
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Programming permanent and transient molecular protection via mechanical stoppering†
Miguel A.
Soto
 a,
Francesco
Lelj
b and
Mark J.
MacLachlan
*acd
a,
Francesco
Lelj
b and
Mark J.
MacLachlan
*acd
aDepartment of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, BC, V6T 1Z1 Canada. E-mail: mmaclach@chem.ubc.ca
bLa.M.I. and LaSCAMM INSTM Sezione Basilicata, Dipartimento di Chimica, Università della Basilicata, via dell'Ateneo Lucano 10, Potenza, 85100 Italy
cQuantum Matter Institute, University of British Columbia, 2355 East Mall, Vancouver, BC, V6T 1Z4 Canada
dWPI Nano Life Science Institute, Kanazawa University, Kanazawa, 920-1192 Japan
First published on 4th October 2019
Abstract
Chemical protection is an essential tool in synthetic chemistry, which involves blocking reactive sites on a molecule through covalent bonds. Physical approaches, such as encapsulation and host-mediated protection, have emerged as interesting alternatives that use steric bulk to inhibit reactivity. Here, we report the protection of a redox-active viologen through its incorporation into mechanically interlocked molecules (MIMs), namely hetero[4]rotaxanes. The viologen was confined inside a host cavity and flanked by two mechanical stoppers, which allowed for permanent and transient protection. Deprotection occurred on-demand via an unstoppering process, triggered by a proton transfer, polarity effect, or a thermal stimulus. We anticipate that permanent and transient mechanical stoppering could be incorporated into devices to function as molecular probes, transport/delivery systems, or stimuli-controlled degradable materials.
Introduction
Since the introduction of the mechanical bond, numerous mechanically interlocked molecules (MIMs) have been synthesized. [2]Rotaxanes and [2]catenanes are the “simplest” and most documented MIMs,1–5 though many other exotic species have been accessed, e.g. hetero[n]rotaxanes,6–9 rotacatenanes,10,11 knot-capped rotaxanes,12 and main-chain poly[n]catenanes.13 Currently, research on MIMs involves the synthesis of even more intricate assemblies,14 while also focusing on the operation of correlated dynamic processes,15 and the quest for function, i.e., MIMs performing as catalysts,16 sensors,17,18 drug carriers,19 molecular electronics,20 and machines.21One intriguing approach to achieve function with MIMs is the concept of mechanical protection,22i.e. the shielding of an environment-susceptible substrate by its confinement in the interior of a macrocycle. In this methodology the physical and chemical properties of a substrate (e.g. melting point, solubility, reactivity or degradation) are dramatically altered, so that it no longer behaves as unprotected.23 Despite the attractiveness of this approach for synthetic chemistry, only a few MIMs have been prepared with the intention of using mechanical protection.24–36 Typically in these examples, the substrate (or a modified form of it) acts as a recognition site for the protecting macrocycle, then the implementation of an appropriate protocol (e.g. stoppering or clipping) generates a MIM where the macrocycle is secured and provides permanent protection to the substrate. In some specific systems, the protection can be reversed through the degradation of one of the MIM components, which requires a specific input such as an organometallic catalyst22 or an enzyme.37
Overall, both protection approaches, permanent and transient, are equally relevant. A permanent effect is appropriate to preserve or enhance the properties and function of a substrate during a process,38,39 whereas transient protection permits the controlled release of the active substrate once the adverse environment has been evaded.37 Here, we present a strategy to protect a molecule both temporarily and permanently by using bespoke stoppering on MIMs. To pursue our concept, we designed two hetero[4]rotaxanes that are depicted in Fig. 1a. These MIMs contain three macrocycles threaded onto a linear species; the central macrocycle functions as a protective unit while the outer macrocycles prevent it from escaping. The outer rings and the thread end-groups act in tandem as mechanical stoppers40 to preserve a protected environment around the substrate.
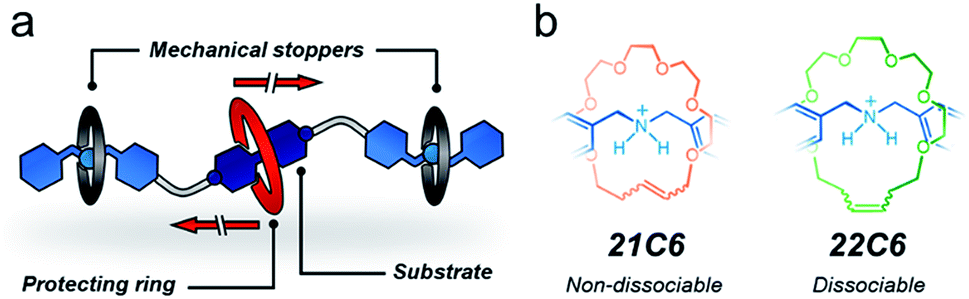 | ||
| Fig. 1 Schematic representation of (a) a hetero[4]rotaxane structure, and (b) permanently interlocked (left) and metastable (right) rotaxanes constructed from a dibenzylammonium unit and crown ether rings. | ||
To program both permanent and transient protection, we relied on positioning metastable and permanently interlocked species as mechanical stoppers. Dasgupta and Wu recently demonstrated that the dibenzylammonium cation can be encircled by [21] and [22]-membered crown ethers (21C6, 22C6) to yield [2]rotaxanes using the clipping method.41 The one-atom difference in the macrocyclic structures (21 vs. 22) strongly affects the stability of the MIMs (Fig. 1b). A [2]rotaxane containing 21C6 is permanently interlocked, so it does not disassemble without the cleavage of a covalent bond. Conversely, the 22C6-containing species is isolable but prone to disassemble upon chemical or physical stimulation. Based on these previous observations, we selected the macrocycles 21C6 and 22C6, and the dibenzylammonium motif for the fabrication of two different mechanical stoppers: a dissociable set and a non-dissociable counterpart.
