
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free C–C bond formation via coupling of nitrile imines and boronic acids†
Keith
Livingstone
a,
Sophie
Bertrand
b,
Jenna
Mowat
a and
Craig
Jamieson
 *a
*a
aDepartment of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral St, Glasgow G1 1XL, UK. E-mail: craig.jamieson@strath.ac.uk
bGlaxoSmithKline Medicines Research Centre, Gunnels Wood Road, Stevenage, Hertfordshire SG1 2NY, UK
First published on 27th September 2019
Abstract
The challenges of developing sustainable methods of carbon–carbon bond formation remains a topic of considerable importance in synthetic chemistry. Capitalizing on the highly reactive nature of the nitrile imine 1,3-dipole, we have developed a novel metal-free coupling of this species with aryl boronic acids. Photochemical generation of a nitrile imine intermediate and trapping with a palette of boronic acids enabled rapid and facile access to a broad library of more than 25 hydrazone derivatives in up to 92% yield, forming a carbon–carbon bond in a metal free fashion. This represents the first reported example of direct reaction between boronic acids and a 1,3-dipole.
Introduction
The formation of carbon–carbon (C–C) bonds is one of the most fundamental transformations in organic synthesis.1 Indeed, efforts to enable this transformation have occupied synthetic chemists for over a century. The vast majority of C–C bond-forming processes require the application of an organometallic species, or metal catalysis. Such reagents and catalysts are often associated with environmental concerns, in addition to their resources being finite in nature.2 Consequently, more recent approaches towards C–C bond formation have emphasized the importance of sustainability in the development of new reaction manifolds.2A member of the nitrillium betaine family of 1,3-dipoles, the nitrile imine (NI) functional group is a versatile intermediate that is generated in situ by the photolysis of a 2,5-substituted tetrazole moiety.3–5 NIs have recently been employed extensively within materials chemistry in polymer cross-linking and surface functionalization, and in biorthogonal chemistry as ligation handles and photo-affinity labels.6–10 Their widespread application is attributable to the traceless, light-activated nature of the formation of the dipole. While NIs are predominantly known for their role in 1,3-dipolar cycloadditions,5a,5c,5d,11 more recent reports have documented the pleiotropic reactivity profile of NIs with a range of nucleophiles, including carboxylic acids and thiols.10,12,13
As part of our studies aimed at further understanding the reactivity of NIs, we were eager to harness the potential of this versatile dipole in the context of light-activated, metal-free C–C bond formation. This study was developed using a mechanistic hypothesis elucidated from consideration of the Petasis–Mannich reaction, a multicomponent transformation which furnishes highly substituted amine derivatives (Scheme 1a).14–16 Following initial formation of an iminium intermediate from a primary or secondary amine and the corresponding aldehyde, coordination of a vinyl or aryl boronic acid to an α-hydroxy moiety of the iminium species generates the “ate” complex of the Petasis–Mannich.15,17 The principal feature of this complex is the increased nucleophilicity of the boronate moiety relative to the initial boronic acid.18 This pre-activation facilitates migration of the pendant R group into the electrophilic iminium center, forming the reaction product. We reasoned that NIs possessed similar properties to the analogous α-hydroxy iminium intermediate formed during the Petasis–Mannich reaction. The C-terminus of the NI may be considered a pseudo-iminium center, while the anionic N-terminus could readily facilitate boronate formation, in an analogous manner to the α-hydroxy moiety of a Petasis–Mannich substrate (Scheme 1b). If successful, this hypothesis would enable the migration of the aryl group of the boronic acid towards the NI dipole, allowing metal-free access to a range of aryl hydrazones, forging a new C–C bond using photochemical promotion.
