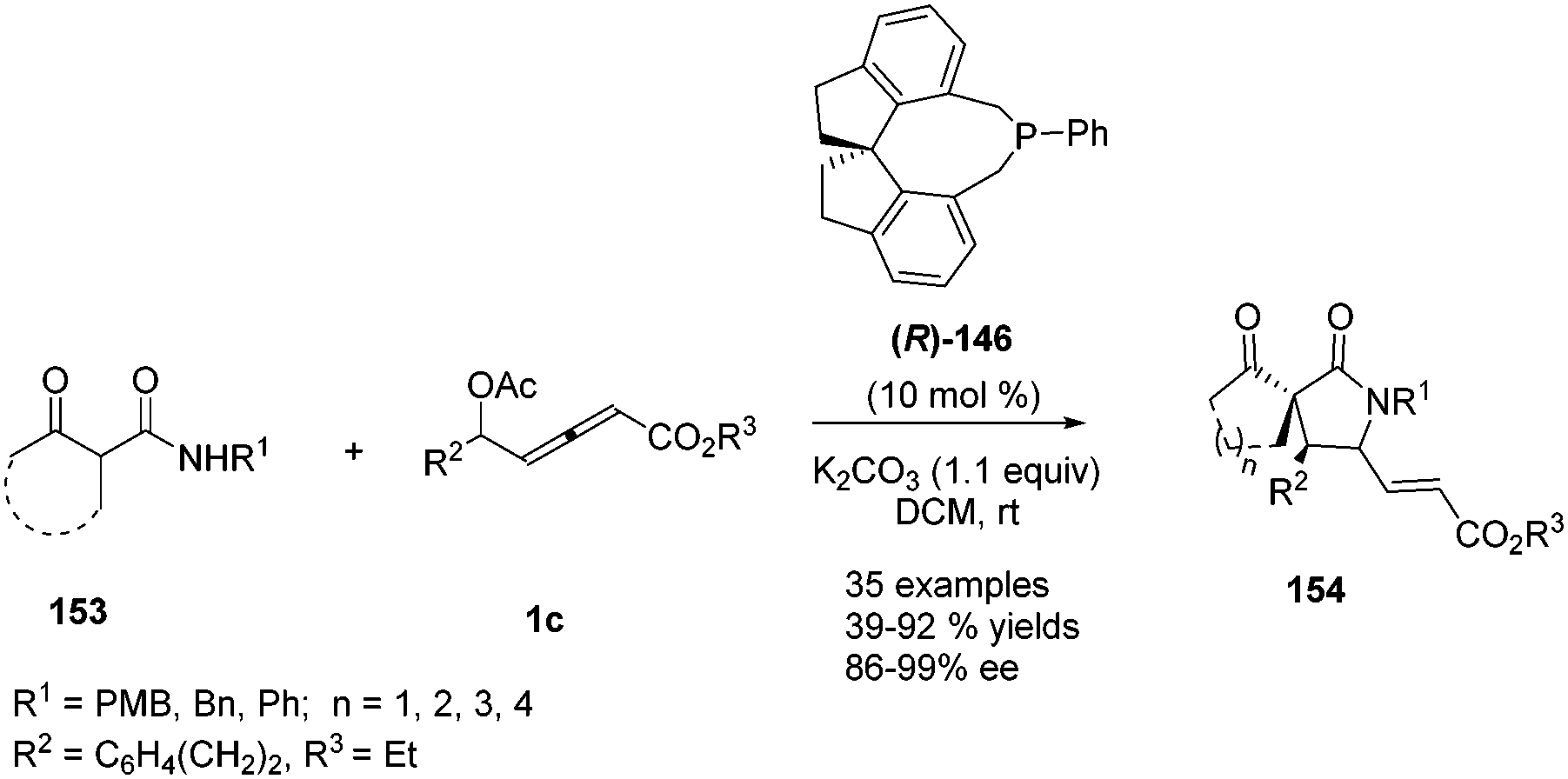
In 2017, Tong and co-workers reported asymmetric (3+2) annulations of δ-acetoxy allenoates 1c with β-carbonyl amides 153. In this study, the authors applied the (R)-SITCP (R)-146, developed by their group, as the chiral phosphine catalyst, providing a robust method for synthesis of various γ-lactams and spirocyclic β-keto lactams 154 with high stereoselectivity (84–97% ee) (Scheme 49).60
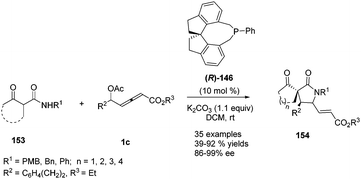 |
| | Scheme 49 Phosphine-catalysed asymmetric (3+2) annulations of δ-acetoxy allenoates with β-carbonyl amides. | |
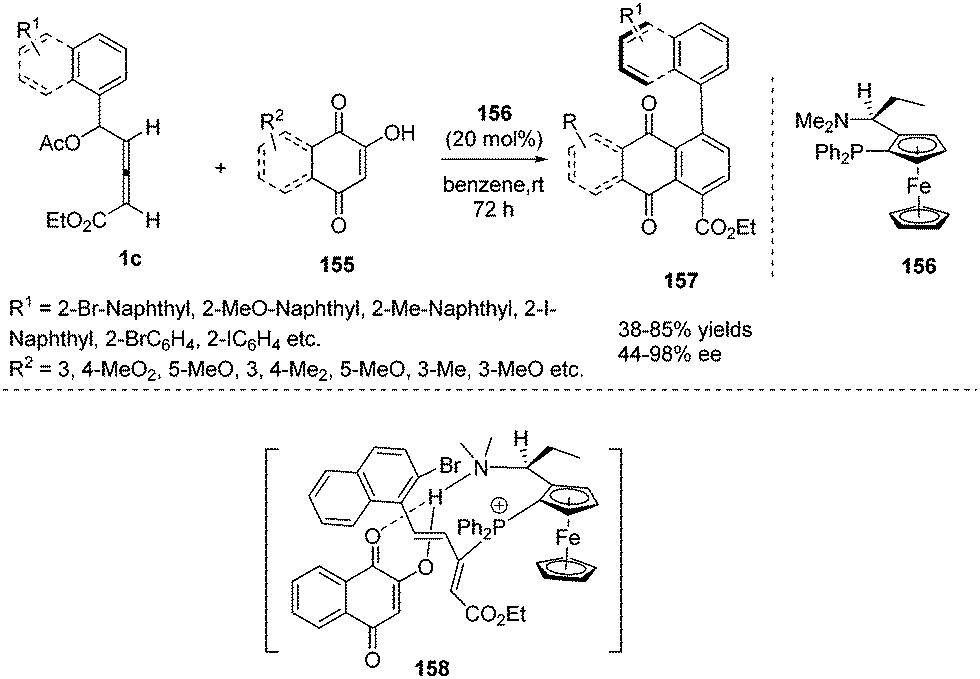
Very recently, Tong and co-workers first reported a phosphine-catalysed atroposelective (4+2) annulation of δ-acetoxy allenoates 1c and 2-hydroxyquinone derivatives 155, affording