We chose the 4,4′-bipyridinium unit, a viologen, as the substrate to protect as these redox-active cations are readily accessible, undergo chemical reduction with a macroscopic readout (color change), and can assemble with several classes of macrocycles, such as pillar[n]arenes, cucurbit[n]urils and crown ethers.42 Considering its tight fit with the viologen derivatives and its relative size and aspect ratio, with respect to 21C6 and 22C6 (see Fig. S1†),43 we selected the crown ether 1,5-dinaphtho[32]crown-8 (DN32C8) as the protective unit.
Results and discussion
With the targeted components identified, we synthesized [1·H2]4+ (Scheme 1), a linear tetracation composed of a 4,4′-bipyridinium core that is spaced from two dibenzylammonium moieties by oxybutylene chains. [1·H2][PF6]4 was prepared in four steps, in 80% overall yield, and characterized by HRMS, NMR, and UV-vis spectroscopy (see ESI†). As with other viologen derivatives, [1·H2]4+ undergoes a one-electron reduction when zinc dust is added to a solution of [1·H2]4+.44,45 We found that this process is fast (<5 min) and leads to a visible color change. Reduction of [1·H2]4+ in acetonitrile (pale yellow solution) generates the blue radical cation [1·H2](3+)˙ that features structured bands in the UV-vis spectrum, centered at 375 and 575 nm (Fig. S2†).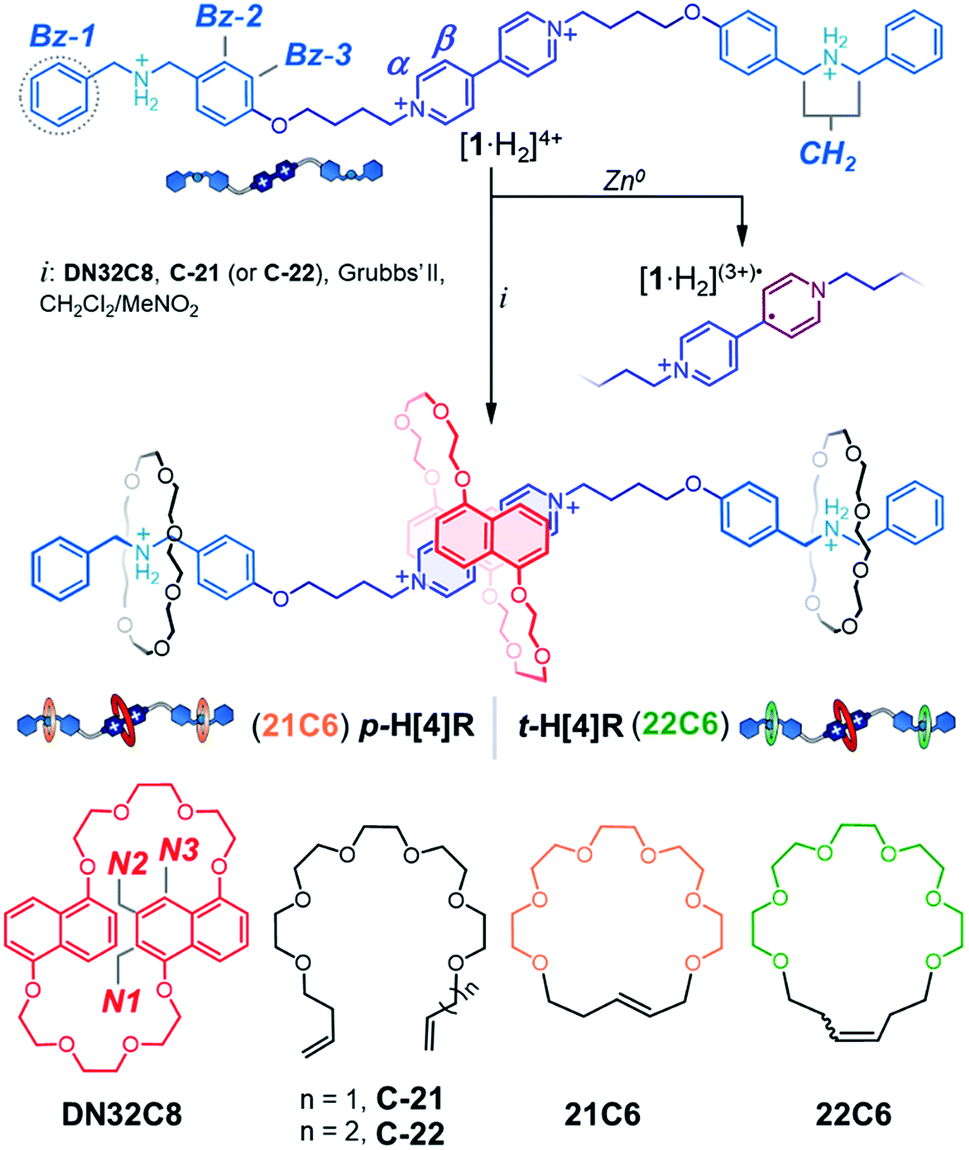 | ||
| Scheme 1 Syntheses of the two designed hetero[4]rotaxane structures and a thread-like radical cation. Chemical structures and proton assignments for the macrocyclic structures and thread unit are also shown. Note that the radical cation is delocalized. | ||
To protect [1·H2]4+ from reduction, we first investigated the self-assembly of [1·H2]4+ with the protecting ring DN32C8, and tethers C-21 and C-22. The addition of one equiv of DN32C8 to an acetonitrile solution of [1·H2][PF6]4 causes a rapid color change from pale yellow to red. This color change is ascribed to the formation of a charge-transfer (CT) complex between the electron-rich DN32C8 and the electron-poor core of [1·H2]4+; this complex produces an absorption band at 475 nm in the UV-vis spectrum. Furthermore the 1H NMR spectrum showed an upfield shift for the α and β resonances of [1·H2]4+ upon mixing with DN32C8 (Δδα = 0.1 ppm and Δδβ = 0.3 ppm), but the dibenzylammonium signals were unaffected (Fig. S3†). These observations suggest that the macrocycle DN32C8 spontaneously threads onto [1·H2]4+ and sits on the bipyridinium core to yield the pseudorotaxane [1·H2⊂DN32C8]4+ [ΔGasso = −(12.8 ± 0.2) kJ mol−1, CD3CN, 25 °C], see Table S1.† These spectroscopic data and complex stability resemble those of other reported [2]pseudorotaxane complexes composed of bipyridinium–DN32C8 pairs.46
