
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed hydroalkylation and hydroalkenylation of 1,3-dienes with hydrazones†
Lei
Cheng
,
Ming-Ming
Li
,
Biao
Wang
,
Li-Jun
Xiao
,
Jian-Hua
Xie
 and
Qi-Lin
Zhou
*
and
Qi-Lin
Zhou
*
State Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: qlzhou@nankai.edu.cn
First published on 27th September 2019
Abstract
Transition-metal-catalyzed hydrofunctionalization of 1,3-dienes is a useful and atom-economical method for constructing allylic compounds. Although substantial progress on hydroalkylation of dienes with stabilized carbon nucleophiles has been made, hydroalkylation of dienes with unstabilized carbon nucleophiles has remained a challenge. In this article, we report a protocol for nickel-catalyzed hydroalkylation of dienes with hydrazones, which serve as equivalents of alkyl carbon nucleophiles. In addition, we developed a protocol for hydroalkenylation of dienes with α,β-unsaturated hydrazones, providing a new method for the synthesis of 1,4-dienes. These hydroalkylation and hydroalkenylation reactions feature mild conditions and a wide substrate scope, and the utility of the reaction products is demonstrated by the preparation of an activator of soluble guanylate cyclase.
Introduction
1,3-Dienes are readily available commodity chemicals and are widely used in the synthesis of valuable compounds, including polymeric materials.1,2 Transition-metal-catalyzed hydrofunctionalization of 1,3-dienes has emerged as a useful and atom-economical method for constructing allylic compounds,3 which are versatile building blocks in organic synthesis.4 In particular, selective addition of carbon nucleophiles such as enols and enolates to dienes (i.e., hydroalkylation of 1,3-dienes) is an efficient method for coupling two simple carbon components via C–C bond formation. In these hydroalkylation reactions, the active catalytic species is usually a metal hydride, which reacts with the diene to generate an electrophilic metal-π-allyl intermediate that couples with the carbon nucleophile. Substantial progress has been made on palladium- and rhodium-catalyzed coupling reactions of dienes with stabilized carbon nucleophiles (Scheme 1a).5 However, the coupling of dienes with unstabilized carbon nucleophiles has remained a challenge. Recently, we developed a protocol for nickel-catalyzed regio- and enantioselective hydroalkylation reactions of dienes with carbon nucleophiles containing a single carbonyl group (Scheme 1b).6 In these reactions, the active catalytic nickel hydride species is generated by oxidative addition reaction between Ni(0) and an alcohol.7 To increase the utility of hydroalkylation reactions of dienes, we have been working on hydroalkylation reactions with different kinds of alkyl carbon nucleophiles. Recently, Li and co-workers reported an elegant umpolung strategy that uses hydrazones as alkyl