
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of an activatable hybrid persistent luminescence nanoprobe for background-free bioimaging-guided investigation of food-borne aflatoxin in vivo†
Jing-Min Liu a,
Xin-Yue Yuanb,
Hui-Lin Liub,
Dai Chenga and
Shuo Wang*ac
a,
Xin-Yue Yuanb,
Hui-Lin Liub,
Dai Chenga and
Shuo Wang*ac
aTianjin Key Laboratory of Food Science and Health, School of Medicine, Nankai University, Tianjin 300071, China. E-mail: elisasw2002@aliyun.com
bBeijing Advanced Innovation Center for Food Nutrition and Human Health, Beijing Technology & Business University (BTBU), Beijing, 100048, China
cState Key Laboratory of Food Nutrition and Safety, Tianjin University of Science and Technology, Tianjin, 300457, China
First published on 8th August 2018
Abstract
The development of in situ and real-time analytical methods for specifically probing food-borne hazardous substances is promising for clarifying their harmful behaviors and related disease mechanisms inside the living body through in situ investigation of their in vivo behaviors. Herein, optical nanoimaging with the ability of in situ non-damage detection and real-time monitoring was introduced for specific recognition of aflatoxin in cellular levels and in vivo via the fluorescence resonance energy transfer (FRET) protocol. Persistent luminescence nanophosphors (PLNPs) with distinct advantages of improved sensitivity and signal-to-noise ratio were employed in in vivo bioimaging as photoluminescence nanoprobes, while copper sulfide nanoparticles were utilized as the quencher. Due to their long-lasting afterglow, PLNPs do not require external illumination before imaging, effectively eliminating the scattering light and autofluorescence from the biological matrix that can occur during in situ excitation. The proposed FRET imaging assay achieved high sensitivity and specificity as well as improved imaging resolution for the target aflatoxin present in vivo. This study will provide insights towards advanced methodology for the applications of bioimaging in food safety, and could potentially provide an advisory roadmap for bioimaging-guided exploration and mediation of food-borne hazards to human health.
1. Introduction
Recently, human disease and food safety issue caused by the presence of harmful compounds in the environment or food-supply is of great concern in modern society.1 Specifically, food contamination, resulting due to the exposure to toxins or pathogens through raw materials or processing environment, is a major problem that can lead to severe food-borne diseases.2 As a global priority, significant efforts have been put towards the establishment of high-performance analytical methods to identify and quantify hazardous substances in food samples; these methods include electrochemical sensing, colorimetric detection, fluorescence sensing, immunoassays, and chromatographic separation.3–7 With the continuous increase in complexity and diversity of food-borne harmful substances, research interest has been focused on the specific behaviors of food-borne harmful substances during digestion in the body as well as their in vivo actions and distributions, so as to better understand the scientific relationship between food contamination and illness, which is important for prevention, diagnosis, and therapy of food-borne diseases. Therefore, in vitro detection of certain food-borne toxins in food samples gives limited useful information in vivo.The development of in situ and real-time analytical methods for specific and sensitive quantification of food-borne hazardous substances ingested in the human body, such as toxins or pathogens, is the key research trend in food science and health fields. This research has been aimed at clarifying and investigating the harmful behaviors of these substances and their related disease mechanisms inside the living body through in situ investigation of their in vivo behaviors. There appears to be a great demand for the development of innovative analytical methods that are capable of in situ collection of intuitive and reliable information of the in vivo behaviors of target toxins to further support food safety inspection research.
Compared with the traditional in vitro analytical methodology, defined as simply the quantification of target toxins or pathogens in certain samples, optical bioimaging with the ability of non-damaging detection and real-time monitoring holds significant potential for the in vivo investigation and mediation of harmful substances in food.8 Originally developed as a noninvasive and nonionizing tool for real-time monitoring and probing of physiological processes and life functions inside the living body, fluorescence bioimaging technology could be easily realized by combination of a camera with adjustable filters to collect certain fluorescence signals from the living body; hence, it has been extensively applied in human disease diagnosis and treatment.9–12 Recent efforts have focused on the fabrication of novel luminescent nanostructures as advanced contrast agents, termed as nano-imaging methodology.13,14 Various nanomaterials with distinctive optical features have been proposed as functional nanoprobes for nano-imaging, such as quantum dots (tunable emission, high quantum yields, ease of surface functionalization, etc.),15 carbon nanodots (green synthesis, good stability, low toxicity, excellent biocompatibility, etc.),16 and upconversion nanoparticles (intense emission, anti-stokes luminescence, low toxicity, high photostability, etc.).17 However, the abovementioned nanoprobes still encounter some problems while applied for in vivo imaging, including insufficient signal-readout resolution, poor tissue penetration performance, and tissue damage caused by the involvement of laser irradiation.18
Persistent luminescence nanophosphors (PLNPs), with continuous afterglow performance across the visible to near-infrared spectral regions for hours to days, have been recognized as a new generation of luminescent nanoprobes.18–23 PLNPs possess advantageous properties, such as super-long and re-excitable afterglow luminescence, excellent structural stability, superior biocompatibility, low toxicity, and facile surface-modification ability. The remarkable super-long afterglow feature enables the PLNPs to be used in in vitro optical sensing as well as in vivo real-time nano-imaging without the need for simultaneous external excitation.19,20 Therefore, PLNPs have attracted much attention as advantageous luminescent nanoprobes and have been the subject of novel research in the biomedical and biological research fields, including sensing24–26 and imaging.27–30
Herein, for the first time, a luminescence bioimaging technique with an activatable nanoprobe was introduced for specific recognition and determination of food-borne toxins both at the cellular level and in vivo via the fluorescence resonance energy transfer (FRET) strategy. Aflatoxin B1 (AFT B1) was chosen as the model analyte. Aflatoxins, which are secondary metabolites mainly produced by the fungi of A. parasiticus and Aspergillus flavus, are known to be highly carcinogenic, hepatotoxic, and mutagenic food-borne toxins.31,32 As the most common aflatoxin, AFT B1 is of great food-safety concern and research interest and hence, in vivo probing of AFT B1 digested in the living body would be of great significance.33,34 In the present FRET assay, persistent luminescence nanophosphors with the distinct advantages of improved sensitivity and signal-to-noise ratio in in vivo bioimaging as well as excellent photostability and biocompatibility, were employed as the photoluminescence nanoprobes, while copper sulfide nanoparticles (CuS) were utilized as the quencher. Then, an activatable FRET nanoprobe was constructed by linking PLNPs with CuS via DNA hybridization, and the aptamer-modified silica on the PLNPs' surface ensured the selective capture of AFT B1 with high capacity. Most importantly, due to the long-lasting afterglow of PLNPs, there is no need for external illumination before imaging, effectively eliminating the scattering light and autofluorescence from the biological matrix that is typically encountered while under in situ excitation. The proposed FRET sensing and imaging assay achieved high sensitivity and specificity as well as improved imaging resolution for the target aflatoxin present in cells and in vivo. This study will provide insights towards the advanced methodology for the applications of bioimaging in food safety detection and provides an advisory roadmap for bioimaging-guided exploration and mediation of food-borne hazards to human health (Fig. 1).