aryl-naphthaquinone atropisomers 157 in high yield and high enantioselectivity. To realize an atroposelective version of this process, the authors first employed a bulky aryl substituent at Cδ of 1c to ensure the conformational stability of 157. Next choosing a suitable chiral phosphine catalyst 156 was necessary to achieve the high enantioselective (4+2) cycloaddition. The authors found that the two functionalities of the catalyst, a tertiary phosphine (Lewis base) and a tertiary amine (Brønsted base), cooperatively enable this process with high regio- and enantioselectivity (Scheme 50).61
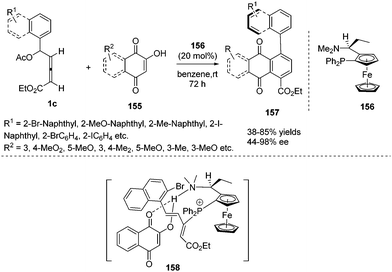 |
| | Scheme 50 Phosphine catalysed atroposelective (4+2) annulations of δ-acetoxy allenoates with 2-hydroxyquinone derivatives. | |
6.2 Chiral phosphine catalysed γ-addition reactions of γ-substituted allenoates
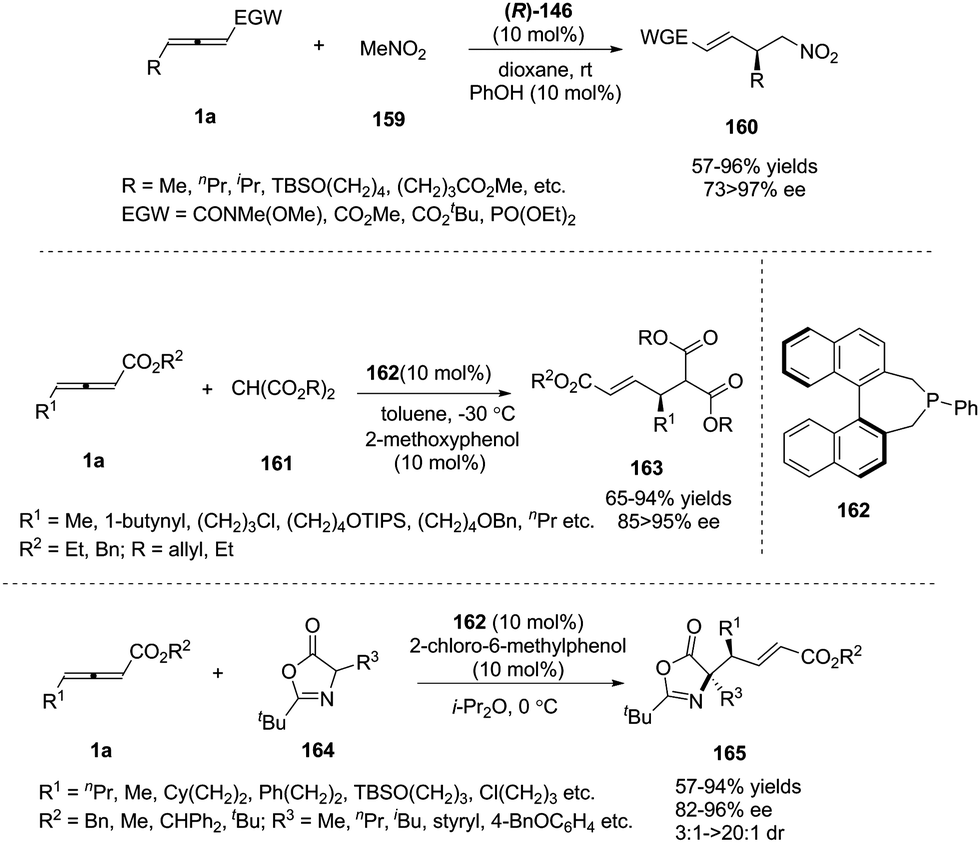
In pioneering investigations, Trost and Lu disclosed that tertiary phosphines could catalyse the additions of certain carbon, nitrogen, and oxygen nucleophiles to the γ-position of 2-butynoates or 2,3-butadienoates;62 for these electrophiles, the γ-carbon of the product was not a stereocenter. In 2009, Fu and co-workers first reported a chiral phosphine-catalysed asymmetric γ-addition by using γ-substituted allenoates and allenamides as substrates, to deliver γ-functionalized butenoates 160 in good to excellent yields along with high regio- and enantioselectivity.63 Next, Fu and co-workers also realized phosphine-catalysed asymmetric γ-additions of malonate esters64 and 1,3-oxazol-5(4H)-one65 (Scheme 51).