Addition of the precursor C-21 or C-22 to a solution containing [1·H2⊂DN32C8]4+ did not interfere with pseudorotaxane formation. Indeed, all components self-sort: DN32C8 encircles the bipyridinium motif, while the glycolic tethers (C-21 or C-22) solely wrap the ammonium stations. This was confirmed by the observation of a downfield shift on the +NH2 resonance (Δδ+NH2 = 0.2 ppm) in the 1H NMR spectrum (Fig. S5†). Addition of second-generation Grubbs' catalyst to any of the mixtures containing DN32C8, [1·H2]4+, and C-21 (or C-22), followed by heating, led to ring-closure of the glycolic tethers, securing the DN32C8 ring. This reaction yielded the transiently (t) and permanently (p) protected hetero[4]rotaxanes t-H[4]R and p-H[4]R in 30% and 37% yield, respectively (see ESI†).47Fig. 2 shows a representative 1H NMR spectrum for one of the isolated species, rotaxane p-H[4]R (t-H[4]R data are in Fig. S6†).
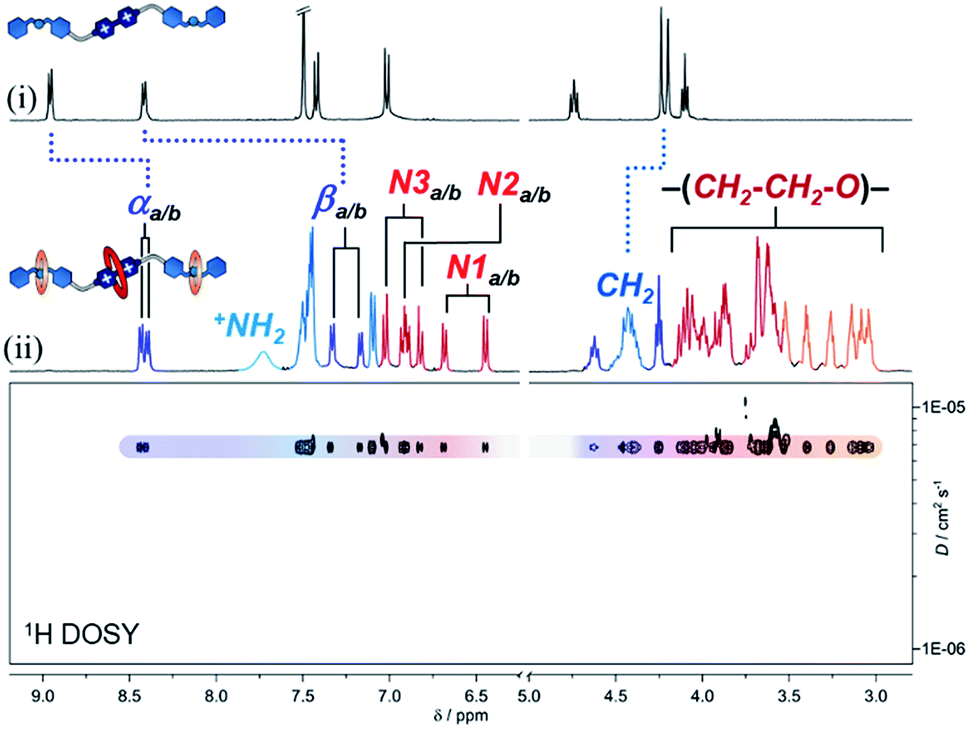 | ||
| Fig. 2 1H NMR spectra (400 MHz, CD3CN) of (i) compound [1·H2][PF6]4 and (ii) hetero[4]rotaxane p-H[4]R. 1H DOSY NMR spectrum is shown in the bottom portion. | ||
Using 2D NMR spectroscopy (Fig. S7–S9†), we identified four principal regions in the 1H NMR spectrum: (i) bipyridinium resonances at 8.43/8.40 ppm (αa/b) and 7.3/7.2 ppm (βa/b); (ii) naphthalene groups at 7.0/6.8 ppm (N1a/b), 6.9 ppm (N2a/b), and 6.7/6.5 ppm (N3a/b); (iii) glycol chains (for the three crown ether rings) at 3.0–4.1 ppm; and (iv) dibenzylammonium signals at 7.7 (+NH2), 7.5–7.1 (Bz1–3) and 4.4 ppm (CH2). Integration of the peaks in the 1H NMR data confirmed the stoichiometry of the assembled MIM to be one thread, two 21C6 rings, and one DN32C8 crown ether. This was supported by HRMS, where the parent ion [1 + DN32C8 + (2 × 21C6) + 2H]4+ was detected at m/z = 462.7604. Furthermore, we proved by diffusion-ordered NMR spectroscopy (DOSY, Fig. 2) that all observed resonances in the 1H data correspond to a single species that diffuses in solution at ca. 6.8 × 10−6 cm2 s−1 (CD3CN, 25 °C), i.e. rotaxane p-H[4]R.