 | ||
| Fig. 1 Schematic of the PLNPs-involved FRET sensing and imaging assay for in vivo determination of aflatoxin. | ||
2. Experimental section
2.1 Preparation of ZGGO persistent luminescence nanophosphors
The well-performing ZGGO PLNPs with a nominal formula of Zn1.25Ga1.5Ge0.25O4:0.5%Cr3+, 2.5%Yb3+, 0.25%Er3+ were prepared by the typical solvothermal method, followed by a short period of calcination in air.35 Aqueous solutions of Ga(NO3)3·9H2O, Zn(NO3)3·6H2O, Cr(NO3)3·9H2O, Er(NO3)3·5H2O, and Yb(NO3)3·5H2O were prepared. GeO2 was dissolved in diluted ammonium hydroxide solution. Certain volumes of aqueous solutions of chromium nitrate, zinc nitrate, yttrium nitrate, erbium nitrate and ammonium germinate, all with the concentration of 0.1 M, were added to the aqueous solution of gallium nitrate (0.3 M), according to the above formula of ZGGO, under vigorous stirring (20 mL). Then, diluted ammonium hydroxide was added to the precipitated precursor to adjust the pH to 8, followed by 10 min-ultrasonic treatment and 2 h-stirring at room temperature. Subsequently, the solution was mixed with an organic solution of toluene (18 mL) and oleic acid (2 mL) to obtain the bi-phasic mixture, and then transferred into a 50 mL Teflon-lined stainless steel autoclave and reacted at 160 °C for 36 h. After cooling to room temperature, the resultant compounds were precipitated from the synthesis solution with excess volume of ethanol, and then washed twice sequentially with water and ethanol, followed by lyophilization. The dried white powder was finally sintered in air at 1000 °C for 2 h, followed by ethanol washing and lyophilization.To obtain the small-sized and monodispersed PLNPs, the as-prepared ZGGO powder was hydroxylated by ultrasonic treatment in NaOH solution (5 mM) for 3 h, followed by vigorous stirring overnight at room temperature. The resulting colloid solution was centrifuged at 4500 rpm for 5 min. The supernatant was collected and concentrated, and then centrifuged at 3500 rpm for 20 min; ZGGO PLNPs with a diameter of ∼40 nm were thus obtained in the supernatant. The resultant small-sized PLNPs were then coated with silica to decorate amine groups onto the PLNP nanoparticles via the TEOS and APTES hydrolysis reaction, according to a modified Stöber sol–gel process.36
2.2 Preparation of CuS nanoparticles
The CuS nanoparticles were synthesized according to the reported sodium citrate-template method.37 Briefly, to a 250 mL aqueous solution containing Cu(NO3)2 (0.25 mmol) and sodium citrate (0.17 mmol), 250 μL of sodium sulfide solution (Na2S, 1 M) was added under stirring at room temperature. Upon the addition of sodium sulfide, the initial pale-blue Cu(NO3)2 solution turned dark-brown immediately. After vigorous stirring for 5 min, the mixed solution was heated to 90 °C for conducting the reaction and stirred for 15 min to obtain a dark-green solution. Then, the reaction mixture was transferred into ice-cold water. The obtained CuS nanoparticles were centrifuged and washed with water and stored in PBS buffer at 4 °C.2.3 Preparation of PLNPs–CuS nanoprobes
The PLNPs–CuS nanoprobes were prepared by linking aptamer-modified PLNPs (apt-PLNPs) with ssDNA-modified CuS nanoparticles (ssDNA–CuS) via DNA hybridization. Referring to a previously-reported method,24 the apt–PLNPs were prepared by EDC/NHS-assisted binding of amino groups (from the silica layer of PLNPs) with the carboxyl groups of the aptamer, while the ssDNA–CuS was prepared via direct binding of thiol–ssDNA onto the CuS surface. To prepare the PLNPs–CuS nanoprobe, 5 mL of apt–PLNPs dispersion (1 mg mL−1, PBS 10 mM, pH 7.4) was gently mixed with 5 mL of ssDNA–CuS dispersion (5 mg mL−1), and stirring was conducted for 3 h in the dark. The resulting mixture was washed with PBS via centrifugation to obtain the PLNPs–CuS hybrid nanoprobe.2.4 In vitro cell luminescence imaging
To image the AFT B1 in cells via the developed FRET-based nanoprobes, 3T3 and HepG2 cell lines were chosen as the control group and treatment group, respectively. The cells were all seeded in 24-well plates and carefully incubated under 5% CO2 at 37 °C. After a 24 h incubation, three groups of HepG2 cell lines were treated with AFT B1 with concentrations of 1, 5 and 10 μM for 3 h, and 3T3 cells were treated with an equal amount of PBS as a control. Subsequently, all the control and treatment groups were incubated with the PLNPs–CuS probe for another 3 h and then washed with PBS 3 times and fixed with 4% paraformaldehyde for 25 min. DAPI was added to stain the nuclei for 5 min. The cells were washed with PBS 3 times before imaging was conducted by an inverted fluorescence microscope (IX81, Olympus, Japan) in U-MWIG3 mode (wide band interference green excitation, with exciter filter BP530-550 and barrier filter BA575IF). The fluorescence signal of the PLNPs were collected.2.5 In vivo luminescence bioimaging