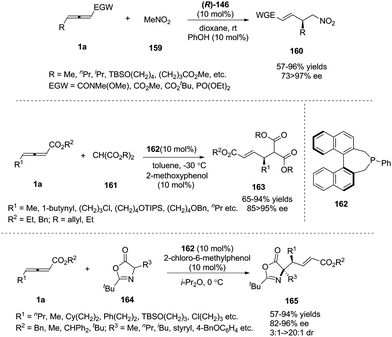 |
| | Scheme 51 Phosphine-catalyzed asymmetric additions of γ-substituted allenoates and carbon electrophiles. | |
Besides carbon electrophiles, the phosphine-catalysed γ-additions could expand to sulfur electrophiles. In 2010, Fu and co-workers reported a high stereoselective γ-addition reaction of alkyl thiols 166 and γ-substituted allenoates 1a in the presence of chiral bisphosphine TangPhos 167 (10 mol%), furnishing the γ-sulfenylated products in 67–89% yield and 85–95% ee.66 Next the authors expanded the γ-addition reaction to aryl thiols by applying Fu's catalyst (Scheme 52).67
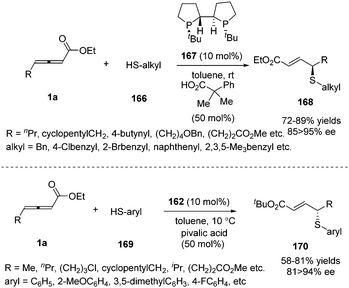 |
| | Scheme 52 Chiral phosphine catalysed γ-addition of aryl thiols. | |
Having made great progress in enantioselective γ-umpolung additions, Fu and co-workers explored the possibility of performing enantioselective γ-umpolung additions of nitrogen nucleophiles. In 2013, they developed an effective asymmetric addition of γ-aminoacrylates when applying trifluoromethylamide 171 as the nucleophile. With their spirophosphine (S)-146 as the chiral catalyst, the desired γ-amino-α,β-unsaturated esters 172 were obtained in excellent yields (68–94%) and good enantioselectivity (82–89% ee) (Scheme 53).68
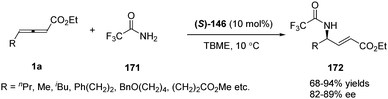 |
| | Scheme 53 Phosphine-catalysed γ-addition of trifluoromethyl-amide to γ-substituted allenoates. | |
With imidazoles as nitrogen nucleophiles, Guo and co-workers disclosed asymmetric γ-addition of heteroaromatic compounds to allenoates in good yields with high enantiomeric ratios and regioselectivity in the presence of (S)-SITCP. The reaction tolerated a broad range of substituted pyrazoles and imidazoles bearing various functional groups. The authors have also realized the synthesis of the key intermediate of muscarinic receptor M3 antagonist to demonstrate the synthetic utility of this strategy.69 Simultaneously, Zhang and co-workers have successfully expanded the scope of nitrogen nucleophiles to 2-oxazolidones by the employment of their chiral phosphine catalysts (LePhos) (Scheme 54).70
 |
| | Scheme 54 Phosphine-catalyzed γ-addition of substituted pyrazoles, imidazoles and oxazolidones. | |
7. Conclusions
In the presence of a nucleophilic phosphine catalyst, the versatile reactivity of γ-substituted allenoates has been greatly explored in the past few years. In this review, the recent advances in the application of γ-substituted allenoates as reactive substrates for the synthesis of a great variety of five, six and polycyclic structures are summarized. Although previous studies involving a 1,3-zwitterionic intermediate have been well developed by Lu and co-workers, most of the recent examples are based on the new reaction partners. In the presence of a phosphine, γ-substituted allenoates can be applied as C2, C3 and C4 synthons, to participate in many kinds of cycloaddition reactions, such as (2+4), (3+2), (3+3), (4+2) or sequential annulation reactions. Besides, phosphine catalysed γ-addition, δ-addition, or Wittig reaction of γ-substituted allenoates have also been revealed. From this point of view, the γ-substituted allenoates are full of promise.
The reactivity and potential utility of γ-substituted allenoates remain exciting and fertile in a more innovative domino cycloaddition process. It is reasonable to believe that new advances in phosphine promoted annulation involving γ-substituted allenoates will come.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the National Natural Science Foundation of China (No. 21871148, 21702189, 21672109 and 21472097), China Postdoctoral Science Foundation (No. 2017M610458, 2018T110737), Postdoctoral Research Grant in Henan Province (No. 001701006), the Natural Science Foundation of Tianjin (15JCYBJC20000) and Zhengzhou University of China for financial support of this research.