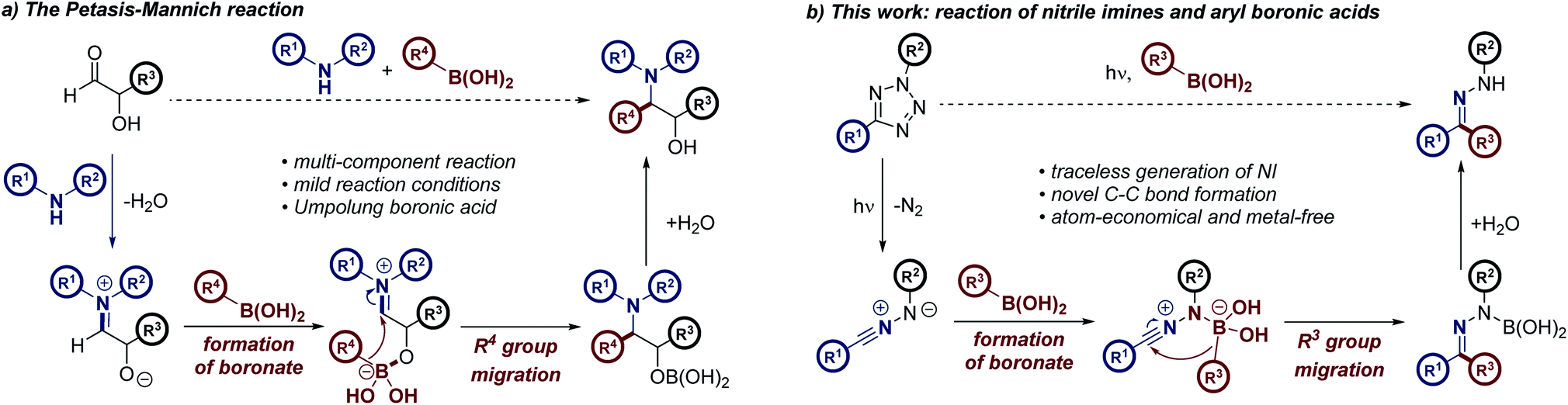 | ||
| Scheme 1 (a) The key steps comprising the mechanism of the Petasis–Mannich multicomponent reaction. (b) Proposed formation of aryl hydrazones via the metal-free coupling of NIs and aryl boronic acids. | ||
Results and discussion
Preliminary studies into the development of the reaction utilized diaryl tetrazole 1a and fluorinated boronic acid 2a (see ESI for full details†). The quantum yields of the photolysis of 2,5-tetrazoles to furnish nitrile imines has previously been reported by several groups.19,20 These values are found to generally be very high (0.5–0.9), and a wide variety of tetrazoles have been shown to undergo photochemical degradation using a simple UV lamp designed for visualising TLC silica plates, therefore indicating the generality of the process. Initial conversions were shown to be highly dependent on the stoichiometry of boronic acid employed, however this effect was considerably less pronounced when exceeding three equivalents of 2a (Table 1, entries 1–3). An additional challenge encountered was the undesired formation of hydrazide 4, the formal hydration product of the intermediate NI. It was thought that the condensation of excess 2a, forming the corresponding boroxine analogue, was responsible for the generation of water within the reaction mixture, leading to hydrazide formation.21 Efforts to prevent the generation of 4 focused on the introduction of drying agents, culminating in the generation of target hydrazone 3a in 61% yield with minimal by-product formation (Table 1, entries 4–6).|
|
||||
|---|---|---|---|---|
| Entry | Eq iv. 2a | Additive | 3a Conv.b [%] | 4 Conv.b [%] |
| a Reaction conditions unless otherwise specified: 1a (1 equiv.), 2a (x equiv.), THF (0.1 M), 270–330 nm light, under N2 at room temperature in a 50 mL quartz flask. b Determined by 19F NMR spectroscopy. c 10 equiv. d 400 mg mmol−1. e Crushed. | ||||
| 1 | 1 | — | 33 | 23 |
| 2 | 3 | — | 53 | 19 |
| 3 | 5 | — | 55 | 11 |
| 4 | 3 | CH(OMe)3c | 38 | 21 |
| 5 | 3 | 3 Å mol. sievesd | 46 | 8 |
| 6 | 3 | 3 Å mol. sievesd,e | 61 | 2 |
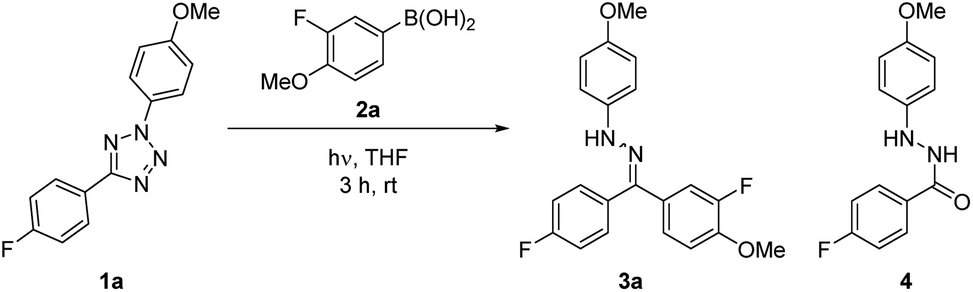
Following on from the identification of these reaction conditions, we sought to exemplify the process by investigating a palette of aryl boronic acids in the reaction (Scheme 2a). The procedure was shown to furnish a broad range of aryl hydrazones in moderate to excellent yields, with a noted correlation on the electronic properties of the boronic acid (cf.3b and 3h). We propose that aryl groups with greater electron density have a greater migratory aptitude during the key step of the process (Scheme 1b), leading to improved conversions. This electronic activation of the substrate may be either conjugative or inductive in nature (cf.3d and 3g). The transformation was also shown to be compatible with both meta- and ortho-substituted aryl boronic acids, with increased steric bulk having a relatively minor impact on reaction yield (cf. 3i, 3j, and 3k). The lower conversion of 3a relative to the isolated yield of 3k is reasoned to be a consequence of the smaller scale used during reaction optimisation, promoting more rapid photolysis of 1a, and subsequent rapid decomposition of the NI. A number of heterocyclic analogues were also synthesized in good to excellent yield (e.g.3l to 3o). Alkyl boronic acids such as cyclopropyl and cyclohexyl systems were also examined, however none of the corresponding hydrazone was obtained, indicating the requirement for an aryl boronic acid derivative for successful reaction.