The 1H NMR spectrum of p-H[4]R shows two major differences from that of pure [1·H2][PF6]4. First, the bipyridinium resonances α and β show an upfield shift 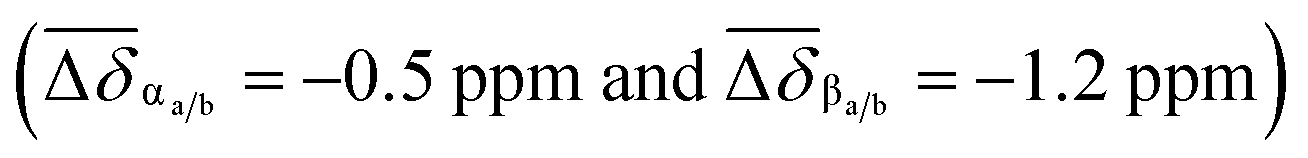 , which is attributed to the confinement of the substrate inside the DN32C8 cavity that leads to shielding from the naphthalene rings. Second, the dibenzylammonium protons (+NH2, Bz, and CH2) moved downfield (Δδ+NH2 = 0.3 ppm) because of hydrogen bonding with the 21C6 rings. The relative position of the [21] and [32]-membered macrocycles, with respect to the thread stations, was supported by NOESY NMR spectroscopy, where through-space couplings were identified for each individual host–guest pair, i.e. dibenzylammonium–21C6 and bipyridinium–DN32C8 (Fig. S8†).
, which is attributed to the confinement of the substrate inside the DN32C8 cavity that leads to shielding from the naphthalene rings. Second, the dibenzylammonium protons (+NH2, Bz, and CH2) moved downfield (Δδ+NH2 = 0.3 ppm) because of hydrogen bonding with the 21C6 rings. The relative position of the [21] and [32]-membered macrocycles, with respect to the thread stations, was supported by NOESY NMR spectroscopy, where through-space couplings were identified for each individual host–guest pair, i.e. dibenzylammonium–21C6 and bipyridinium–DN32C8 (Fig. S8†).
Interestingly, the set of aromatic signals for the bipyridinium–DN32C8 pair splits into two groups (a and b) in the 1H NMR spectrum. According to a 2D EXSY experiment (Fig. S9†), both a and b sets are exchanging at room temperature, indicating the appearance of two isomers that presumably interconvert by an anti/syn isomerization of DN32C8 (Scheme S4†). A [3]rotaxane that does not contain this macrocycle features a single set of resonances in the NMR spectrum (see Fig. S10†), thus confirming that both isomers of p-H[4]R emerge as a consequence of the dynamics on DN32C8.48
After characterizing the hetero[4]rotaxane molecules, we interrogated them against chemical reduction (Fig. 3a). Separate solutions of p-H[4]R and t-H[4]R in CH3CN were loaded with zinc dust and stirred under a N2 atmosphere for one day. We did not observe any visible change. UV-vis spectroscopy showed that both solutions preserved their characteristic CT absorption band, at 475 nm, and the radical cation [1·H2](3+)˙ was not detected (Fig. S11†), confirming that DN32C8 effectively protects the bipyridinium substrate in p-H[4]R and t-H[4]R. The lack of reactivity of the viologen within the rotaxanes is in stark contrast to [1·H2]4+, which reacts rapidly (<5 min) with zinc metal under the same conditions.