Notes and references
- Selected reviews, see:
(a) A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703–4832 CrossRef CAS PubMed;
(b) T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC.
- Selected reviews about phosphine catalysed reaction of allenoates, see:
(a) X. Lu, C. Zhang and Z. Xu, Acc. Chem. Res., 2001, 34, 535–544 CrossRef CAS PubMed;
(b) B. J. Cowen and S. J. Miller, Chem. Soc. Rev., 2009, 38, 3102–3116 RSC;
(c) Y. C. Fan and O. Kwon, Chem. Commun., 2013, 49, 11588–11619 RSC;
(d) P. Xie and Y. Huang, Eur. J. Org. Chem., 2013, 6213–6226 CrossRef CAS;
(e) C. Gomez, J.-F. Betzer, A. Voituriez and A. Marinetti, ChemCatChem, 2013, 5, 1055–1065 CrossRef CAS;
(f) Z. Wang, X. Xu and O. Kwon, Chem. Soc. Rev., 2014, 43, 2927–2940 RSC;
(g) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed.
- C. Zhang and X. Lu, J. Org. Chem., 1995, 60, 2906–2908 CrossRef CAS.
- Selected examples:
(a) X.-F. Zhu, C. E. Henry and O. Kwon, J. Am. Chem. Soc., 2007, 129, 6722–6723 CrossRef CAS PubMed;
(b) Y. Xia, Y. Liang, Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y. Li and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 3470–3471 CrossRef CAS PubMed;
(c) N. N. B. Kumar and K. C. K. Swamy, Tetrahedron Lett., 2008, 49, 7135–7138 CrossRef CAS;
(d) Z. Lu, S. Zheng, X. Zhang and X. Lu, Org. Lett., 2008, 10, 3267–3270 CrossRef CAS PubMed;
(e) X. Meng, Y. Huang and R. Chen, Org. Lett., 2009, 11, 137–140 CrossRef CAS PubMed;
(f) Y.-W. Sun, X.-Y. Guan and M. Shi, Org. Lett., 2010, 12, 5664–5667 CrossRef CAS PubMed;
(g) L. Zhou, C. Wang, C. Yuan, H. Liu, C. Zhang and H. Guo, Org. Lett., 2018, 20, 6591–6595 CrossRef CAS PubMed;
(h) H. Wang, J. Zhang, Y. Tu and J. Zhang, Angew. Chem., Int. Ed., 2019, 58, 5422–5426 CrossRef CAS PubMed.
- S. G. Pyne, K. Schafer, B. W. Skelton and A. H. White, Chem. Commun., 1997, 2267–2268 RSC.
- Z. Xu and X. Lu, Tetrahedron Lett., 1999, 40, 549–552 CrossRef CAS.
- X.-F. Zhu, C. E. Henry and O. Kwon, Tetrahedron, 2005, 61, 6276–6282 CrossRef CAS.
- B. Zhang, Z. He, S. Xu, G. Wu and Z. He, Tetrahedron, 2008, 64, 9471–9479 CrossRef CAS.
- C. Gomez, M. Gicquel, J.-C. Carry, L. Schio, P. Retailleau, A. Voituriez and A. Marinetti, J. Org. Chem., 2013, 78, 1488–1496 CrossRef CAS PubMed.
- M. Gicquel, C. Gomez, M. Concepcion, G. Alvarez, O. Pamlard, V. Guérineau, E. Jacquet, J. Bignon, A. Voituriez and A. Marinetti, J. Med. Chem., 2018, 61, 9386–9392 CrossRef CAS PubMed.
- R. Ma, G. Song, Q. Xi, L. Yang, E.-Q. Li and Z. Duan, Chin. J. Org. Chem., 2019, 39, 2196–2202 CrossRef.
- E. Li, H. Jin and Y. Huang, ChemistrySelect, 2018, 3, 12007–12010 CrossRef CAS.
- D. Wang, W. Liu, Y. Hong and X. Tong, Org. Lett., 2018, 20, 5002–5005 CrossRef CAS PubMed.