 | ||
| Scheme 2 (a) Boronic acid substrate scope. (b) Nitrile imine substrate scope using (2b) as the reaction partner, and examples of elevated yields when employing 4-methoxyphenylboronic acid (2c) as the reaction partner. aReaction conditions: tetrazole (0.25 mmol), boronic acid (0.75 mmol), and 3 Å mol. sieves (100 mg) under N2 in THF (2.5 mL), stirred under 270–330 nm light for 16 h in a 50 mL quartz flask. bIsolated as the ketone. | ||
Having established the scope of the aryl boronic acid partner, the scope of the 2,5-diaryl tetrazole component was then assessed (Scheme 2b). Boronic acid 2b was selected as a model substrate for this study as the moderate conversion of hydrazone 3e during the previous phase of the study would highlight the impact of the tetrazole species on reaction yield. In most cases, modification of either aryl ring of the NI precursor was found to have a negligible impact on conversion (e.g.3p to 3t). Both electron-rich and electron-donating substituents were compatible, with a small reduction in yield when employing electron-rich substituents (cf.3r and 3s). The reaction was also found to be tolerant of meta- and ortho-substitution of the C-aryl ring of the tetrazole (cf.3v and 3x), despite its proximity to the site of bond migration during the key step of the reaction.
Representative yields of two tetrazole substrates from this study applied in conjunction with a more electron-rich boronic acid (2c) are shown in examples 3u and 3w. Based on this, it can be inferred that similar enhancements in yield would be observed in all of the above examples when employing a more reactive boronic acid. The majority of substrates were isolated as stereochemical mixtures of both the Z and E hydrazone derivatives. This was reasoned to be a consequence of the application of UV light via the photoisomerization of hydrazone products upon exposure to the reaction conditions (vide infra). The major isomer has been assigned as the aryl group of the boronic acid in an antiperiplanar arrangement with the lone pair of the sp2 hybridised nitrogen. This is based both on our mechanistic rationale for the reaction, and previous literature precedent for nitrillium-based 1,3-dipoles.22 Interestingly, ortho-substitution of either the boronic acid and tetrazole substrates resulted in improvements in stereoselectivity in selected examples (e.g.3j and 3x).