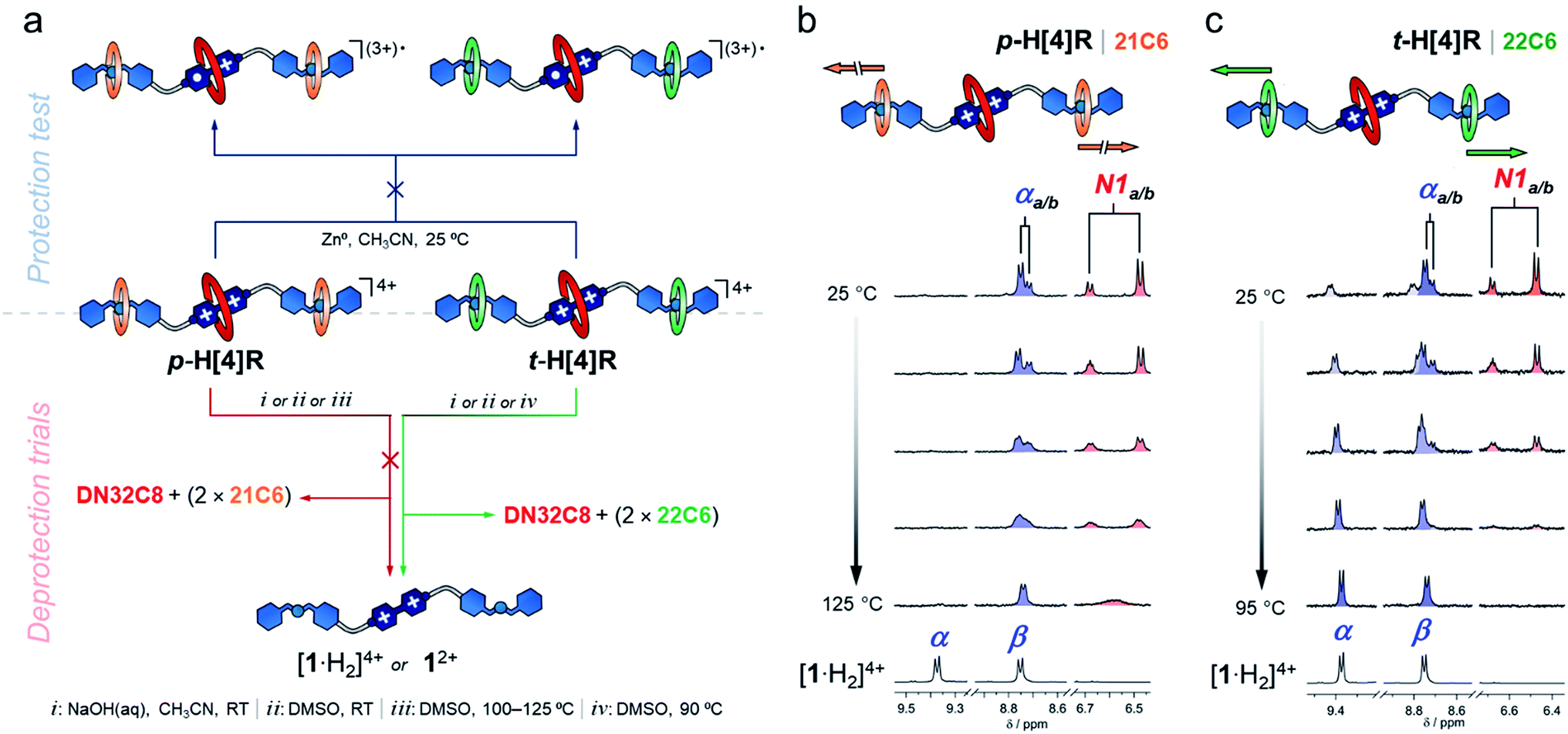 | ||
| Fig. 3 (a) A scheme showing the tests performed on p-H[4]R and t-H[4]R: deprotection attempt (top) and controlled unstoppering (bottom). (b and c) Partial 1H VT-NMR spectra (400 MHz, DMSO-d6) of (b) p-H[4]R and (c) t-H[4]R. | ||
Next, we attempted to deprotect both hetero[4]rotaxanes using controlled stimuli. The deprotection step should occur via disassembly of (at least) one of the mechanical stoppers by the disruption of the intercomponent hydrogen bonding. To disable these interactions, we tried three approaches: (i) deprotonation of the ammonium groups; (ii) use of a high polarity solvent; and (iii) application of a thermal stimulus (Fig. 3a). These triggers were first tested on rotaxane p-H[4]R and then on t-H[4]R.
An CD3CN solution of p-H[4]R was treated with one equiv of base (1 M NaOH(aq)) at room temperature. According to the recorded NMR experiment (Fig. S12†), the addition of base did not cause proton transfer; the +NH2 resonance was still observed at 7.7 ppm and other p-H[4]R signals were unaffected. Furthermore, the molecular ion [1 + DN32C8 + (2 × 21C6) + 2H]4+ was identified by HRMS (m/z = 463.0509) while none of the individual components (1, DN32C8 or 21C6) were detected. Thus, deprotonation of p-H[4]R is inaccessible, probably because of the tight fit between the dibenzylammonium unit and the 21C6 macrocycle. Switching from CH3CN to DMSO (a highly competitive solvent) did not produce a different result, and only a change of the anti/syn-DN32C8 isomers ratio was observed, from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in CD3CN to 2:1 in DMSO-d6. Disassembly of p-H[4]R was not achieved even after several weeks at room temperature in DMSO (see Fig. S14†).
:1 in CD3CN to 2:1 in DMSO-d6. Disassembly of p-H[4]R was not achieved even after several weeks at room temperature in DMSO (see Fig. S14†).