-
(a) A. Cerveri, O. N. Faza, C. S. López, S. Grilli, M. Monari and M. Bandini, J. Org. Chem., 2019, 84, 6347–6355 CrossRef CAS PubMed;
(b) L. Birbaum, L. Gillard, H. Gérard, H. Oulyadi, G. Vincent, X. Moreau, M. De Paolis and I. Chataigner, Chem. – Eur. J., 2019, 25, 13688–13693 CrossRef CAS PubMed.
- X.-C. Zhang, S.-H. Cao, Y. Wei and M. Shi, Chem. Commun., 2011, 47, 1548–1550 RSC.
- A. Jose, K. C. S. Lakshmi, E. Suresh and V. Nair, Org. Lett., 2013, 15, 1858–1861 CrossRef CAS PubMed.
- W. Luo, H. Hu, S. Nian, L. Qi, F. Ling and W. Zhong, Org. Biomol. Chem., 2017, 15, 7523–7526 RSC.
- X. Meng, Y. Huang, H. Zhao, P. Xie, J. Ma and R. Chen, Org. Lett., 2009, 11, 991–994 CrossRef CAS PubMed.
- R. Ma, S. Xu, X. Tang, G. Wu and Z. He, Tetrahedron, 2011, 67, 1053–1061 CrossRef CAS.
- J. Zheng, Y. Huang and Z. Li, Org. Lett., 2013, 15, 5064–5067 CrossRef CAS PubMed.
- E. Li, M. Chang, L. Liang and Y. Huang, Eur. J. Org. Chem., 2015, 710–714 CrossRef CAS.
- S. Ma, A. Yu and X. Meng, Org. Biomol. Chem., 2018, 16, 2885–2892 RSC.
- Z. Gan, Y. Gong, Y. Chu, E.-Q. Li, Y. Huang and Z. Duan, Chem. Commun., 2019, 55, 10120–10123 RSC.
-
(a) G. S. Creech and O. Kwon, Org. Lett., 2008, 10, 429–432 CrossRef CAS PubMed;
(b) T. Dudding, O. Kwon and E. Mercier, Org. Lett., 2006, 8, 3643–3646 CrossRef CAS PubMed.
- S. Xu, L. Zhou, R. Ma, H. Song and Z. He, Chem. – Eur. J., 2009, 15, 8698–8702 CrossRef CAS PubMed.
- H. Xiao, Z. Chai, R.-S. Yao and G. Zhao, J. Org. Chem., 2013, 78, 9781–9790 CrossRef CAS PubMed.
- A. Jose, A. J. Jayakrishnan, K. C. S. Lakshmi, S. Varughese and V. Nair, Org. Biomol. Chem., 2015, 13, 3589–3592 RSC.
- X. Kong, L. Liu, S. Luo, S. Fan, H. Qian and H. Xiao, Synlett, 2018, 1244–1248 CAS.
- J. Xing, Y. Lei, Y.-N. Gao and M. Shi, Org. Lett., 2017, 19, 2382–2385 CrossRef CAS PubMed.
- Selected examples, see:
(a) T. Wang and S. Ye, Org. Lett., 2010, 12, 4168–4171 CrossRef CAS PubMed;
(b) Y. S. Tran and O. Kwon, J. Am. Chem. Soc., 2007, 129, 12632–12633 CrossRef CAS PubMed;
(c) X.-F. Zhu, J. Lan and O. Kwon, J. Am. Chem. Soc., 2003, 125, 4716–4717 CrossRef CAS PubMed.
- E. Li, Y. Huang, L. Liang and P. Xie, Org. Lett., 2013, 15, 3138–3141 CrossRef CAS PubMed.
- M. Gicquel, C. Gomez, P. Retailleau, A. Voituriez and A. Marinetti, Org. Lett., 2013, 15, 4002–4005 CrossRef CAS PubMed.
- Q. Zhang, L. Yang and X. Tong, J. Am. Chem. Soc., 2010, 132, 2550–2551 CrossRef CAS PubMed.
- Y. Zhang and X. Tong, Org. Lett., 2017, 19, 5462–5465 CrossRef CAS PubMed.
- H. Zhao, X. Meng and Y. Huang, Chem. Commun., 2013, 49, 10513–10515 RSC.