carbanion equivalents in C–C coupling reactions with various electrophiles.8 Inspired by this work, we have now developed a protocol for nickel-catalyzed hydroalkylation reactions of dienes with hydrazones as alkyl carbon nucleophiles. In addition, when α,β-unsaturated hydrazones were used, we unexpectedly develop an efficient hydroalkenylation of dienes, providing 1,4-skipped diene products with high yield and regioselectivity (Scheme 1c).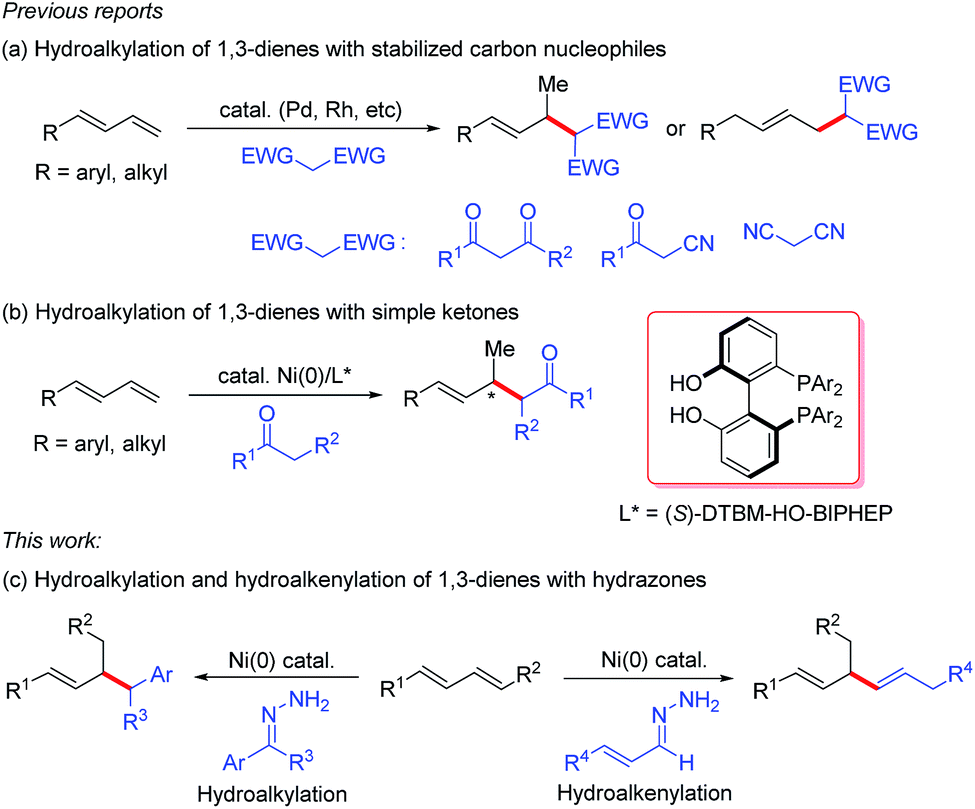 | ||
| Scheme 1 Transition-metal-catalyzed allylic C–C bond formation using 1,3-dienes and carbon nucleophiles. | ||
Results and discussion
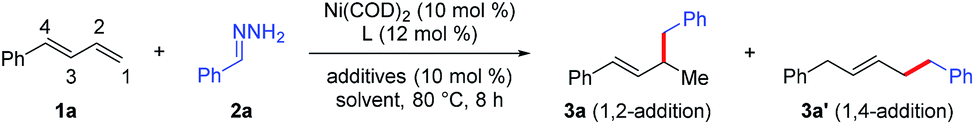
We began by investigating the reaction of phenylbutadiene (1a) and phenyl hydrazone (2a, 1.5 equiv.) using a Ni(COD)2/DTBM-SEGPHOS catalyst under the previously reported hydroalkylation conditions (Table 1).6 Reaction in EtOH in the presence of catalytic tBuOLi (10 mol%) afforded 1,2-hydroalkylation product 3a in 13% yield and 1,4-hydroalkylation product 3a′ in 1% yield (entry 1). In an attempt to improve the yield of 3a and prevent the 1,4-hydroalkylation reaction, we explored various other ligands (entries 2–9). We were delighted to find that the electron-deficient ligand tris[4-(trifluoromethyl)phenyl]phosphine not only gave the best yield of 3a (85%) but also showed high regioselectivity (1,2-/1,4-hydroalkylation > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, entry 9). Control experiments showed that tBuOLi was crucial for the reaction; the yield and the regioselectivity were lower in the absence of the base (entry 10), but increasing the amount of tBuOLi to 1 equiv. resulted in no improvement in the yield or selectivity (entry 11). Screening of other alcohol solvents showed that EtOH was the best choice (compare entry 9 with entries 12–14). Finally, when the amount of the diene substrate was increased to 2 equiv., the yield of the hydroalkylation reaction increased to 92% (entry 15).
:1, entry 9). Control experiments showed that tBuOLi was crucial for the reaction; the yield and the regioselectivity were lower in the absence of the base (entry 10), but increasing the amount of tBuOLi to 1 equiv. resulted in no improvement in the yield or selectivity (entry 11). Screening of other alcohol solvents showed that EtOH was the best choice (compare entry 9 with entries 12–14). Finally, when the amount of the diene substrate was increased to 2 equiv., the yield of the hydroalkylation reaction increased to 92% (entry 15).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Additive | Solvent | Yieldb (%) | 3a/3a′c |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.15 mmol), Ni(COD)2 (0.01 mmol), ligand (0.012 mmol for monodentate ligand, 0.01 mmol for bidentate ligand), additive (0.01 mmol), solvent (0.75 mL) at 80 °C for 8 h. b 1H NMR yields of major isomer 3a using 1,3,5-trimethoxybenzene as internal standard. c Regioselectivity (ratio of 1,2- and 1,4-hydrogenation products) was determined by 1H NMR analysis of reaction mixture. d Additives: 1.0 equivalent. e 1a (0.2 mmol), 2a (0.1 mmol). f Isolated yield in the parentheses. DPPP = 1,3-bis(diphenylphosphino)propane; DPPB = 1,4-bis(diphenylphosphino)butane; DPPPe = 1,5-bis(diphenylphosphino)pentane; DPPF = 1,1′-bis(diphenylphosphino)ferrocene. | |||||