All animal experiments were conducted using the adult athymic BALB/c (BALB/c-nu) mice (16–20 g), purchased from Beijing HFK Bioscience Co., LTD. (Beijing, China). All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Nankai University and experiments were approved by the Animal Ethics Committee of Tianjin. All animal procedures were performed using chloral hydrate anaesthesia (200 μL, 4%) to ensure minimized suffering of the mice. The PLNPs–CuS (1 mg mL−1, dispersed in 10 mM PBS) were excited for 10 min with a 324 nm UV light source (6 W) before injection, and 1 min red LED light (650 ± 10 nm) illumination was performed on the injected mice before acquiring the afterglow imaging photographs. In vivo luminescence imaging results of the mice were obtained by employing a Berthold NightOWL LB 983 Imaging System without excitation sources. The exposure time was fixed at 120 s with an emission filter of 700 nm.3. Results and discussion
3.1 Preparation and characterization of PLNPs–CuS nanoprobes
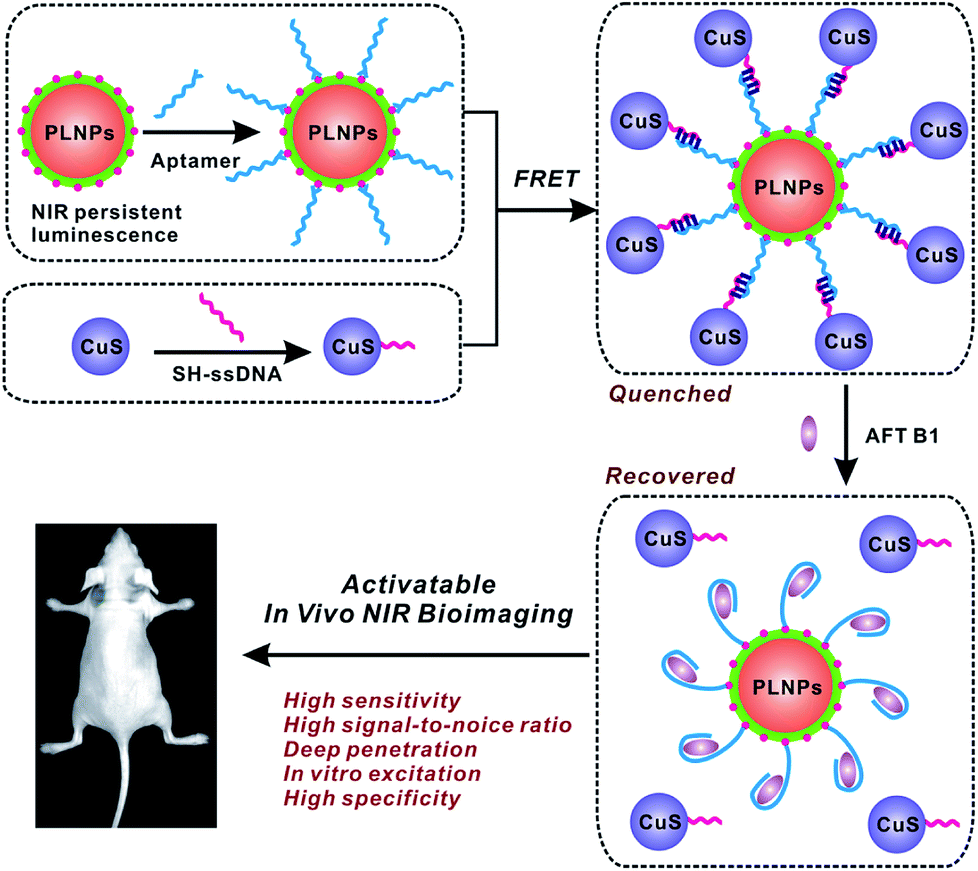
In the present study, the activatable PLNPs–CuS hybrid nanoprobes were fabricated as a contrast agent for in vitro and in vivo targeted imaging of AFT B1. ZGGO persistent luminescence nanoparticles with super-long afterglow and red light renewability were employed as a NIR emission center to ensure autofluorescence-free bioimaging, while the CuS nanoparticles were used as a quencher due to their broad absorption that effectively overlapped with the emission of the PLNPs. The PLNPs–CuS hybrid nanoprobes were constructed through DNA hybridization that occurred between the aptamer immobilized on the PLNPs' surface and the ssDNA linked onto the CuS surface, resulting in effective FRET that quenched the luminescence. In the presence of target AFT B1, the high affinity of the aptamer to target broke the DNA hybrid, which led to the separation of PLNPs with CuS, accompanied by the recovery of luminescence. As a result, a luminescence-activatable nano-platform was constructed to further improve the signal-to-noise ratio and sensitivity of bioimaging.The ZGGO PLNPs were synthesized by the typical two-phase solvothermal reaction method, followed by a short of period calcination in air; CuS nanoparticles were prepared via the citrate-template method. The well-acknowledged ZGGO PLNPs as bioimaging contrast agents gave an intense NIR luminescence centered at 690 nm and a wide excitation across the UV to visible spectra, and demonstrated super-long afterglow approaching 500 h (Fig. 2A and S1†). Moreover, the luminescence showed outstanding renewability that could be reactivated by red LED light (650 ± 10 nm), verifying the applicability of ZGGO PLNPs for long-term in vivo imaging and tracking (Fig. S2†). The XRD analysis (Fig. S3†) revealed the spinel phase of Zn2GeO4 (JCPDS 25-1018) and ZnGa2O4 (JCPDS 38-1240), which is consistent with previous study.35
 | ||
| Fig. 2 (A) Excitation and emission spectra of PLNPs and absorption spectra of CuS nanoparticles. (B) Afterglow property of PLNPs and PLNPs–CuS nanoprobes in the presence of AFT B1. (C) The luminescence quenching of CuS NPs to PLNPs. (D) Comparison of the quenching effect of AuNRs and CuS NPs on the luminescence of PLNPs. | ||
The obtained CuS NPs have wide absorption and significant overlap with the emission of PLNPs, favoring an effective FRET process. Further modification of the aptamer or ssDNA produced limited influence on the optical property of PLNPs or CuS. Incubating ssDNA–CuS of various concentrations with apt–PLNPs caused the gradual quenching of luminescence, giving almost 95% luminescence quenching to achieve minimal background signal of the activatable nanoprobe (Fig. 2C). Although gold nanorods (AuNRs) could also efficiently quench the luminescence of ZGGO PLNPs, CuS was chosen as the quencher for the activatable nanoprobe, in consideration of the potential toxicity of CTAB that was adsorbed on the AuNRs as a stabilizer (Fig. 2D).