- E. Li and Y. Huang, Chem. – Eur. J., 2014, 20, 3520–3527 CrossRef CAS PubMed.
- E. Li and Y. Huang, Chem. Commun., 2014, 50, 948–950 RSC.
- Selected examples, see:
(a) P. Jakubec, D. M. Cockfield and D. J. Dixon, J. Am. Chem. Soc., 2009, 131, 16632–16633 CrossRef CAS PubMed;
(b) D. B. Martin and C. D. Vanderwal, Angew. Chem., Int. Ed., 2010, 49, 2830–2832 CrossRef CAS PubMed;
(c) C. Hundsdörfer, H.-J. Hemmerling, C. Götz, F. Totzke, P. Bednarski, M. L. Borgne and J. Jose, Bioorg. Med. Chem., 2012, 2282–2289 CrossRef PubMed.
- E. Li, P. Jia, L. Liang and Y. Huang, ACS Catal., 2014, 4, 600–603 CrossRef CAS.
- J. Zheng, Y. Huang and Z. Li, Org. Lett., 2013, 15, 5758–5761 CrossRef CAS PubMed.
- Y. Jia, X. Tang, G. Cai, R. Jia, B. Wang and Z. Miao, Eur. J. Org. Chem., 2015, 4720–4725 CrossRef CAS.
- N. Li, P. Jia and Y. Huang, Chem. Commun., 2019, 55, 10976–10979 RSC.
- S. Xu, L. Zhou, S. Zeng, R. Ma, Z. Wang and Z. He, Org. Lett., 2009, 11, 3498–3501 CrossRef CAS PubMed.
- Z. Qin, R. Ma, S. Xu and Z. He, Tetrahedron, 2013, 69, 10424–10430 CrossRef CAS.
- J. Hu, W. Dong, X.-Y. Wu and X. Tong, Org. Lett., 2012, 14, 5530–5533 CrossRef CAS PubMed.
- T. Xu, D. Wang, W. Liu and X. Tong, Org. Lett., 2019, 21, 1944–1947 CrossRef CAS PubMed.
- Q.-F. Zhou, K. Zhang and O. Kwon, Tetrahedron Lett., 2015, 56, 3273–3276 CrossRef CAS PubMed.
- G. Zhu, Z. Chen, Q. Jiang, D. Xiao, P. Cao and X. Zhang, J. Am. Chem. Soc., 1997, 119, 3836–3837 CrossRef CAS.
- Selected examples see:
(a) B. J. Cowen and S. J. Miller, J. Am. Chem. Soc., 2007, 129, 10988–10989 CrossRef CAS PubMed;
(b) H. Xiao, Z. Chai, C.-W. Zheng, Y.-Q. Yang, W. Liu, J.-K. Zhang and G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 4467–4470 CrossRef CAS PubMed;
(c) M. Sampath and T.-P. Loh, Chem. Sci., 2010, 1, 739–742 RSC;
(d) H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS PubMed.
- D. Wang, Y. Lei, Y. Wei and M. Shi, Chem. – Eur. J., 2014, 20, 15325–15329 CrossRef CAS PubMed.
- C. E. Henry, Q. Xu, Y. C. Fan, T. J. Martin, L. Belding, T. Dudding and O. Kwon, J. Am. Chem. Soc., 2014, 136, 11890–11893 CrossRef CAS PubMed.
- A. J. Smaligo, S. Vardhineedi and O. Kwon, ACS Catal., 2018, 8, 5188–5192 CrossRef CAS PubMed.
-
(a) N. Fukawa, T. Osaka, K. Noguchi and K. Tanaka, Org. Lett., 2010, 12, 1324–1327 CrossRef CAS PubMed;
(b) K. Yavari, S. Moussa, B. Ben Hassine, P. Retailleau, A. Voituriez and A. Marinetti, Angew. Chem., Int. Ed., 2012, 51, 6748–6752 CrossRef CAS PubMed;
(c) Y. Sawada, S. Furumi, A. Takai, M. Takeuchi, K. Noguchi and K. Tanaka, J. Am. Chem. Soc., 2012, 134, 4080–4083 CrossRef CAS PubMed;
(d) K. Yavari, P. Retailleau, A. Voituriez and A. Marinetti, Chem. – Eur. J., 2013, 19, 9939–9947 CrossRef CAS PubMed;
(e) P. Aillard, P. Retailleau, A. Voituriez and A. Marinetti, Chem. Commun., 2014, 50, 2199–2201 RSC.