| 1 | DTBM-SegPhos | t BuOLi | EtOH | 13 | 13:1 |
| 2 | DPPP | t BuOLi | EtOH | 18 | 9:1 |
| 3 | DPPB | t BuOLi | EtOH | 65 | 18:1 |
| 4 | DPPPe | t BuOLi | EtOH | 62 | 18:1 |
| 5 | DPPF | t BuOLi | EtOH | 58 | 9:1 |
| 6 | PCy3 | t BuOLi | EtOH | 33 | 3:1 |
| 7 | PPh3 | t BuOLi | EtOH | 64 | 11:1 |
| 8 | P(4-OMeC6H4)3 | t BuOLi | EtOH | 81 | 9:1 |
| 9 | P(4-CF3C6H4)3 | t BuOLi | EtOH | 85 | >20:1 |
| 10 | P(4-CF3C6H4)3 | None | EtOH | 30 | 6:1 |
| 11d | P(4-CF3C6H4)3 | t BuOLi | EtOH | 85 | >20:1 |
| 12 | P(4-CF3C6H4)3 | t BuOLi | MeOH | 28 | 11:1 |
| 13 | P(4-CF3C6H4)3 | t BuOLi | n PrOH | 80 | >20:1 |
| 14 | P(4-CF3C6H4)3 | t BuOLi | i PrOH | 71 | 16:1 |
| 15e | P(4-CF3C6H4)3 | t BuOLi | EtOH | 95 (92)f | >20:1 |
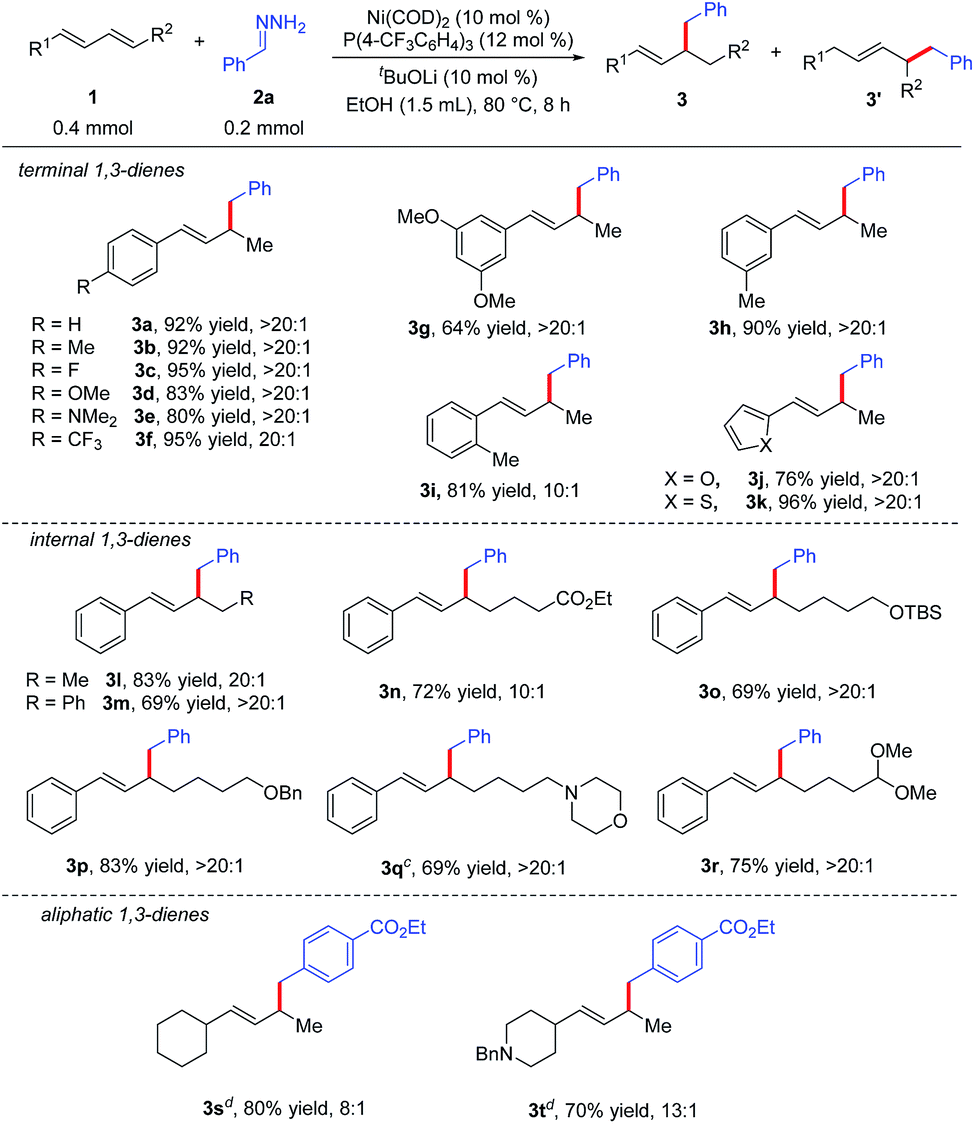
Under the optimal reaction conditions (Table 1, entry 15), we evaluated various 1,3-dienes 1 in reactions with aryl hydrazone (2a or 2g) (Table 2). Terminal dienes with para-substituted aromatic rings (1b–1f) afforded the desired hydroalkylation products in high yields (80–95%) with excellent regioselectivity (≥20:1); dienes with electron-donating substituents such as OMe (1d) and NMe2 (1e) gave slightly lower yields than dienes with electron-withdrawing substituents. Terminal dienes with meta- and ortho-substituted aromatic rings (1g–1i) also reacted smoothly with 2a to afford the corresponding hydroalkylation products, although diene 1g, which has two meta OMe groups, gave only a moderate yield of 3g (64%). Diene 1i, which has an ortho Me group, showed lower regioselectivity (10:1). In addition, furyl diene 1j and thienyl diene 1k also worked well in the reaction with 2a, affording good yields of the expected products with excellent regioselectivity. We were delighted to find that internal aromatic dienes 1l–1r were also suitable substrates; a variety of functional groups in the internal dienes, including an ester (1n), ethers (1o and 1p), an amine (1q), and an acetal (1r), were tolerated under the reaction conditions. It is worth mentioning that the Z/E configuration of the aromatic dienes had no effect on the yield or regioselectivity of the reaction (see ESI†). In addition to aromatic dienes, aliphatic dienes 1s and 1t were also examined in the reaction with 2g and found to afford 1,2-hydroalkylation products 3s and 3t, in good yields (80% and 70%, respectively) with good regioselectivities (8:1 and 13:1, respectively) when the diphosphine ligand DPPPe was used.