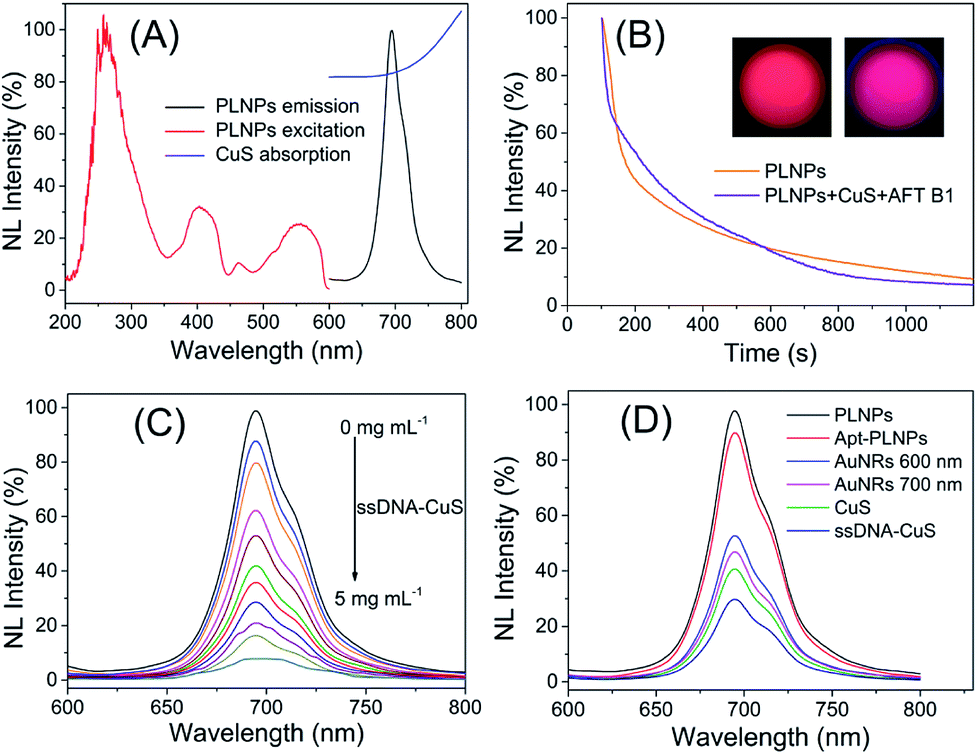
The as-prepared apt–PLNPs and ssDNA–CuS have a relatively uniform nanostructure with a size of 54 ± 6 nm and 7.2 ± 0.6 nm, respectively. The change in the hydrodynamic diameters and zeta potentials revealed the surface modification of PLNPs with aptamers as well as the synthesis of PLNPs–CuS, of which particle size increased to 85 nm (Fig. 3A). Moreover, the HRTEM characterization provided direct evidence of the successful integration of the PLNPs–CuS nanoprobe with the core/satellite-like morphology (Fig. 3E). EDX mapping showed the presence of Zn, Ga, Ge, and O (from ZGGO PLNPs) and Cu and S (from CuS nanoparticles), further confirming the fabrication of the hybrid nanostructures (Fig. 3D).
 | ||
| Fig. 3 (A) Comparison of zeta potential and particle size of PLNPs, apt–PLNPs, and PLNPs–CuS. (B) The structural stability of PLNPs–CuS and the luminescence stability of PLNPs in PBS. (C) The structural stability of PLNPs–CuS and the luminescence stability of PLNPs in serum. (D) EDX mapping of the PLNPs–CuS nanoprobe with the scale bar of 1 μm. (E) The typical HRTEM photograph of CuS nanoparticles (a and b), PLNPs (c), and PLNPs–CuS (d). | ||
Comparison of the persistent luminescence of PLNPs with PLNPs–CuS–AFT B1 demonstrated that the luminescence quenching and recovery process had limited influence on the afterglow performance of PLNPs (Fig. 2B). Continuous monitoring of the luminescence of apt–PLNPs treated in serum and PBS overnight revealed that no more than 5% decrease in the luminescence intensity was observed, indicating good photostability of the apt–PLNPs. Furthermore, incubation of the PLNPs–CuS nanoprobe in serum and PBS for 24 h resulted in no significant change in size distribution, proving the excellent stability of the PLNPs–CuS nanoprobes under a physiological environment (Fig. 3B and C).