- M. Gicquel, Y. Zhang, P. Aillard, P. Retailleau, A. Voituriez and A. Marinetti, Angew. Chem., Int. Ed., 2015, 54, 5470–5473 CrossRef CAS PubMed.
- D. Wang, G.-P. Wang, Y.-L. Sun, S.-F. Zhu, Y. Wei, Q.-L. Zhou and M. Shi, Chem. Sci., 2015, 6, 7319–7325 RSC.
- Selected examples, see:
(a) T. Wang, X. Han, F. Zhong, W. Yao and Y. Lu, Acc. Chem. Res., 2016, 49, 1369–1378 CrossRef CAS PubMed;
(b) K. Li, T. P. Gonçalves, K.-W. Huang and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 5427–5431 CrossRef CAS PubMed.
- H. Xiao, Z. Chai, C.-W. Zheng, Y.-Q. Yang, W. Liu, J.-K. Zhang and G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 4467–4470 CrossRef CAS PubMed.
-
(a) X. Dong, L. Liang, E. Li and Y. Huang, Angew. Chem., Int. Ed., 2015, 54, 1621–1624 CrossRef CAS PubMed;
(b) H. Jin, Q. Zhang, E. Li, P. Jia, N. Li and Y. Huang, Org. Biomol. Chem., 2017, 15, 7097–7101 RSC.
- E. Li, H. Jin, P. Jia, X. Dong and Y. Huang, Angew. Chem., Int. Ed., 2016, 55, 11591–11594 CrossRef CAS PubMed.
- C. Ni, J. Chen, Y. Zhang, Y. Hou, D. Wang, X. Tong, S.-F. Zhu and Q.-L. Zhou, Org. Lett., 2017, 19, 3668–3671 CrossRef CAS PubMed.
- X. Chen, D. Gao, D. Wang, T. Xu, W. Liu, P. Tian and X. Tong, Angew. Chem., Int. Ed., 2019, 58, 15334–15338 CrossRef CAS PubMed.
-
(a) B. M. Trost and C.-J. Li, J. Am. Chem. Soc., 1994, 116, 3167–3168 CrossRef CAS;
(b) B. M. Trost and C.-J. Li, J. Am. Chem. Soc., 1994, 116, 10819–10820 CrossRef CAS;
(c) C. Zhang and X. Lu, Synlett, 1995, 645–646 CrossRef CAS;
(d) B. M. Trost and G. R. Dake, J. Org. Chem., 1997, 62, 5670–5671 CrossRef CAS.
- S. W. Smith and G. C. Fu, J. Am. Chem. Soc., 2009, 131, 14231–14233 CrossRef CAS PubMed.
- R. Sinisi, J. Sun and G. C. Fu, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20652–20654 CrossRef CAS PubMed.
- M. Kalek and G. C. Fu, J. Am. Chem. Soc., 2015, 137, 9438–9442 CrossRef CAS PubMed.
- J. Sun and G. C. Fu, J. Am. Chem. Soc., 2010, 132, 4568–4569 CrossRef CAS PubMed.
- Y. Fujiwara, J. Sun and G. C. Fu, Chem. Sci., 2011, 2, 2196–2198 RSC.
- R. J. Lundgren, A. Wilsily, N. Marion, C. Ma, Y. K. Chung and G. C. Fu, Angew. Chem., Int. Ed., 2013, 52, 2525–2528 CrossRef CAS PubMed.
- H. Wang and C. Guo, Angew. Chem., Int. Ed., 2019, 58, 2854–2858 CrossRef CAS PubMed.
- H. Qiu, X. Chen and J. Zhang, Chem. Sci., 2019, 10, 10510–10515 RSC.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a and
You
Huang
a and
You
Huang