
Hydrazone chemistry assisted DNAzyme for the analysis of double targets†
Juan
Zhang  *a
*a
*a
Abstract
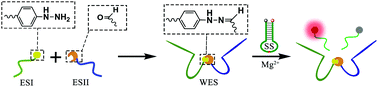
In this work, a hydrazone chemistry assisted DNAzyme has been designed and constructed. The introduction of hydrazone chemistry increases the versatility of DNAzymes. With superior catalytic capability, the hydrazone chemistry assisted DNAzyme has been successfully applied for the analysis of double targets. Taking 5-hydroxymethylfurfural (HMF) and lipopolysaccharide (LPS) as samples, the hydrazone chemistry assisted DNAzyme can be used for the detection of different combinations of targets. Moreover, because hydrazone chemistry is popular in nature, this work may also provide a new insight for the development of DNAzymes and their multifunctionality.
 Please wait while we load your content...
Please wait while we load your content...