To create a more severe environment, we heated a solution of p-H[4]R in DMSO-d6 from 25 to 125 °C and analyzed it by 1H NMR spectroscopy. Fig. 3b shows the partial aromatic region of the collected spectra (full data in Fig. S15†). Below ∼100 °C, both p-H[4]R isomers were clearly identified; holding T at 100 °C for one hour did not damage p-H[4]R. Elevating the temperature above 115 °C only caused coalescence of the resonances assigned to the isomers, indicating that their interconversion becomes faster at higher temperatures. We calculated an exchange rate of 110 s−1 at the coalescence temperature (see ESI†). Furthermore, a p-H[4]R rotaxane solution prepared in DMSO, and loaded with zinc dust, did not degrade after storage for six months under a N2 atmosphere (Fig. S19†). Together, our observations suggest that p-H[4]R is permanently stoppered and therefore the bipyridinium substrate is permanently protected. This protection effect is ascribed to (1) the steric shield provided by DN32C8, and (2) the electronic stabilization given by this macrocycle to the viologen substrate; cyclic voltammetry experiments (Fig. S20†) revealed a stabilization of ∼13 kJ mol−1 with respect to the unprotected species [1·H2]4+. Noteworthy, there is only one way to deprotect p-H[4]R: by breaking covalent bonds. Rotaxane p-H[4]R was fully disassembled by controlled ring-opening of the 21C6 macrocycles, which ultimately released the protecting unit (Fig. S21†).
In contrast, we expected that incorporating 22C6 rings into the hetero[4]rotaxane structure would allow transient protection, and breakage of covalent bonds would not be required. We therefore applied the same tested stimuli to t-H[4]R. Addition of one equivalent of base (NaOH(aq)) to a CH3CN solution of t-H[4]R produced an immediate change in the color of the solution from red to pale yellow, and the characteristic CT band in the UV-vis spectrum disappeared. This indicated that the proton transfer process occurred rapidly and caused two subsequent events: unstoppering followed by the release of the protecting ring. Although the process was too fast (<1 min) to detect the deprotonated form of t-H[4]R by 1H NMR spectroscopy, we observed the free thread 12+ and macrocycles 21C6 and DN32C8 in solution by both NMR spectroscopy and HRMS (Fig. S22–S23†).
Dissolving t-H[4]R in a polar solvent also led to rapid unstoppering. After dissolving t-H[4]R in DMSO-d6 at 25 °C, we observed three new sets of resonances in the 1H NMR spectrum (Fig. 3c) that correspond to the free components 22C6, DN32C8 and [1·H2]4+. Integration of the 1H NMR data suggests that 20% of the complex was rapidly disassembled. Heating the sample up to 95 °C led to full unstoppering, so that the rotaxane t-H[4]R resonances were no longer evident, while the bipyridinium signals at 9.4 (α) and 8.7 (β) ppm reached maximum intensity. As well, the free species DN32C8, [1·H2]4+ and 22C6, were all found in solution by HRMS (see Fig. S25†).
After we proved that t-H[4]R undergoes controlled unstoppering, we attempted to use this deprotection as a means to control the reactivity of the viologen. A solution of t-H[4]R in CH3CN was loaded with zinc dust, sparged with N2, and left to settle for one hour. Within this period, we did not observe any change, neither macroscopic nor spectroscopic. This implies that the DN32C8 unit stays in place protecting the viologen, an effect that cannot be achieved in systems that are under dynamic assembly/disassembly.45 Conversely, upon addition of an alkaline solution (1 M NaOH(aq), 1.0 equiv.) the mixture rapidly changed color from red to blue. We ascribe this to the unstoppering/deprotection/reduction sequence that gives rise to [1·H2](3+)˙ in solution (Fig. S26†) along with residual DN32C8 and 22C6 units.
Heating t-H[4]R in DMSO, in the presence of zinc metal, rendered similar results, although the unstoppering followed by deprotection appeared to be gradual (Fig. 4a). After heating for one minute at 90 °C, we observed a drop in the intensity of the characteristic CT band at 475 nm, and two additional bands assigned to [1·H2](3+)˙ (375 and 575 nm) were present (Fig. 4b). Continuous heating at the same temperature produced a concomitant increase of these bands. Interestingly, the deprotection/reduction process was detected at the macroscopic scale. The pale red solution that contained t-H[4]R gradually transformed into a blue mixture in ca. five minutes at 90 °C (Fig. 4c), due to the presence of [1·H2](3+)˙, i.e. once the unstoppering process was completed.