3.2 Determination of AFT B1 via the FRET nanoprobes
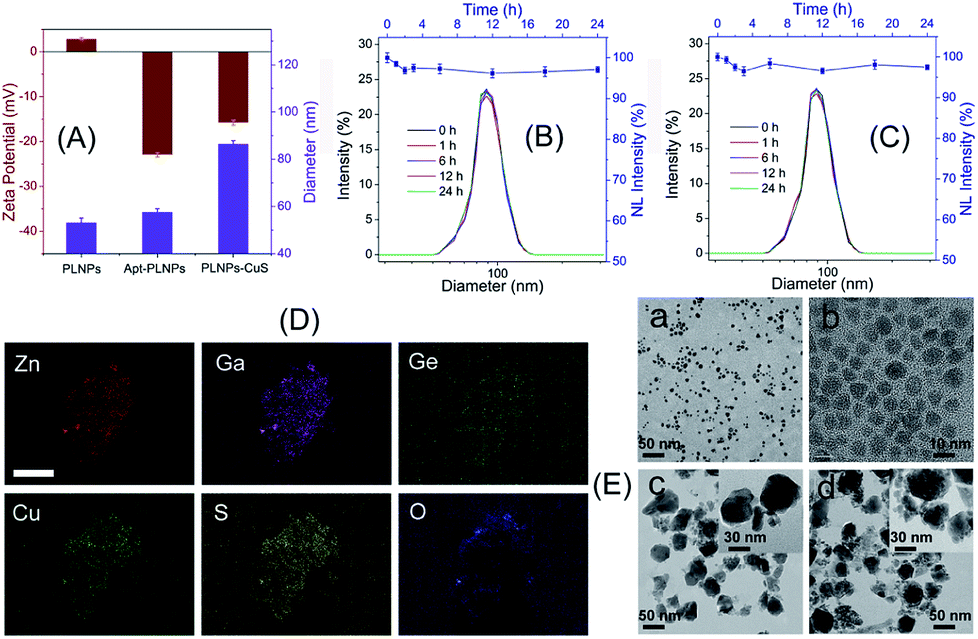
In the design of the FRET nanoprobes, the target toxin would compete with the ssDNA to bind with the aptamer and break up the conjugation of the aptamer–ssDNA hybridization that bridged the PLNPs and CuS, thus recovering the initial luminescence. The results shown in Fig. 4 indicated that the luminescence of PLNPs–CuS gradually increased as the amount of AFT B1 increased, giving a linear response in the range of 0.1–2.4 μM to AFT B1 and a low detection limit of 0.03 μM (3 s). The pH effect on the response of the PLNPs–CuS nanoprobe to AFT B1 was assessed both in PBS and Tris–HCl buffer of various pH; the results revealed that neutral pH conditions gave the best fluorescence turn-on performance (Fig. S4†). | ||
| Fig. 4 (A) Luminescence recovery of the AFT B1 to PLNPs–CuS nanoprobe. (B) Linear response of AFT B1 determination. (C and D) Specificity of AFT B1 determination via the PLNPs–CuS nanoprobe. | ||
To examine the specificity of the FRET nanoprobe, various interferents, such as toxins, amino acids, and proteins, were tested on the nanoprobe under the same experimental conditions. The PLNPs–CuS gave a negligible response to all the interferents due to the high selectivity and affinity of the aptamer to AFT B1. A co-existing interference assay using three mixtures (M1: mixture of toxins; M2: mixture of proteins; M3: mixture of amino acids) further verified the excellent specificity of the developed FRET nanoprobes (Fig. 4C). All the above results proved that the developed activatable FRET nanoprobe was applicable for selective and sensitive determination of AFT B1 in samples, which laid the foundation for imaging AFT B1 in a biofluid or intracellular environment.
3.3 Toxicity of the PLNPs–CuS nanoprobes
Prior to the utilization of the proposed PLNPs–CuS FRET nanoprobes for in vivo luminescence imaging, their potential toxicity was thoroughly assessed. In vitro cytotoxicity of the PLNPs–CuS was examined with the regular MTT assay. The three typical cell lines, Balb/3T3, HepG2 and MCF-7 were separately incubated with the hybrid nanoprobes with various concentrations in the range of 50–1000 μg mL−1 for 24 h. The following MTT counting showed that the viability was more than 83% after incubation, thus testifying the low cytotoxicity of the hybrid nanoprobes (Fig. 5A).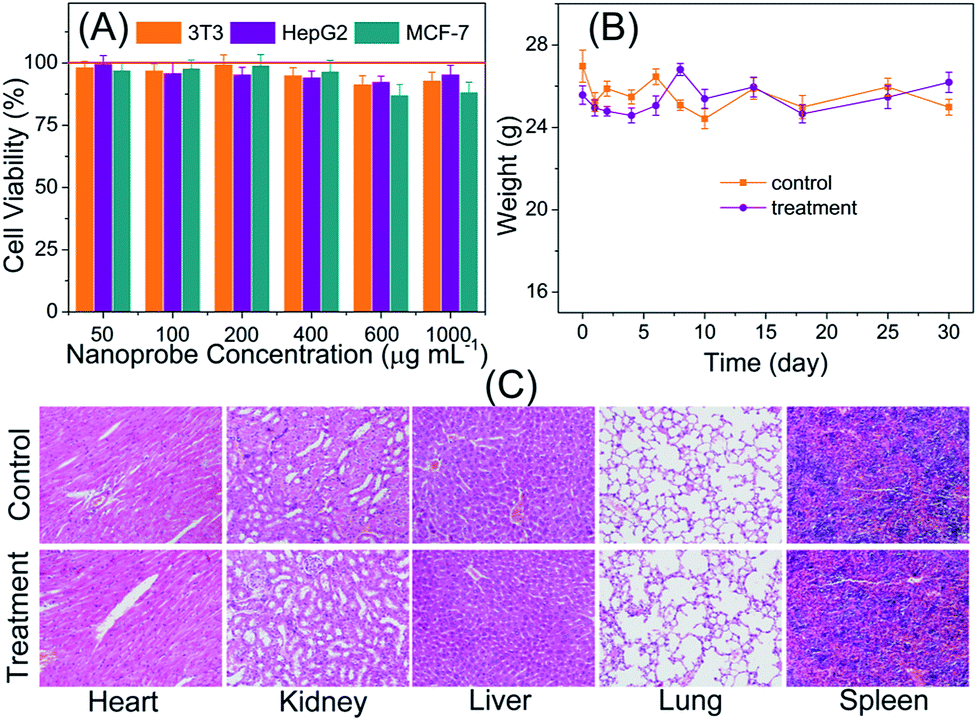 | ||
| Fig. 5 Evaluation of the toxicity of the fabricated nanoprobes: (A) In vitro viability of 3T3, HepG2, and MCF-7 cell lines incubated with PLNPs–CuS nanoprobes of various concentrations for 1 day. (B) Body weight changes of the nude mice (normal nude mice and HeLa tumor-bearing nude mice) with/without injection by the imaging probes (0.8 mL, 1 mg mL−1). (C) Representative hematoxylin and eosin stained images of major organs including heart, liver, lung, spleen, and kidney collected from the nanoprobe (0.4 mL, 1.0 mg mL−1) injected mice and the control mice (injected with PBS) at 7 days after administration. | ||