| a Reaction conditions: 1 (0.4 mmol), 2a (0.2 mmol), Ni(COD)2 (0.02 mmol), P(4-CF3C6H4)3 (0.024 mmol), tBuOLi (0.02 mmol), EtOH (1.5 mL) at 80 °C for 8 h. Isolated yields. Regioselectivity was the ratio of 3 to 3′, which was determined by 1H NMR analysis of the product. b Z/E configuration of aromatic dienes had no effect on the yield and selectivity of the reaction (see ESI). c 1 (0.2 mmol), 2a (0.3 mmol). d E-isomer of aliphatic diene as starting material, DPPPe as the ligand. |
|---|
|
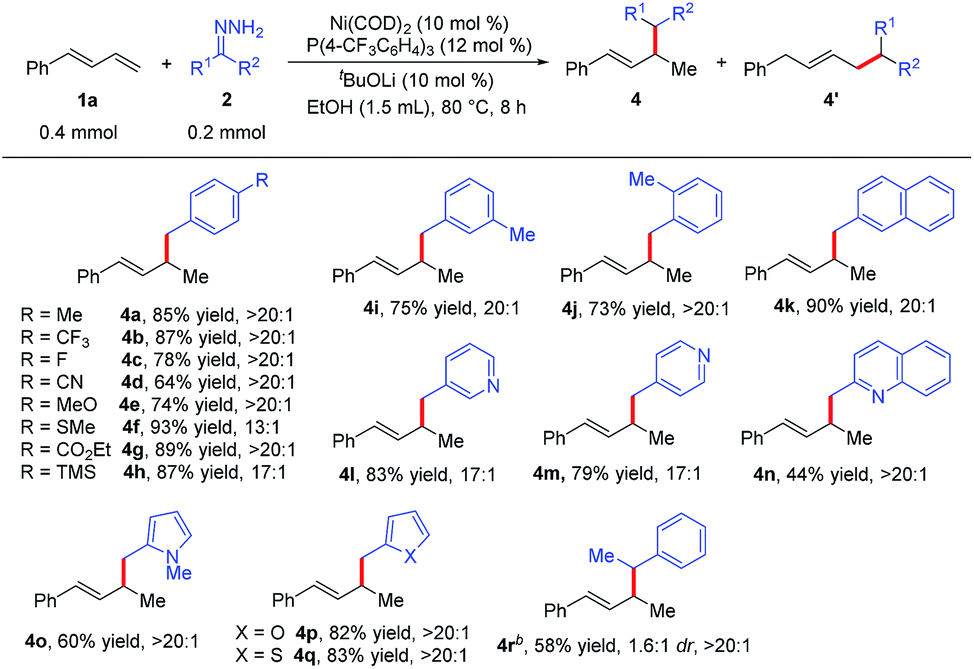
We next examined the scope of the reaction with respect to the hydrazone substrate by carrying out reactions of various hydrazones 2 with phenylbutadiene (1a) (Table 3). Many substituted benzaldehyde hydrazones gave good to high yields, and excellent functional group compatibility was observed, demonstrating the benefit of using hydrazones as nucleophiles in this hydroalkylation reaction. For example, hydrazone 2g, which has an ester group that would not be tolerated in reactions with organometallic reagents, gave corresponding product 4g in 89% yield with >20:1 regioselectivity. Base-sensitive trimethylsilyl-substituted hydrazone 2h did not decompose under the reaction conditions. The remarkable compatibility of the reaction with substrates containing heterocycles (pyridine, quinoline, pyrrole, furan, and thiophene; 2l–2q) showed the potential utility of this protocol for the synthesis of natural products. In addition, the hydrazone derived from aromatic ketone 2r was also a viable substrate, although the reaction conditions had to be modified slightly and the diastereoselectivity was low. Note, however, that no hydroalkylation products were obtained from reactions with hydrazones derived from aliphatic ketones or aldehydes.