One of the most attractive aspects of this novel transformation is the absence of any exogenous reagents other than the input tetrazole and boronic acid. As a means of further demonstrating the applicability of this emerging process, we applied the method to the transition-metal free synthesis of fenofibrate, a marketed hypolipidemic agent (Scheme 3a).23 The combination of 4-hydroxyphenylboronic acid and tetrazole 1c using the optimized conditions formed the intermediate hydrazone 3z, which was hydrolysed and combined with alkyl bromide 6 to generate the target molecule in excellent overall yield. This approach represents a viable alternative to existing synthetic routes towards this compound, which often require additional steps or excessive quantities of harmful reagents.24
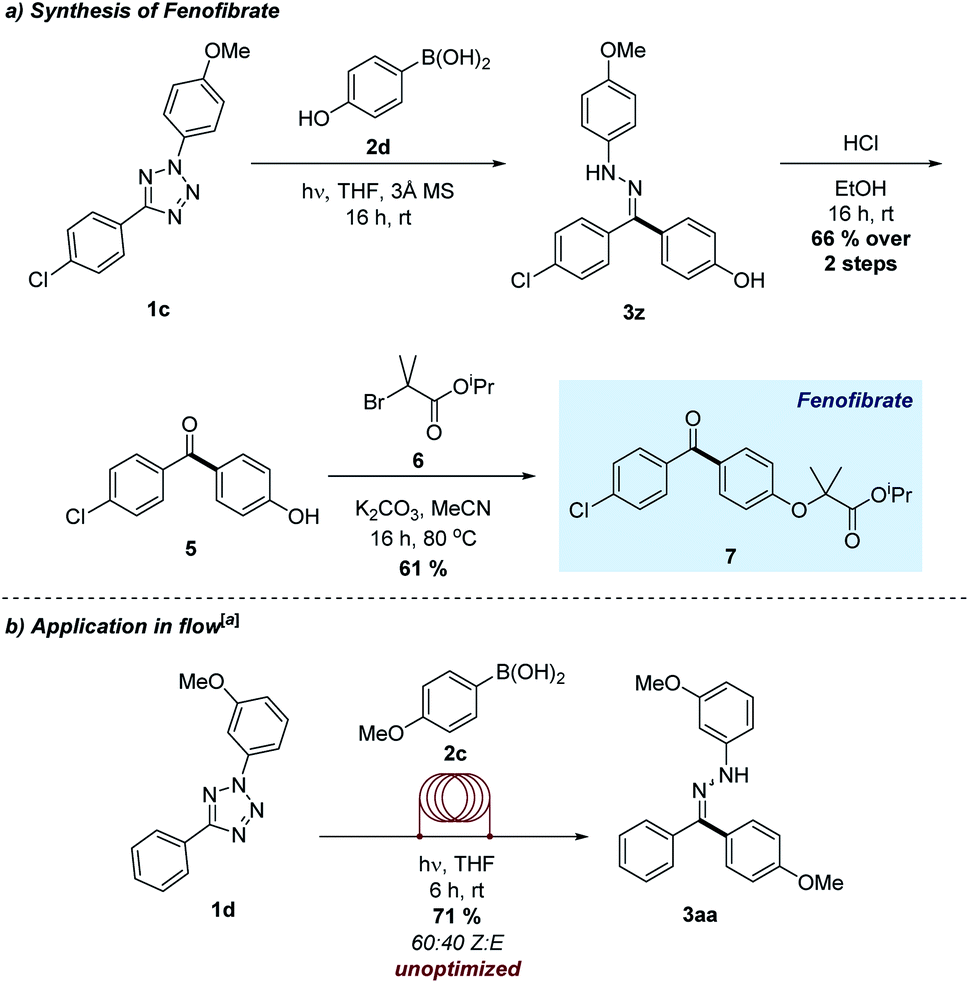 | ||
| Scheme 3 (a) Synthesis of a marketed pharmaceutical agent. (b) Application in a flow reactor. [a]0.75 mmol scale. | ||
Given the practical difficulties often associated with performing light–driven reactions on a large scale, we were interested in the application of the transformation using a flow chemistry reactor. Flow methods are particularly powerful when combined with photochemistry, as it may afford more efficient exposure to the light source when compared to a traditional reaction vessel.25 Pleasingly, photolysis of tetrazole 1d in the presence of boronic acid 2c using a flow reaction manifold successfully furnished the desired hydrazone 3aa in good yields (Scheme 3b). Furthermore, this transformation was accomplished on a larger scale and in shorter time compared to the batch process.
While the photolysis of 2,5-tetrazoles is often viewed as an attractive method of NI generation, we also sought to demonstrate an alternative approach. The treatment of hydrazonyl chloride 8 with potassium phosphate26 in the presence of boronic acid 2c was found to yield the desired hydrazone 3b in excellent yield (Scheme 4), with an enriched Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E ratio, which again suggests isomerization when using UV light. Indeed, subjecting this sample to the photochemical conditions outlined above resulted in a 55:45 mix of geometric isomers. In addition to this, the base promoted reaction manifold was also employed on a 5 mmol scale to furnish 3b, with a similar (72%) isolated yield obtained. A comprehensive investigation into the application of hydrazonyl chlorides within this reaction manifold is currently underway within our laboratory, and will be reported in due course.
:E ratio, which again suggests isomerization when using UV light. Indeed, subjecting this sample to the photochemical conditions outlined above resulted in a 55:45 mix of geometric isomers. In addition to this, the base promoted reaction manifold was also employed on a 5 mmol scale to furnish 3b, with a similar (72%) isolated yield obtained. A comprehensive investigation into the application of hydrazonyl chlorides within this reaction manifold is currently underway within our laboratory, and will be reported in due course.