The in vivo toxicity of the FRET nanoprobes was assessed by the histopathology assay. The same amount of PLNPs–CuS solutions (0.4 mL, 1 mg mL−1) and PBS were intravenously injected into the treatment group and control group of mice vein. One week post injection, the major organs including spleen, heart, lung, liver, and kidney were collected and analyzed to identify the possible histological changes; the results revealed no significant inflammatory lesions or organ damage related to the PLNPs–CuS nanoprobe treatment (Fig. 5C).
Furthermore, the long-term in vivo toxicity was investigated by 30 day-continuous monitoring of the change in body weight of the tumor-bearing mice and healthy mice injected with PLNPs–CuS (0.8 mL, 1 mg mL−1) as treatment groups and compared with the control groups injected with PBS. The results showed that the mice viability was 100%, and there was no significant difference between the control group and the treated group in terms of body weight after a one-month period (Fig. 5B). Collectively, the above results proved the low toxicity of the PLNPs–CuS hybrid nanoprobes.
3.4 Intracellular assay
Intracellular assay was performed to further study the luminescence recovery of PLNPs–CuS inside the cells. All the control (3T3 and HepG2) and treatment (HepG2 treated with AFT B1) groups were incubated with the PLNPs–CuS probe at 37 °C for 3 h. A remarkable luminescence recovery was detected in the treatment group due to the presence of AFT B1, whereas no recovery was observed in the control groups. Moreover, the recovered luminescence intensity increased with the concentration of AFT B1 that was used to treat the HepG2 cell lines before the addition of nanoprobes (Fig. 6). The above results inferred that the PLNPs–CuS nanoprobes could be readily internalized into the cancer cells and successfully activated by the target toxins inside the cells, offering the ability to selectively image AFT B1 in vitro.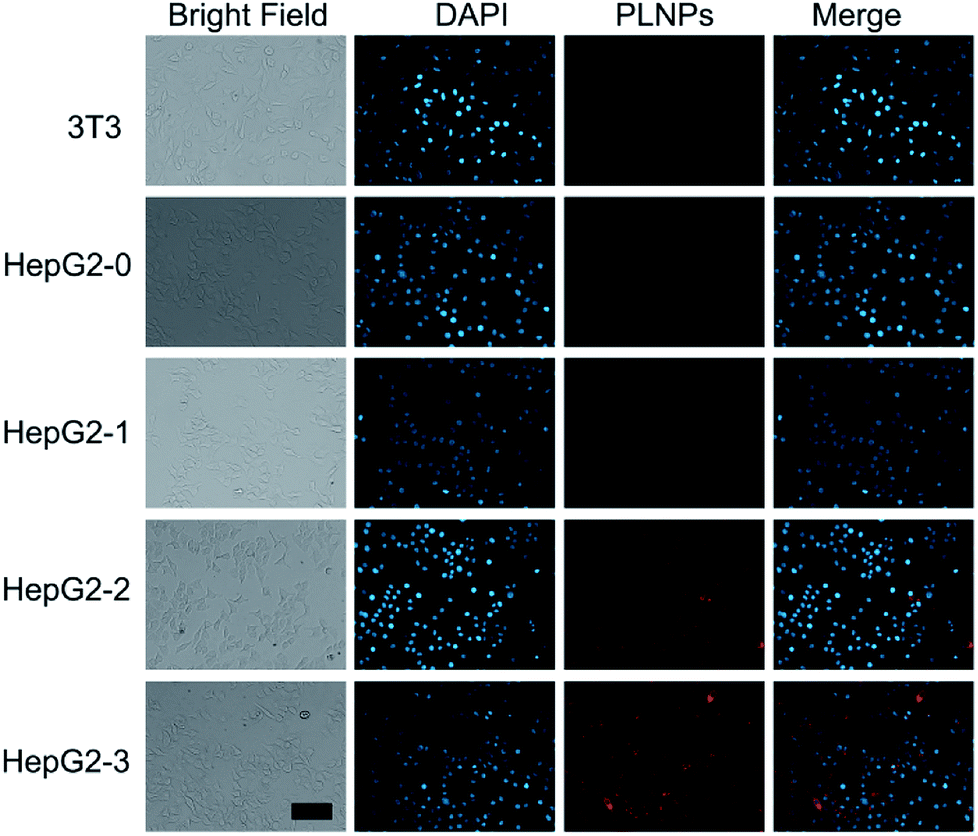 | ||
| Fig. 6 Intracellular assay of PLNPs–CuS nanoprobes for 3T3 cells and HepG2 cells treated with different amounts (0 μM, 5 μM, 10 μM, and 20 μM) of AFT B1. The scale bar represents 40 μm for all images. | ||
3.5 In vivo bioimaging
The above inspiring results of cell imaging assay encouraged us to apply PLNPs–CuS for in vivo imaging of AFT B1 to probe its behaviors and bio-distribution inside the living body. The PLNPs–CuS nanoprobe (0.4 mL, 1 mg mL−1) was pre-excited with a 324 nm-UV lamp for 10 min and intravenously administrated to the mice through the tail vein. Then, 1 min-red LED light (650 ± 10 nm) illumination was performed on the injected mice each time before acquiring the afterglow imaging photographs. Since there was no need for in situ excitation, no auto-fluorescence background was observed.In the control groups (healthy mice feed with normal food), minor luminescence signal was detected across the whole mouse body, indicating that the activatable nanoprobe was stable in the living body and would hardly be activated by non-specific interaction with biomolecules during circulation, thus offering a low probe background signal in the activatable optical imaging. In the treatment group (healthy mice fed with food containing 10 μg of AFT B1), significant luminescence