| a Reaction conditions: 1a (0.4 mmol), 2 (0.2 mmol), Ni(COD)2 (0.02 mmol), P(4-CF3C6H4)3 (0.024 mmol), tBuOLi (0.02 mmol), EtOH (1.5 mL) at 80 °C for 8 h. Isolated yields. Regioselectivity was the ratio of 4 to 4′, which was determined by 1H NMR analysis of the product. b 1a (0.2 mmol), 2r (0.3 mmol), Ni(COD)2 (0.02 mol), PBn3 (0.024 mmol), tBuOLi (0.02 mmol), EtOH (1.0 mL) at 80 °C for 8 h. |
|---|
|
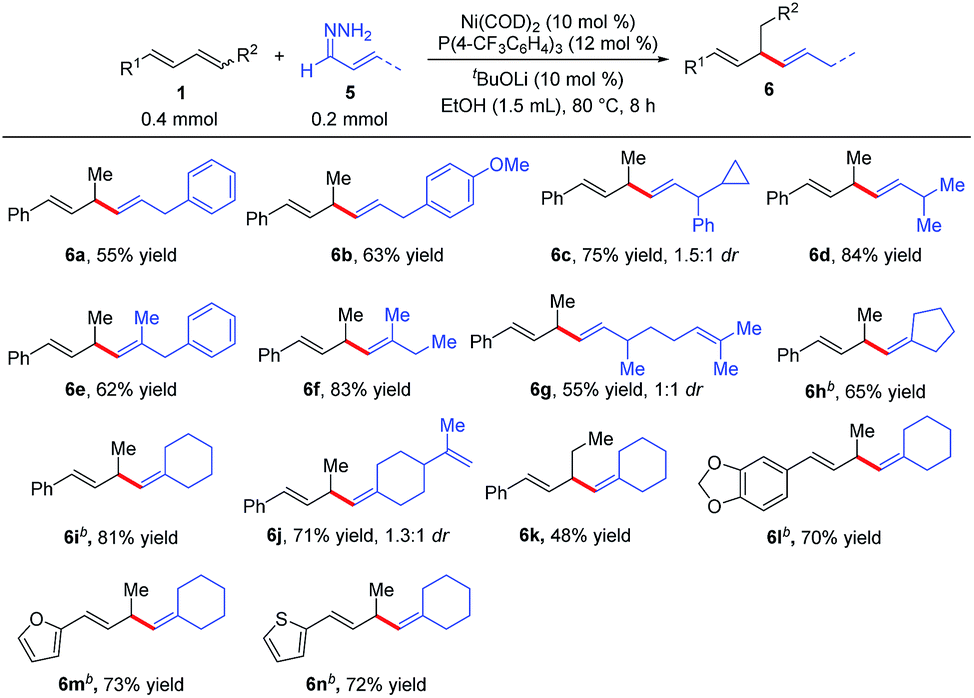
We also explored reactions between 1,3-dienes 1 and α,β-unsaturated hydrazones 5 (Table 4). Surprisingly, a completely different hydroalkenylation product, 1,4-diene 6a, was obtained from the reaction of 3-phenylallylidenehydrazine (5a) with phenylbutadiene (1a); clearly, the double bond of 5a had migrated during the reaction. 1,4-Dienes, which are widely found in natural products and are important building blocks in organic synthesis,9 are usually prepared by transition-metal-catalyzed allylic substitution reactions with stoichiometric amounts of alkenylmetal reagents.10 The reaction between 1a and 5a avoids the preparation of alkenylmetal reagents and can occur under mild conditions, thus providing an effective method for the construction of 1,4-dienes.11 As shown in Table 4, a variety of substituted 1,4-dienes were synthesized by reactions of 1,3-dienes with hydrazones. α,β-Unsaturated hydrazones derived from cinnamaldehyde (6a–6c and 6e), acyclic enals (6d, 6f, and 6g), and cyclic enals (6h–6n) were amenable to this protocol, particularly noteworthy are the reactions of the natural-product-derived enals citral (6g) and perillaldehyde (6j). In addition, the internal diene 6k, and the heteroaromatic dienes 6m and 6n also worked well, giving 1,4-diene products.