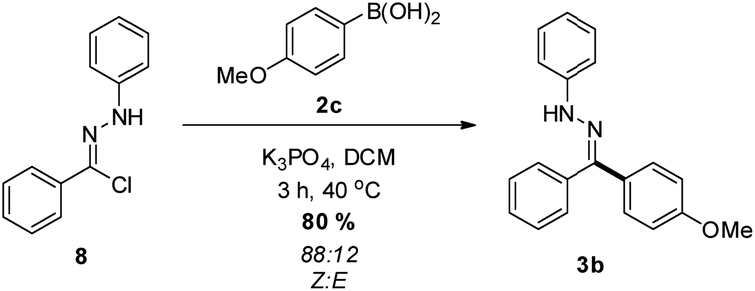 | ||
| Scheme 4 The use of hydrazonyl chlorides as an NI precursor. | ||
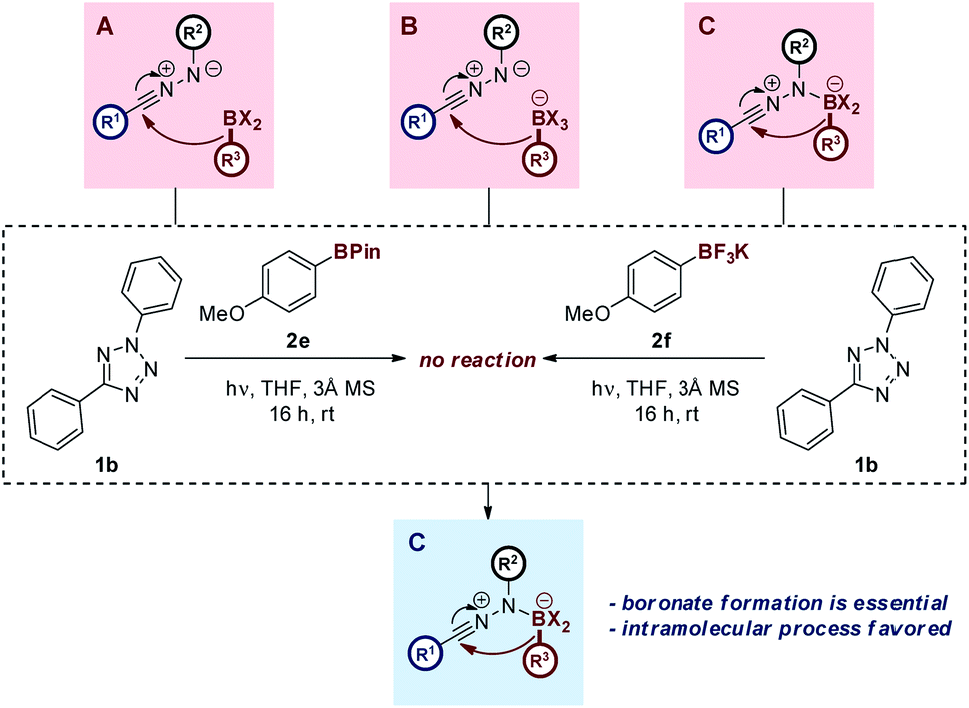
The proposed reaction mechanism is believed to proceed in an analogous manner to the Petasis–Mannich reaction, as outlined above (Scheme 1). This hypothesis is supported by the increased reactivity of electron-rich boronic acid substrates within the reaction manifold, a feature that is shared by the Petasis–Mannich.17a,27 Control experiments involving either the pinacol ester or potassium trifluoroborate analogue of boronic acid 2c have failed to yield the expected hydrazone (Scheme 5). Attempts to form the more electrophilic ArBF2 species by treatment of the potassium trifluoroborate analogue with TMSCl29 led to multiple unidentifiable products. The incompatibility of the pinacol ester substrate provides evidence towards the formation of an anionic nucleophilic boron species during the reaction mechanism, owing to the established reluctance of boron-pinacol esters to form boronates (paths B and C, Scheme 5).28 The migration of the aryl group from the boronate to the NI is also likely to favour an intramolecular process, as deduced from the lack of product formation when employing the highly nucleophilic trifluoroborate analogue (path C, Scheme 5).18 Additional spectroscopic and computational studies are on-going in order to further delineate the mechanism of reaction.