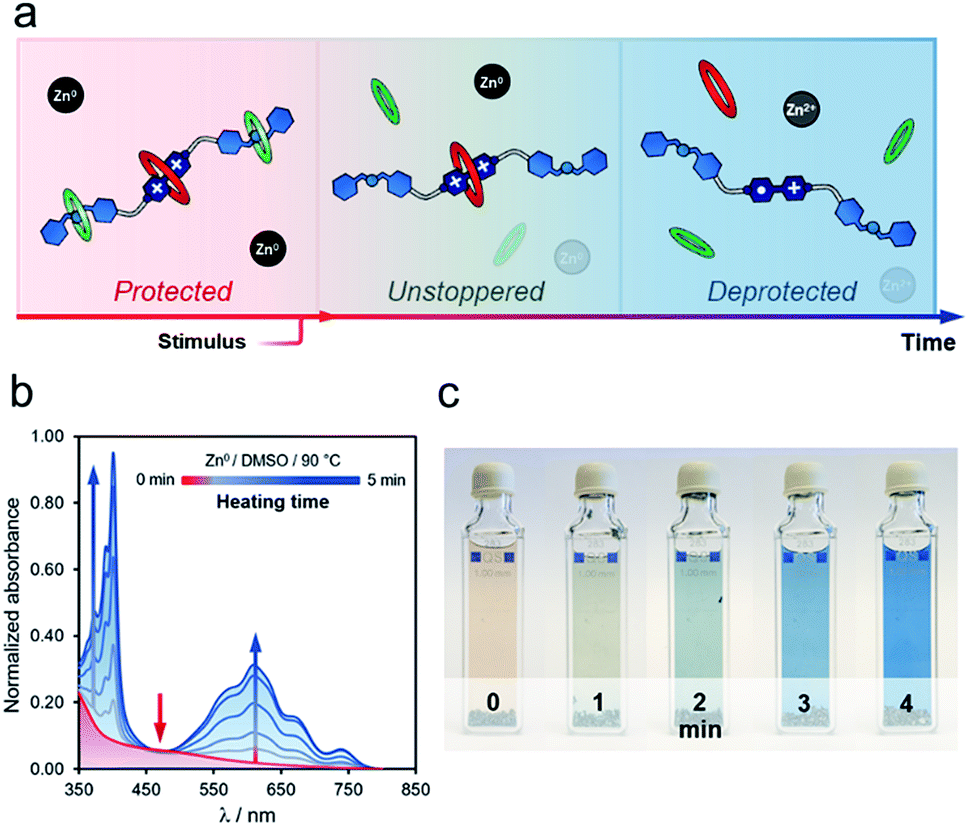 | ||
| Fig. 4 (a) Gradual deprotection/reduction sequence. Experiment conducted on rotaxane t-H[4]R (DMSO, 90 °C) and followed at macroscopic scale and by UV-vis spectroscopy (b and c). | ||
Conclusions
In summary, we have synthesized two hetero[4]rotaxane MIMs that resist chemical reduction. Both molecules bear two key components that, in synergy, make protection viable: (1) a central ring that offers steric bulk and stabilizes the sensitive substrate, and (2) a couple of outer macrocycles that prevent the protecting unit from escaping, i.e. the mechanical stoppers.By selecting between two stoppers, comprising [21] or [22]-membered rings, we programmed permanent and transient protection on p-H[4]R and t-H[4]R, respectively. p-H[4]R does not disassemble even when exposed to severe environments, whereas t-H[4]R dissociates to leave the sensitive substrate exposed, on demand. We consider that mechanical stoppering, as a general concept, could be valuable for the design of, e.g., other protected MIMs bearing relevant substrates, delivery systems, and stimuli-controlled degradable materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
MAS thanks CONACYT for a postdoctoral fellowship. MJM thanks NSERC for Discovery and CREATE grants. The input provided by Dr Charlotte Boott during the preparation of this manuscript is also acknowledged.References
- H. W. Gibson, M. C. Bheda and P. T. Engen, Prog. Polym. Sci., 1994, 19, 843–945 CrossRef CAS.
- C. Dietrich-Buchecker, C. M. Jimenez-Molero, V. Sartor and J. P. Sauvage, Pure Appl. Chem., 2003, 75, 1383–1393 CAS.
- M. J. Langton and P. D. Beer, Acc. Chem. Res., 2014, 47, 1935–1949 CrossRef CAS PubMed.
- E. M. G. Jamieson, F. Modicom and S. M. Goldup, Chem. Soc. Rev., 2018, 47, 5266–5311 RSC.
- X.-Q. Wang, W.-J. Li, W. Wang and H.-B. Yang, Chem. Commun., 2018, 54, 13303–13318 RSC.
- W. Jiang, H. D. F. Winkler and C. A. Schalley, J. Am. Chem. Soc., 2008, 130, 13852–13853 CrossRef CAS PubMed.
- Z.-J. Zhang, H.-Y. Zhang, H. Wang and Y. Liu, Angew. Chem., Int. Ed., 2011, 50, 10834–10838 CrossRef CAS PubMed.
- C. Ke, R. A. Smaldone, T. Kikuchi, H. Li, A. P. Davis and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 381–387 CrossRef CAS.
- S.-J. Rao, Q. Zhang, J. Mei, X.-H. Ye, C. Gao, Q.-C. Wang, D.-H. Qu and H. Tian, Chem. Sci., 2017, 8, 6777–6783 RSC.
- G. Barin, A. Coskun, D. C. Friedman, M. A. Olson, M. T. Colvin, R. Carmielli, S. K. Dey, O. A. Bozdemir, M. R. Wasielewski and J. F. Stoddart, Chem.–Eur. J., 2011, 17, 213–222 CrossRef CAS.
- R. Hayashi, P. Slavík, Y. Mutoh, T. Kasama and S. Saito, J. Org. Chem., 2016, 81, 1175–1184 CrossRef CAS PubMed.
- D. A. Leigh, L. Pirvu, F. Schaufelberger, D. J. Tetlow and L. Zhang, Angew. Chem., Int. Ed., 2018, 57, 10484–10488 CrossRef CAS PubMed.
- Q. Wu, P. M. Rauscher, X. Lang, R. J. Wojtecki, J. J. de Pablo, M. J. A. Hore and S. J. Rowan, Science, 2017, 358, 1434–1439 CrossRef CAS PubMed.
- M. T. Nguyen, D. P. Ferris, C. Pezzato, Y. Wang and J. F. Stoddart, Chem, 2018, 4, 2329–2344 CAS.
- K. Zhu, G. Baggi and S. J. Loeb, Nat. Chem., 2018, 10, 625–630 CrossRef CAS.
- D. A. Leigh, V. Marcos and M. R. Wilson, ACS Catal., 2014, 4, 4490–4497 CrossRef CAS.
- B. R. Mullaney, A. L. Thompson and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11458–11462 CrossRef CAS.