| a Reaction conditions: 1a (0.4 mmol), α,β-unsaturated hydrazones 4 (0.2 mmol), Ni(COD)2 (0.02 mmol), P(4-CF3C6H4)3 (0.024 mmol), tBuOLi (0.02 mmol), EtOH (1.5 mL) at 80 °C for 8 h. Isolated yields. Regioselectivity was determined by 1H NMR analysis. b 1a (0.2 mmol), α,β-unsaturated hydrazones 4 (0.4 mmol). |
|---|
|
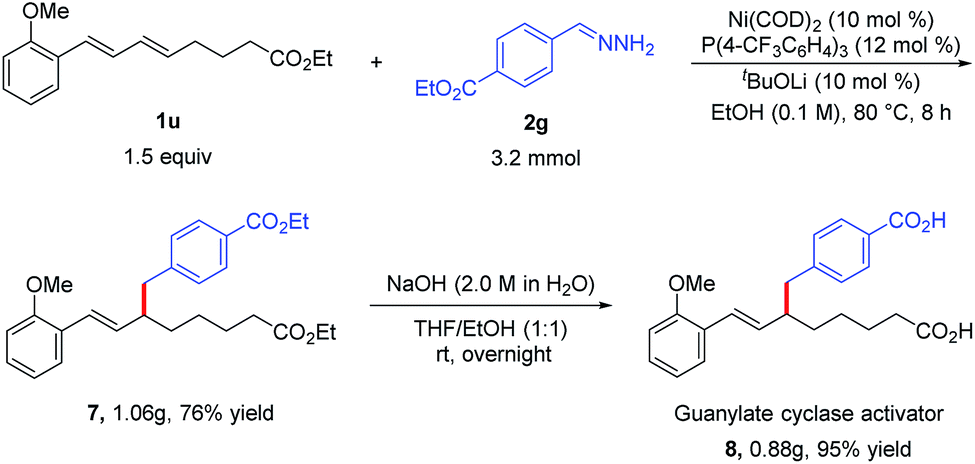
To demonstrate the utility of the protocol, we synthesized dicarboxylic acid 8, a soluble guanylate cyclase activator that is used to treat cardiovascular diseases.12 Specifically, the reaction of diene 1u and hydrazone 2g under the standard conditions afforded hydroalkylation product 7 (76%), which could be hydrolyzed to afford desired dicarboxylic acid 8 in total 72% yield (Scheme 2). This two-step Ni-catalyzed hydroalkylation process represents a considerable improvement over the previously reported synthesis of 8, which requires multiple steps.12a
 | ||
| Scheme 2 Synthesis of guanylate cyclase activator 8. | ||
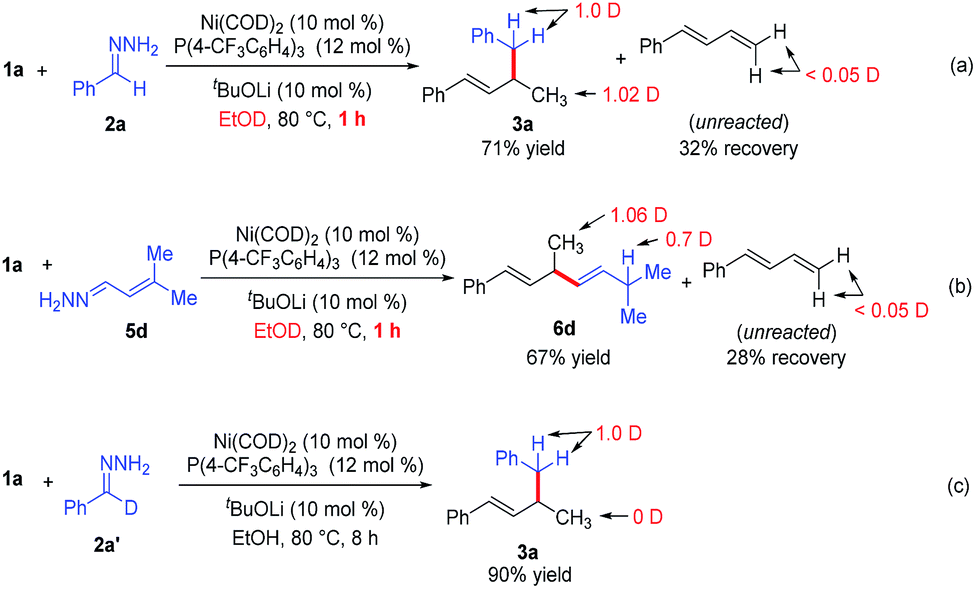
To gain insight into the reaction mechanism, we conducted deuterium-labeling experiments in EtOD (Scheme 3a–3c). The hydroalkylation and hydroalkenylation reactions were carried out in EtOD and stop the reactions at 1 hour. In the hydroalkylation (Scheme 3a), one deuterium is incorporated into the methyl group, one deuterium into the benzyl position of product 3a. In the hydroalkenylation (Scheme 3b), one deuterium is incorporated into the methyl group, and 0.7 deuterium into the newly formed allylic position of product 6d. Notably, the unreacted diene 1a only contains <0.05 deuterium in both reactions, which implies that the addition of Ni–H bond into the diene is irreversible in the reaction. Finally, when deuterated hydrazone 2a′ was allowed to react with phenyldiene 1a, deuterium was observed only at the benzyl position of product 3a (Scheme 3c), which means that the reaction did not involve cleavage of the C–H bond at the benzyl position of hydrazone.13
 | ||
| Scheme 3 Deuterium-labeling experiments. | ||
On the basis of the aforementioned experimental results and previous reports,6–8 we propose the mechanism shown in Scheme 4. First, oxidative addition of EtOH to Ni(0) generates Ni–H intermediate A, which adds to diene 1a to afford π-allylnickel intermediate B. A ligand exchange reaction between B and the diazene anion generated from hydrazone 2a forms intermediate C. In the presence of base in the protic medium, expulsion of nitrogen and protonation of the intermediate C occur to provide intermediate D.14 A reductive elimination reaction of D delivers hydroalkylation product 3a and regenerates the Ni(0) catalyst.15 Similar steps are involved in the hydroalkenylation reaction. Specifically, reaction of the diazene anion generated from α,β-unsaturated hydrazone 5d with π-allylnickel intermediate B forms intermediate C′. Expulsion of nitrogen from C′via a 1,5-sigmatropic migration gives alkenyl-Ni intermediate D′,14d,15 which undergoes a reductive elimination reaction to form hydroalkenylation product 6d.