 | ||
| Scheme 5 Preliminary mechanistic investigations. | ||
Conclusions
In conclusion, we have demonstrated the novel reaction of NIs with aryl boronic acids in a traceless, light-activated procedure. This transformation has been shown to be highly general, furnishing over 25 hydrazone analogues in moderate to excellent yields. The method can be utilized in the synthesis of pharmaceutically relevant compounds, and may also be employed at scale, either via the application of flow chemistry, or through the use of an alternative NI source such as the hydrazonyl chloride. The transformation serves as a powerful example of the utility of photochemistry and metal-free syntheses in the pursuit of robust and sustainable methods of C–C bond formation.Experimental
General procedure for the synthesis of aryl hydrazones 3a–3z
To an oven-dried quartz round-bottom flask (50 mL) equipped with a stirrer bar was added 3 Å molecular sieves (400 mg mmol−1), tetrazole (1 equiv.), and boronic acid (3 equiv.). The mixture was dissolved in THF (10 mL mmol−1), purged with N2 and irradiated under a UV lamp (270–330 nm) with stirring for 16 hours. The reaction mixture was diluted with ethyl acetate, filtered through Celite and rinsed with additional ethyl acetate. The crude solution was concentrated under vacuum and purified by column chromatography.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank financial support from the EPSRC and GSK, as well as the EPSRC UK National Mass Spectrometry Facility at Swansea University for analyses. We are also indebted to Mr Andrew Moxen for technical assistance.Notes and references
- W. Caruthers and I. Coldham, Modern Methods of Organic Synthesis, Cambridge University Press, Cambridge, 4th edn, 2004 Search PubMed.
- (a) J. R. Ludwig and C. S. Schindler, Chem, 2017, 2, 313 CrossRef CAS; (b) T. P. Umile, Catalysts for Sustainability: Goals, Challenges and Impacts, CRC Press, Boca Raton, 2015 CrossRef.
- (a) A. Padwa, 1,3-Dipolar Cycloaddition Chemistry, Wiley, New York, 1984, ch. 7 Search PubMed; (b) G. Betrand and C. Wentrup, Angew. Chem. Int. Ed. Engl., 1994, 33, 527 ( Angew. Chem. , 1994 , 106 , 549 ) CrossRef.
- J. S. Clovis, A. Eckell, R. Huisgen and R. Sustmann, Chem. Ber., 1967, 100, 60 CrossRef CAS.
- For recent examples, see (a) Y. Wang, C. I. Rivera Vera and Q. Lin, Org. Lett., 2007, 9, 4155 CrossRef CAS PubMed; (b) S. Stewart, R. Harris and C. Jamieson, Synlett, 2014, 25, 2480 CrossRef CAS; (c) D. Pla, D. S. Tan and D. Y. Gin, Chem. Sci., 2014, 5, 2407 RSC; (d) R. Remy and C. G. Bochet, Eur. J. Org. Chem., 2018, 316 CrossRef CAS.
- For reviews on NI applications, see (a) F. Langa and F. Oswald, C. R. Chim., 2006, 9, 1058 CrossRef CAS; (b) R. K. V. Lim and Q. Lin, Acc. Chem. Res., 2011, 44, 828 CrossRef CAS PubMed; (c) A. Herner and Q. Lin, Top. Curr. Chem., 2016, 1, 374 Search PubMed; (d) G. Delaittre, A. S. Goldmann, J. O. Mueller and C. Barner-Kowollik, Angew. Chem. Int. Ed. Engl., 2015, 54, 11388 ( Angew. Chem. , 1994 , 127 , 11548 ) CrossRef CAS PubMed.
- (a) J. O. Mueller, N. K. Guimard, K. K. Oehlenschlaeger, F. G. Schmidt and C. Barner-Kowollik, Polym. Chem., 2014, 5, 1447 RSC; (b) A. Hufendiek, A. Carlmark, M. A. R. Meier and C. Barner-Kowollik, ACS Macro Lett., 2016, 5, 139 CrossRef CAS; (c) C. Wang, M. M. Zieger, A. Schenzel, M. Wegener, J. Willenbacher, C. Barner-Kowollik and C. N. Bowman, Adv. Funct. Mater., 2017, 27, 1605317 CrossRef; (d) C. Heiler, J. T. Offenloch, E. Blasco and C. Barner-Kowollik, ACS Macro Lett., 2017, 6, 56 CrossRef CAS.
- (a) M. Dietrich, G. Delaittre, J. P. Blinco, A. J. Inglis, M. Bruns and C. Barner-Kowollik, Adv. Funct. Mater., 2012, 22, 304 CrossRef CAS; (b) L. Stolzer, A. Vigovskaya, C. Barner-Kowollik and L. Fruk, Chem.–Eur. J., 2015, 21, 14309 CrossRef CAS PubMed; (c) A. de los Santos Pereira, N. Y. Kostina, M. Bruns, C. Rodriguez-Emmenegger and C. Barner-Kowollik, Langmuir, 2015, 31, 5899 CrossRef CAS PubMed; (d) B. Vonhören, O. Roling, C. Buten, M. Körsgen, H. F. Arlinghaus and B. J. Ravoo, Langmuir, 2016, 32, 2277 CrossRef PubMed; (e) C. Buten, S. Lamping, M. Körsgen, H. F. Arlinghaus, C. Jamieson and B. J. Ravoo, Langmuir, 2018, 34, 2132 CrossRef CAS PubMed.