was observed almost in the whole body and the most intense signal was observed in the liver, indicating the effective activation by the AFT B1 present in the living body that induced FRET inhibition. As time passed, the recovered luminescence was mainly focused on the liver site, which is probably a consequence of AFT B1 accumulation in the liver. The above results indicated that the proposed PLNPs–CuS FRET nanoprobe could be specifically activated by the target AFT B1 toxins present in vivo, favoring highly sensitive in vivo luminescence imaging with an improved signal-to-noise ratio.
To further confirm the luminescence bioimaging results, a bio-distribution study was performed via elemental analysis and ex vivo luminescence measurements. For the control groups (n = 5), ICP-MS elemental analysis of Ga (from PLNPs) and Cu (from CuS) within the organs that were collected 16 h and 24 h post injection implied that the nanoprobes mainly accumulated in the liver and spleen, which are the typical reticuloendothelial system organs, and increased with time. For the control groups and treatment groups (n = 5), elemental analysis of the organs collected 48 h post injection gave similar results. In contrast, low luminescence signal was detected in the liver of the control groups, while significant luminescence was found in the treatment groups, because the accumulated AFT B1 specifically activated the PLNPs–CuS nanoprobe by inhibiting the FRET process and recovering the luminescence. All the above results conclude that the proposed FRET nanoprobe is applicable for in vivo probing and investigation of food-borne toxins via activatable luminescence imaging (Fig. 7).
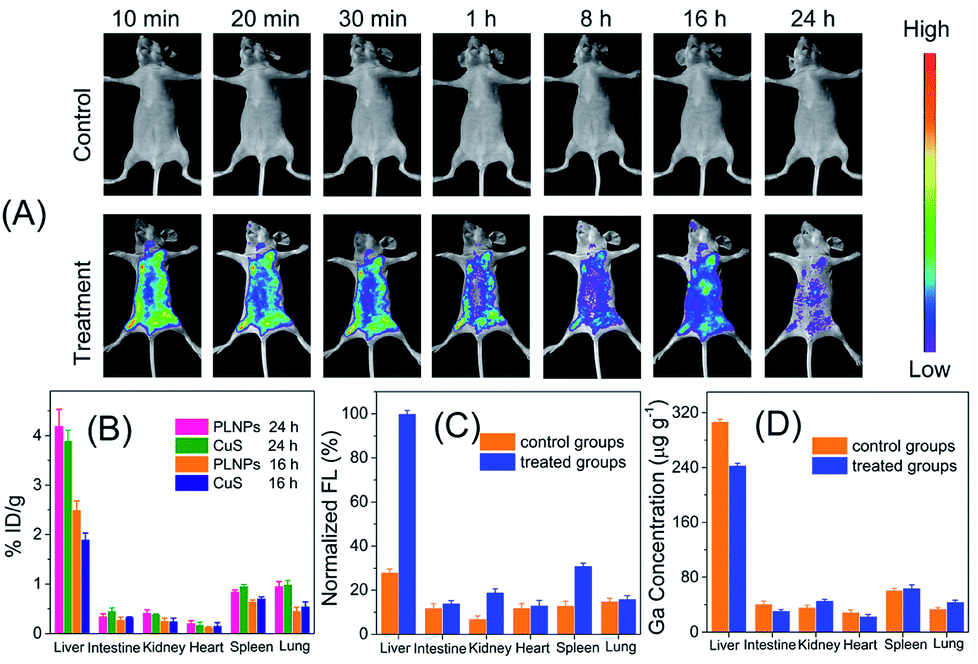 | ||
| Fig. 7 (A) In vivo luminescence imaging of nanoprobe (0.4 mL, 1.0 mg mL−1) injected mice with the control group and treatment group. (B) Bio-distribution of the nanoprobe in mice of control group against time. (C and D) Bio-distribution of the nanoprobe in mice measured by luminescence (C) and elemental analysis (D). | ||
4. Conclusions
In the present study, for the first time, NIR fluorescence bioimaging techniques have been successfully applied for in vivo probing of food-borne toxins inside the living body using the activatable FRET nanoprobe that employed PLNPs as an emission center and CuS as a quencher. The aptamer-involved FRET assay ensured specific determination of AFT B1 both in vitro and in vivo. The long-lasting afterglow and reactivatable luminescence of the PLNPs achieved long-term bioimaging with a high signal-to-noise ratio and improved imaging resolution for the target aflatoxin present in vivo. This study will open up a new path for the applications of bioimaging in food safety detection and provide an advisory roadmap of bioimaging-guided exploration and mediation of food-borne hazards to human health.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Key R&D Program of China (No. 2018YFD0400703), the Fundamental Research Funds for the Central Universities, and the China Postdoctoral Science Foundation (No. 2017M621059).Notes and references
- R. Romero-Gonzalez, Anal. Methods, 2015, 7, 7193–7201 RSC.