 | ||
| Scheme 4 A proposed mechanism. | ||
Conclusions
In summary, we have developed a protocol for nickel-catalyzed hydroalkylation reactions of dienes with hydrazones, and this protocol provides a new method for hydroalkylation reactions using hydrazones as alkyl carbon nucleophiles. In addition, we developed a protocol for hydroalkenylation of dienes with α,β-unsaturated hydrazones, providing a new synthetic route to 1,4-dienes. These hydrofunctionalization reactions feature mild conditions and a wide substrate scope, and the products are versatile building blocks for organic synthesis, as demonstrated by the preparation of soluble guanylate cyclase activator 8. Works on the development of asymmetric version of the reactions and exploration of their mechanisms are currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China, (No. 21790332, 21532003) and the “111” project (B06005) of the Ministry of Education of China for financial support.Notes and references
- For selected reviews on 1,3-dienes as building blocks, see: (a) S. Reymond and J. Cossy, Chem. Rev., 2008, 108, 5359 CrossRef CAS PubMed; (b) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Rev., 2015, 115, 5301 CrossRef CAS PubMed; (c) M. Büschleb, S. Dorich, S. Hanessian, D. Tao, K. B. Schenthal and L. E. Overman, Angew. Chem., Int. Ed., 2016, 55, 4156 CrossRef PubMed; (d) M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026 CrossRef CAS PubMed; (e) Y. Xiong, Y.-W. Sun and G.-Z. Zhang, Tetrahedron Lett., 2018, 59, 347 CrossRef CAS.
- For reviews on polymerization of 1,3-dienes, see: (a) L. Friebe, O. Nuyken and W. Obrecht, Adv. Polym. Sci., 2006, 204, 1 CrossRef CAS; (b) G. Ricci, A. Sommazzi, F. Masi, M. Ricci, A. Boglia and G. Leone, Coord. Chem. Rev., 2010, 254, 661 CrossRef CAS; (c) A. Valente, A. Mortreux, M. Visseaux and P. Zinck, Chem. Rev., 2013, 113, 3836 CrossRef CAS PubMed; (d) D. Takeuchi, Stereoselective polymerization of conjugated dienes, in Encyclopedia of Polymer Science and Technology, Wiley, New York, 2013, pp. 1–25 Search PubMed.
- Reviews on hydrofunctionalizations of dienes: (a) L.-B. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596 CrossRef CAS PubMed; (b) E. McNeill and T. Ritter, Acc. Chem. Res., 2015, 48, 2330 CrossRef CAS PubMed; (c) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333 CrossRef CAS PubMed; (d) W.-Y. Ai, R. Zhong, X.-F. Liu and Q. Liu, Chem. Rev., 2019, 119, 2876 CrossRef CAS PubMed; (e) D. Wei and C. Darcel, Chem. Rev., 2019, 119, 2550 CrossRef CAS PubMed.
- Applications of allylic substitution reaction: (a) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed; (b) B. M. Trost, J. Org. Chem., 2004, 69, 5813 CrossRef CAS PubMed; (c) B. M. Trost and J. E. Schultz, Synthesis, 2019, 51, 1 CrossRef CAS; (d) Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855 CrossRef CAS PubMed.
- For selected hydroalkylations of diene, see: (a) G. Hata, K. Takahashi and A. Miyake, J. Org. Chem., 1971, 36, 2116 CrossRef CAS; (b) A. Leitner, J. Larsen, C. Steffens and J. F. Hartwig, J. Org. Chem., 2004, 69, 7552 CrossRef CAS PubMed; (c) M. J. Goldfogel and S. J. Meek, Chem. Sci., 2016, 7, 4079 RSC; (d) M. J. Goldfogel, C. C. Roberts, R. S. Manan and S. J. Meek, Org. Lett., 2017, 19, 90 CrossRef CAS PubMed; (e) X.-H. Yang and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1774 CrossRef CAS PubMed; (f) N. J. Adamson, K. C. E. Wilbur and S. J. Malcolmson, J. Am. Chem. Soc., 2018, 140, 2761 CrossRef CAS PubMed; (g) S. Park, N. J. Adamson and S. J. Malcolmson, Chem. Sci., 2019, 10, 5176 RSC.