- (a) W. Song, Y. Wang, J. Qu, M. M. Madden and Q. Lin, Angew. Chem. Int. Ed. Engl., 2008, 47, 2832 ( Angew. Chem. , 2008 , 120 , 2874 ) CrossRef CAS PubMed; (b) W. Song, Y. Wang, J. Qu and Q. Lin, J. Am. Chem. Soc., 2008, 130, 9654 CrossRef CAS PubMed; (c) W. Song, Y. Wang, Z. Yu, C. I. Rivera Vera, J. Qu and Q. Lin, ACS Chem. Biol., 2010, 5, 875 CrossRef CAS PubMed; (d) Z. Yu, Y. Pan, Z. Wang, J. Wang and Q. Lin, Angew. Chem. Int. Ed. Engl., 2012, 51, 10600 ( Angew. Chem. , 2012 , 124 , 10752 ) CrossRef CAS PubMed; (e) Y.-J. Lee, B. Wu, J. E. Raymond, Y. Zeng, X. Fang, K. L. Wooley and W. R. Liu, ACS Chem. Biol., 2013, 8, 1664 CrossRef CAS PubMed; (f) D. N. Kamber, L. A. Nazarova, Y. Liang, S. A. Lopez, D. M. Patterson, H.-W. Shih, K. N. Houk and J. A. Prescher, J. Am. Chem. Soc., 2013, 135, 13680 CrossRef CAS PubMed; (g) Z. Yu and Q. Lin, J. Am. Chem. Soc., 2014, 136, 4153 CrossRef CAS PubMed.
- (a) Z. Li, L. Qian, L. Li, J. C. Bernhammer, H. V. Huynh, J.-S. Lee and S. Q. Yao, Angew. Chem. Int. Ed. Engl., 2016, 55, 2002 ( Angew. Chem. , 2016 , 128 , 2042 ) CrossRef CAS PubMed; (b) A. Herner, J. Marjanovic, T. M. Lewandowski, V. Marin, M. Patterson, L. Miesbauer, D. Ready, J. Williams, A. Vasudevan and Q. Lin, J. Am. Chem. Soc., 2016, 138, 14609 CrossRef CAS PubMed; (c) K. Cheng, J.-S. Lee, P. Hao, S. Q. Yao, K. Ding and Z. Li, Angew. Chem. Int. Ed. Engl., 2017, 56, 15044 ( Angew. Chem. , 2017 , 129 , 15240 ) CrossRef CAS PubMed; (d) Y. Tian, M. P. Jacinto, Y. Zeng, Z. Yu, J. Qu, W. R. Lui and Q. Lin, J. Am. Chem. Soc., 2017, 139, 6078 CrossRef CAS PubMed.
- For selected examples, see (a) A. Eckell, R. Huisgen, R. Sustmann, G. Wallbillich, D. Grashey and E. Spindler, Chem. Ber., 1967, 100, 2192 CrossRef CAS; (b) H. Meier and H. Heimgartner, Helv. Chim. Acta, 1985, 68, 1283 CrossRef CAS; (c) M. P. Sibi, L. M. Stanley and C. P. Jasperse, J. Am. Chem. Soc., 2005, 127, 8276 CrossRef CAS PubMed; (d) C. Spiteri, S. Keeling and J. E. Moses, Org. Lett., 2010, 12, 3368 CrossRef CAS PubMed; (e) A. Singh, A. L. Loomer and G. P. Roth, Org. Lett., 2012, 14, 5266 CrossRef CAS PubMed.
- (a) S. Zhao, J. Dai, M. Hu, C. Liu, R. Meng, X. Liu, C. Wang and T. Luo, Chem. Commun., 2016, 52, 4702 RSC; (b) C. Heiler, J. Offenloch, E. Blasco and C. Barner-Kowollik, ACS Macro Lett., 2017, 6, 56 CrossRef CAS.
- (a) J. A. Zahra, B. A. Abu Thaher, M. M. Abadelah and R. Boese, Org. Biomol. Chem., 2003, 1, 822 RSC; (b) Y. Zhang, W. Liu and Z. K. Zhao, Molecules, 2014, 19, 306 CrossRef PubMed.
- N. A. Petasis and I. Akritopoulou, Tetrahedron Lett., 1993, 34, 583 CrossRef CAS.