- M. Denis, J. Pancholi, K. Jobe, M. Watkinson and S. M. Goldup, Angew. Chem., Int. Ed., 2018, 57, 5310–5314 CrossRef CAS.
- R. Barat, T. Legigan, I. Tranoy-Opalinski, B. Renoux, E. Péraudeau, J. Clarhaut, P. Poinot, A. E. Fernandes, V. Aucagne, D. A. Leigh and S. Papot, Chem. Sci., 2015, 6, 2608–2613 RSC.
- A. Coskun, J. M. Spruell, G. Barin, W. R. Dichtel, A. H. Flood, Y. Y. Botros and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 4827–4859 RSC.
- G. De Bo, M. A. Y. Gall, S. Kuschel, J. De Winter, P. Gerbaux and D. A. Leigh, Nat. Nanotechnol., 2018, 13, 381–385 CrossRef CAS.
- K. Zhu, G. Baggi, V. N. Vukotic and S. J. Loeb, Chem. Sci., 2017, 8, 3898–3904 RSC.
- E. A. Neal and S. M. Goldup, Chem. Commun., 2014, 50, 5128–5142 RSC.
- A. H. Parham, B. Windisch and F. Vögtle, Eur. J. Org. Chem., 1999, 1999, 1233–1238 CrossRef.
- J. E. H. Buston, J. R. Young and H. L. Anderson, Chem. Commun., 2000, 905–906 RSC.
- M. Franz, J. A. Januszewski, D. Wendinger, C. Neiss, L. D. Movsisyan, F. Hampel, H. L. Anderson, A. Görling and R. R. Tykwinski, Angew. Chem., Int. Ed., 2015, 54, 6645–6649 CrossRef CAS.
- L. D. Movsisyan, M. Franz, F. Hampel, A. L. Thompson, R. R. Tykwinski and H. L. Anderson, J. Am. Chem. Soc., 2016, 138, 1366–1376 CrossRef CAS.
- M. Gauthier and F. Coutrot, Eur. J. Org. Chem., 2019, 21, 3391–3395 CrossRef.
- Y. Akae, Y. Koyama, S. Kuwata and T. Takata, Chem.–Eur. J., 2014, 20, 17132–17136 CrossRef CAS.
- R. Eelkema, K. Maeda, B. Odell and H. L. Anderson, J. Am. Chem. Soc., 2007, 129, 12384–12385 CrossRef CAS PubMed.
- A. Mateo-Alonso, P. Brough and M. Prato, Chem. Commun., 2007, 1412–1414 RSC.
- J. J. Gassensmith, J. M. Baumes and B. D. Smith, Chem. Commun., 2009, 6329–6338 RSC.
- D. M. D'Souza, D. A. Leigh, L. Mottier, K. M. Mullen, F. Paolucci, S. J. Teat and S. Zhang, J. Am. Chem. Soc., 2010, 132, 9465–9470 CrossRef PubMed.
- K. Sugiyasu, Y. Honsho, R. M. Harrison, A. Sato, T. Yasuda, S. Seki and M. Takeuchi, J. Am. Chem. Soc., 2010, 132, 14754–14756 CrossRef CAS PubMed.
- K. Nakazono and T. Takata, Chem.–Eur. J., 2010, 16, 13783–13794 CrossRef CAS PubMed.
- C. B. Caputo, K. Zhu, V. N. Vukotic, S. J. Loeb and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 960–963 CrossRef CAS.
- A. Fernandes, A. Viterisi, F. Coutrot, S. Potok, D. A. Leigh, V. Aucagne and S. Papot, Angew. Chem., Int. Ed., 2009, 48, 6443–6447 CrossRef CAS PubMed.
- E. Arunkumar, C. C. Forbes, B. C. Noll and B. D. Smith, J. Am. Chem. Soc., 2005, 127, 3288–3289 CrossRef CAS PubMed.
- M. R. Craig, M. G. Hutchings, T. D. W. Claridge and H. L. Anderson, Angew. Chem., Int. Ed., 2001, 40, 1071–1074 CrossRef CAS.
- M. A. Soto and M. J. MacLachlan, Org. Lett., 2019, 21, 1744–1748 CrossRef CAS PubMed.
- S. Dasgupta and J. Wu, Chem. Sci., 2012, 3, 425–432 RSC.
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC.
- I. R. Fernando, Y. Mo and G. Mezei, CrystEngComm, 2014, 16, 7320–7333 RSC.
- T. Endo, K. Ageishi and M. Okawara, J. Org. Chem., 1986, 51, 4309–4310 CrossRef CAS.
- X. Chi, M. Xue, Y. Yao and F. Huang, Org. Lett., 2013, 15, 4722–4725 CrossRef CAS PubMed.
- I. R. Fernando, S. G. Bairu, G. Ramakrishna and G. Mezei, New J. Chem., 2010, 34, 2097–2100 RSC.
- For C-22, ring-closing metathesis (RCM) yielded a mixture of cis/trans-22C6 macrocycles within the assembled MIM t-H[4]R (evidenced by 1H NMR spectroscopy). In contrast, RCM of C-21 produced only one isomer (trans-21C6), which correlates well with previous reports that involved 21C6, 22C6 and the dibenzylammonium motif (see ref. 41).
- L. Chen, H.-Y. Zhang and Y. Liu, J. Org. Chem., 2012, 77, 9766–9773 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03744f |
| This journal is © The Royal Society of Chemistry 2019 |