- L. Cheng, M.-M. Li, L.-J. Xiao, J.-H. Xie and Q.-L. Zhou, J. Am. Chem. Soc., 2018, 140, 11627 CrossRef CAS PubMed.
- (a) L.-J. Xiao, L. Cheng, W.-M. Feng, M.-L. Li, J.-H. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2018, 57, 461 CrossRef CAS PubMed; (b) L.-J. Xiao, M.-C. Ye and Q.-L. Zhou, Synlett, 2019, 30, 361 CrossRef CAS.
- (a) H. Wang, X.-J. Dai and C.-J. Li, Nat. Chem., 2017, 9, 374 CrossRef CAS PubMed; (b) X.-J. Dai, H. Wang and C.-J. Li, Angew. Chem., Int. Ed., 2017, 56, 6302 CrossRef CAS PubMed; (c) N. Chen, X.-J. Dai, H. Wang and C.-J. Li, Angew. Chem., Int. Ed., 2017, 56, 6260 CrossRef CAS PubMed; (d) L. Lv, D. Zhu, J. Tang, Z. Qiu, C.-C. Li, J. Gao and C.-J. Li, ACS Catal., 2018, 8, 4622 CrossRef CAS; (e) D. Zhu, L. Lv, C.-C. Li, S. Ung, J. Gao and C.-J. Li, Angew. Chem., Int. Ed., 2018, 57, 16520 CrossRef CAS PubMed.
- (a) M. S. F. L. K. Jie, M. K. Pasha and M. S. K. Syed-Rahmatullah, Nat. Prod. Rep., 1997, 14, 163 RSC; (b) Y. Shinohara, F. Kudo and T. Eguchit, J. Am. Chem. Soc., 2011, 133, 18134 CrossRef CAS PubMed; (c) P. Winter, W. Hiller and M. Christmann, Angew. Chem., Int. Ed., 2012, 51, 3396 CrossRef CAS PubMed; (d) W. Tang and E. V. Prusov, Angew. Chem., Int. Ed., 2012, 51, 3401 CrossRef CAS PubMed.
- (a) N. Miyaura, T. Yano and A. Suzuki, Tetrahedron Lett., 1980, 21, 2865 CrossRef CAS; (b) H. Matsushita and E. Negishi, J. Am. Chem. Soc., 1981, 103, 2882 CrossRef CAS; (c) F. K. Sheffy and J. K. Stille, J. Am. Chem. Soc., 1983, 105, 7173 CrossRef CAS; (d) Y. Lee, K. Akiyama, D. G. Gillingham, M. K. Brown and A. H. Hoveyda, J. Am. Chem. Soc., 2008, 130, 446 CrossRef CAS PubMed; (e) K. Akiyama, F. Gao and A. H. Hoveyda, Angew. Chem., Int. Ed., 2010, 49, 419 CrossRef CAS PubMed; (f) J. Y. Hamilton, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2013, 135, 994 CrossRef CAS PubMed.
- For reviews of hydroalkenylation of 1,3-dienes: (a) T. V. RajanBabu, Chem. Rev., 2003, 103, 2845 CrossRef CAS PubMed; (b) T. V. RajanBabu, Synlett, 2009, 853 CrossRef CAS PubMed; (c) G. Hilt, Eur. J. Org. Chem., 2012, 4441 CrossRef CAS.
- (a) C. Alonso-Alija, M. Heil, D. Flubacher, P. Naab, J.-P. Stasch, F. Stasch, K. Dembowsky, E. Perzborn and E. Stahl, US Pat., US 20080058314 A1, Bayer Aktiengesellschaft, USA, 2008; (b) M. Follmann, N. Griebenow, M. G. Hahn, I. Hartung, F. J. Mais, J. Mittendorf, M. Schaefer, H. Schirok, J. P. Stasch, F. Stoll and A. Stoll, Angew. Chem., Int. Ed., 2013, 52, 9442 CrossRef CAS PubMed.
- See ESI† for more details about the mechanism studies.
- (a) H. H. Szmant, Angew. Chem., Int. Ed., 1968, 7, 120 CrossRef CAS; (b) H. H. Szmant and C. E. Alciaturi, J. Org. Chem., 1977, 42, 1081 CrossRef CAS; (c) H. H. Szmant, A. Birke and M. P. Lau, J. Am. Chem. Soc., 1977, 99, 1863 CrossRef CAS; (d) R. O. Hutchins, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 8, pp. 327–362 Search PubMed.
- The isomerization of double bond commonly occurs in reduction of α,β-unsaturated hydrazones : (a) R. A. Sneen and N. P. Matheny, J. Am. Chem. Soc., 1964, 86, 5503 CrossRef CAS; (b) E. J. Taylor and C. Djerassi, J. Am. Chem. Soc., 1976, 98, 2275 CrossRef CAS; (c) R. O. Hutchins and N. R. Natale, J. Org. Chem., 1978, 43, 2299 CrossRef CAS; (d) Ref. 14d .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04177j |
| This journal is © The Royal Society of Chemistry 2019 |