- (a) N. R. Candeias, F. Montalbano, P. M. S. D. Cal and P. M. P. Gois, Chem. Rev., 2010, 110, 6169 CrossRef CAS PubMed; (b) P. Wu, M. Givskov and T. E. Nielsen, Chem. Rev., 2019, 119 DOI:10.1021/acs.chemrev.9b00214.
- (a) N. A. Petasis and I. A. Zavialov, J. Am. Chem. Soc., 1997, 119, 445 CrossRef CAS; (b) G. K. Surya Prakash, M. Mandal, S. Schweizer, N. A. Petasis and G. A. Olah, Org. Lett., 2000, 2, 3173 CrossRef PubMed; (c) M. Sugiura, K. Hirano and S. Kobayashi, J. Am. Chem. Soc., 2004, 126, 7182 CrossRef CAS PubMed; (d) C. W. G. Au and S. G. Pyne, J. Org. Chem., 2006, 71, 7097 CrossRef CAS PubMed.
- (a) N. R. Candeias, L. F. Veiros, C. A. M. Afonso and P. M. P. Gois, Eur. J. Org. Chem., 2009, 1859 CrossRef CAS; (b) J. Tao and S. Li, Chin. J. Chem., 2010, 28, 41 CrossRef CAS; (c) R. Y. Souza, G. A. Bataglion, D. A. C. Ferreira, C. C. Gatto, M. N. Eberlin and B. A. D. Neto, RSC Adv., 2015, 5, 76337 RSC.
- G. Berionni, B. Maji, P. Knochel and H. Mayr, Chem. Sci., 2012, 3, 878 RSC.
- E. Sato, Y. Kanaoka and A. Padwa, J. Org. Chem., 1982, 47, 4256 CrossRef CAS.
- V. Lohse, P. Leihkauf, C. Csongar and G. Tomaschewski, J. Prakt. Chem., 1988, 330, 406–414 CrossRef CAS.
- W. A. Marinaro, L. J. Schieber, E. J. Munson, V. W. Day and V. J. Stella, J. Pharm. Sci., 2012, 101, 3190 CrossRef CAS PubMed.
- (a) L. P. H. Yang and G. M. Keating, Am. J. Cardiovasc. Drugs, 2009, 9, 401 CrossRef CAS PubMed; (b) F. Lalloyer and B. Staels, Arterioscler., Thromb., Vasc. Biol., 2010, 30, 894 CrossRef CAS PubMed.
- A. Hegarty, Acc. Chem. Res., 1980, 13, 448 CrossRef CAS.
- R. Vardanyan and V. Hruby, Synthesis of Best-Seller Drugs, Elsevier, New York, 2016, pp. 419–458 Search PubMed.
- F. Politano and G. Oksdath-Mansilla, Org. Process Res. Dev., 2018, 22, 1045 CrossRef CAS.
- For selected examples of NI formation using hydrazonyl chlorides, see: (a) R. Huisgen, M. Seidel, J. Sauer, J. W. McFarland and G. Wallbillich, J. Org. Chem., 1959, 24, 892 CrossRef CAS; (b) A. F. Hegarty, M. P. Cashman and F. L. Scott, J. Chem. Soc. D, 1971, 684 RSC; (c) G. Zecchi, L. Garanti and G. Molteni, Heterocycles, 1995, 40, 777 CrossRef; (d) L. Tamborini, Y. Chen, C. A. Foss, A. Pinto, A. G. Horti, S. F. Traynelis, C. De Micheli, R. C. Mease, K. B. Hansen, P. Conti and M. G. Pomper, J. Med. Chem., 2016, 59, 11110 CrossRef CAS PubMed; (e) V. V. Voronin, M. S. Ledovskaya, E. G. Gordeev, K. S. Rodygin and V. P. Ananikov, J. Org. Chem., 2018, 83, 3819 CrossRef CAS PubMed.
- (a) M. Follman, F. Graul, T. Schäfer, S. Kopec and P. Hamley, Synlett, 2005, 1009 CrossRef; (b) M. V. Shevchuk, A. E. Sorochinsky, V. P. Khilya, V. D. Romanenko and V. P. Kukhar, Synlett, 2010, 73 CAS.
- J. Takagi, K. Takahashi, T. Ishiyama and N. Miyaura, J. Am. Chem. Soc., 2002, 124, 8001 CrossRef CAS PubMed.
- E. Vedejs, R. W. Chapman, S. C. Fields, S. Lin and M. R. Schrimpf, J. Org. Chem., 1995, 60, 3020 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental protocols and complete compound characterization. See DOI: 10.1039/c9sc03032h |
| This journal is © The Royal Society of Chemistry 2019 |