- H. L. Liu, G. Z. Fang, Q. L. Deng and S. Wang, Biosens. Bioelectron., 2015, 74, 313–317 CrossRef PubMed.
- A. Singh, S. Poshtiban and S. Evoy, Sensors, 2013, 13, 1763–1786 CrossRef PubMed.
- P. Koedrith, T. Thasiphu, K. Tuitemwong, R. Boonprasert and P. Tuitemwong, Sens. Mater., 2014, 26, 711–736 Search PubMed.
- L. Rotariu, F. Lagarde, N. Jaffrezic-Renault and C. Bala, TrAC, Trends Anal. Chem., 2016, 79, 80–87 CrossRef.
- A. Vasilescu and J. L. Marty, TrAC, Trends Anal. Chem., 2016, 79, 60–70 CrossRef.
- E. C. Alocilja and S. M. Radke, Biosens. Bioelectron., 2003, 18, 841–846 CrossRef PubMed.
- K. Tvrdy and M. S. Strano, Nat. Nanotechnol., 2012, 7, 8–9 CrossRef PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef PubMed.
- C.-H. Quek and K. W. Leong, Nanomaterials, 2012, 2, 92–112 CrossRef PubMed.
- T. Ueno and T. Nagano, Nat. Methods, 2011, 8, 642–645 CrossRef PubMed.
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef PubMed.
- B. R. Smith and S. S. Gambhir, Chem. Rev., 2017, 117, 901–986 CrossRef PubMed.
- X. He, K. Wang and Z. Cheng, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol., 2010, 2, 349–366 CrossRef PubMed.
- P. Zrazhevskiy, M. Sena and X. Gao, Chem. Soc. Rev., 2010, 39, 4326–4354 RSC.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef PubMed.
- J. Zhou, Z. Liu and F. Y. Li, Chem. Soc. Rev., 2012, 41, 1323–1349 RSC.
- S. K. Singh, RSC Adv., 2014, 4, 58674–58698 RSC.
- T. Maldiney, A. Bessière, J. Seguin, E. Teston, S. K. Sharma, B. Viana, A. J. J. Bos, P. Dorenbos, M. Bessodes, D. Gourier, D. Scherman and C. Richard, Nat. Mater., 2014, 13, 418–426 CrossRef PubMed.
- Z. Pan, Y.-Y. Lu and F. Liu, Nat. Mater., 2012, 11, 58–63 CrossRef PubMed.
- J.-M. Liu, Y.-Y. Liu, D.-D. Zhang, G.-Z. Fang and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 29939–29949 CrossRef PubMed.
- Q. l. M. de Chermont, C. Chaneac, J. Seguin, F. Pelle, S. Maitrejean, J.-P. Jolivet, D. Gourier, M. Bessodes and D. Scherman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9266–9271 CrossRef PubMed.
- Y. Li, M. Gecevicius and J. Qiu, Chem. Soc. Rev., 2016, 45, 2090–2136 RSC.
- L.-J. Chen, S.-K. Sun, Y. Wang, C.-X. Yang, S.-Q. Wu and X.-P. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 32667–32674 CrossRef PubMed.
- L. Zhang, J. Lei, J. Liu, F. Ma and H. Ju, Biomaterials, 2015, 67, 323–334 CrossRef PubMed.
- B.-Y. Wu, H.-F. Wang, J.-T. Chen and X.-P. Yan, J. Am. Chem. Soc., 2011, 133, 686–688 CrossRef PubMed.
- S.-Q. Wu, C.-X. Yang and X.-P. Yan, Adv. Funct. Mater., 2017, 27, 1604992 CrossRef.
- E. Teston, Y. Lalatonne, D. Elgrabli, G. Autret, L. Motte, F. Gazeau, D. Scherman, O. Clément, C. Richard and T. Maldiney, Small, 2015, 11, 2696–2704 CrossRef PubMed.
- J. Shi, X. Sun, J. Li, H. Man, J. Shen, Y. Yu and H. Zhang, Biomaterials, 2015, 37, 260–270 CrossRef PubMed.
- A. Abdukayum, J.-T. Chen, Q. Zhao and X.-P. Yan, J. Am. Chem. Soc., 2013, 135, 14125–14133 CrossRef PubMed.
- Z. Es'haghi, H. R. Beheshti and J. Feizy, J. Sep. Sci., 2014, 37, 2566–2573 CrossRef PubMed.
- H. E. Ok, F. Tian, E. Y. Hong, O. Paek, S.-H. Kim, D. Kim and H. S. Chun, Toxins, 2016, 8, 371 CrossRef PubMed.
- R. Zheng, H. Xu, W. Wang, R. Zhan and W. Chen, Anal. Bioanal. Chem., 2014, 406, 3031–3039 CrossRef PubMed.
- R. Zekavati, S. Safi, S. J. Hashemi, T. Rahmani-Cherati, M. Tabatabaei, A. Mohsenifar and M. Bayat, Microchim. Acta, 2013, 180, 1217–1223 CrossRef.
- Y.-J. Li and X.-P. Yan, Nanoscale, 2016, 8, 14965–14970 RSC.
- Y. Liu, J.-M. Liu, D. Zhang, K. Ge, P. Wang, H. Liu, G. Fang and S. Wang, J. Agric. Food Chem., 2017, 65, 8229–8240 CrossRef PubMed.
- X. Ran, F. Pu, J. Ren and X. Qu, Chem. Commun., 2015, 51, 2675–2678 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05555f |
| This journal is © The Royal Society of Chemistry 2018 |